

Abstract
Homologous recombination occurring among misaligned repeated sequences is a significant source of the molecular rearrangements resulting in human genetic disease. Studies of the Charcot-Marie-Tooth disease locus on chromosome 17 have implicated the involvement of an ancient DNA transposon of the mariner family (Hsmar2) in the initiation of double-strand break events leading to homologous recombination. In this study, the genomic locations of 109 Hsmar2 elements were determined by primed in situ labeling (PRINS) using primers designed to match the right and left inverted terminal repeats (ITRs) of the transposon. Although the resolution of the PRINS technique is ∼400 chromosomal Giemsa bands, the data presented here provide the first large-scale mapping study of these elements, which may be involved in initiation of homologous recombination events in the human genome.
The human genome is composed not only of DNA sequences coding for genes but also of a significant amount of noncoding or “junk” DNA. However, this noncoding DNA is not a static collection of insignificant DNA sequences. There appears, rather, to be a state of genome flux occurring among these sequences even in the span of a single human generation. Genome evolution models involving gene duplication and exon shuffling have been bolstered by the identification of active L1 elements in the human genome that can transpose not only portions of their own open reading frames but also fragments of surrounding coding sequence to new locations (Kzazian and Moran 1998). Short pericentromeric repeats on both human and other primate chromosomes may be involved in the transposition of duplicated exons or even entire genes to new locations (Eichler et al. 1997). In addition to these possibilities, the most extensively documented method of gene sequence rearrangement in the genomes of eukaryotes is homologous recombination. This kind of rearrangement is often mediated by region specific low-copy repeated sequences (Lupski 1998). Analysis of a well-documented hotspot for homologous recombination in the human genome revealed the presence of a mariner DNA transposon near the site of strand exchange for this recombination event (Kiyosawa and Chance 1996; Reiter et al. 1996). It was proposed that this element, and possibly copies of it located elsewhere in the genome, could be involved in the stimulation of homologous recombination among repeated sequences through the introduction of double-strand breaks in DNA near the region of strand exchange (Kiyosawa and Chance 1996; Reiter et al. 1996, 1998).
The presence of mariner DNA transposons of the irritans subfamily (Hsmar2) in the human genome is a rather recent discovery (Oosumi et al. 1995; Kiyosawa and Chance 1996; Reiter et al. 1996; Robertson and Martos 1997). Multiple copies of Hsmar2 elements have been identified in the EST (dbEST), nonredundant (nr), and high-throughput genomic sequencing (htgs) databases (Robertson and Martos 1997). Many of these copies are deleted for one inverted terminal repeat (ITR) and, therefore, are unlikely to bind a transposase, which appears to require two ITRs in concert for binding and cleavage. Although these elements appear to be inactive remnants of a functional mariner that entered the primate lineage some 80 million years ago, studies of homologous recombination at the Charcot-Marie-Tooth disease type 1A locus on chromosome 17p12 suggest that the presence of an Hsmar2 element within two flanking 24-kb repeats (CMT1A-REP) may stimulate unequal crossing-over events between misaligned CMT1A-REP elements (Kiyosawa and Chance 1996; Reiter et al. 1996, 1997, 1998). These unequal crossing-over events can be resolved as either a 1.5-Mb duplication resulting in Charcot-Marie-Tooth disease or a 1.5-Mb deletion resulting in hereditary neuropathy with liability to pressure palsies (Pentao et al. 1992; Chance et al. 1994). Chromosomal duplications and deletions mediated by large region-specific low-copy-number repeats may prove to be a common mechanism of chromosomal rearrangement leading to disease in the human genome (Lupski 1998) and have been implicated in the molecular mechanisms of a growing number of human genetic disorders such as Smith-Magenis syndrome (Chen et al. 1997), Prader-Willi/Angelman syndrome (PWS/AS) (Christian et al. 1995; Robinson et al. 1998), and Williams-Bueren syndrome (WS) (Urban et al. 1996; Pérez Jurado et al. 1998). Although a direct role for mariner elements in initiating recombination via the stimulation of double-strand breaks remains unclear, we were interested in determining if other Hsmar2 elements containing both a left and right ITR could be found at sites in the human genome where structural changes occur by an unequal crossing-over mechanism. To determine the genomic locations of elements containing both a left and right ITR among the ∼1000 copies of Hsmar2 in the human genome we localized these elements to specific chromosomal bands by primed in situ (PRINS) labeling.
RESULTS
Identifying the Locations of Hsmar2 Elements in the Human Genome
To locate Hsmar2 elements in the human genome we designed 50-bp oligonucleotides from the 3′ and 5′ ends of the consensus sequence for Hsmar2 (GenBank accession no. U49974). These oligonucleotides were used as primers for the fluorescence in situ hybridization (FISH)-related technique known as PRINS (Luke and Shepelsky 1998). The oligonucleotides were annealed to a male human metaphase chromosomal spread and extended with a mixture containing biotin-labeled deoxy-uracil triphosphate (dUTP). The biotin-labeled nucleotides were then detected using a thyramid detection system and viewed under the fluorescence microscope (Fig. 1A). Each chromosome was analyzed independently from 15 different chromosomal spreads to determine the number of signals per chromosome and their positions. Figure 1B shows this type of analysis for chromosome 17, which contains the CMT1A-REP region, the location of the element we previously called MITE for mariner insect transposon-like element. A total of 109 signals were detected using the left ITR oligonucleotide and 108 signals using the right ITR oligonucleotide. These signals are visually summarized in Figure 1C as the culmination of all signals detected on each chromosome. Note that the transposable elements appear to be spread evenly throughout the genome. This is apparent both by visual inspection of integration sites (Fig. 1C) and by correlation of the number of signals per chromosome with chromosome length. However, no elements were detected on the Y chromosome by this method. Table 1 summarizes the cytogenetic locations of all 109 positions of signals detected by PRINS, presumably representing one or more Hsmar2 elements, using both the left and right ITR oligonucleotide primers.
Figure 1.
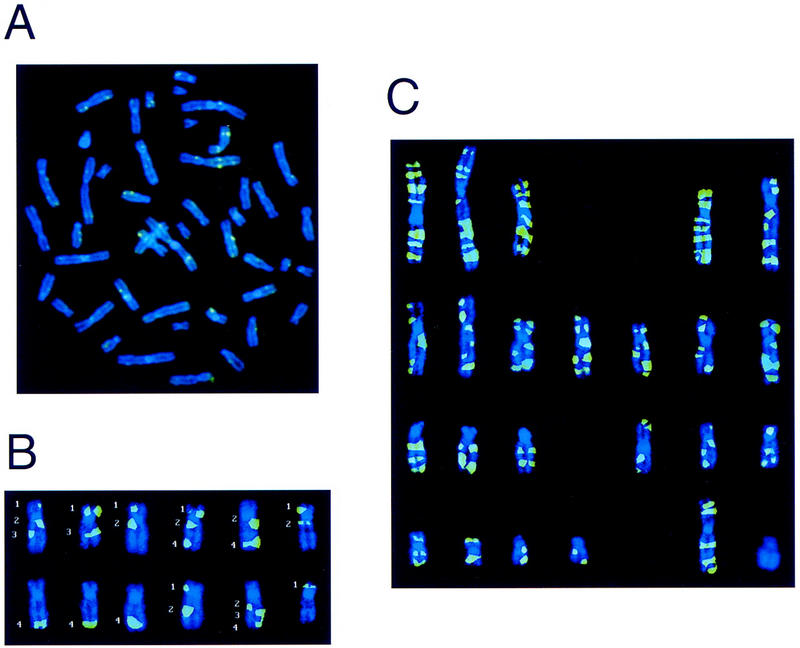
PRINS signals using the left and right ITRs of Hsmar2. (A) Raw PRINS data. This is an unenhanced chromosomal spread of the biotin-labeled left ITR. The green signals represent locations of Hsmar2 elements that contain left ITRs that match the consensus for Hsmar2 with enough homology to promote annealing and priming for the PRINS signal. (B) An example of the analysis of PRINS signals on chromosome 17. Twelve independent PRINS experiments are represented here by the 12 individual chromosome 17 images. The green signals are PRINS labeling using the left ITR. Note that not all chromosomes show all of the signals and that some chromosomes only show a signal for one homolog. This limitation of the technique requires the analysis of multiple chromosomal spreads. The signals indicated by numbers 1–4 are located at 17p12, 17q12, 17q22, and 17q24-25, respectively, at a resolution of 400 chromosomal Giemsa bands. (C) Summary of all left and right ITR PRINS data. This is a karyotype representing all loci that contain Hsmar2 elements by PRINS. The image was constructed by combining the PRINS signals for each individual chromosome into a composite image that was then added to other chromosomal images and arranged into the karyotype.
Table 1.
Cytogenetic Locations of Hsmar2 Elements Detected by PRINS
| Chromosome | No. of Signals | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 7 | 1p36 | 1p35 | 1p32 | 1p22 | 1q22–23 | 1q31–32 | 1q41–42 | ||
| 2 | 9 | 2p24–25 | 2p16 | 2p14 | 2q12 | 2q21 | 2q24 | 2q32 | 2q34 | 2q36 |
| 3 | 7 | 3p24–25 | 3p21 | 3p13 | 3q13 | 3q21 | 3q25–26 | 3q28 | ||
| 4 | 8 | 4p16 | 4p15 | 4p12 | 4q12–13 | 4q22–24 | 4q28 | 4q31 | 4q34 | |
| 5 | 6 | 5p15 | 5p13–14 | 5q11–12 | 5q14 | 5q23–31 | 5q34 | |||
| 6 | 6 | 6p24–25 | 6p21 | 6q12 | 6q16 | 6q24 | 6q26–27 | |||
| 7 | 7 | 7p21 | (7p14–15) | 7p12 | 7q11 | 7q21 | 7q31 | 7q32 | 7q35–36 | |
| 8 | 5 | 8p23 | 8p12–21 | 8q13–21 | 8q23 | 8q24 | ||||
| 9 | 5 | 9p23–24 | 9p12–13 | 9q12 | 9q22 | 9q33–34 | ||||
| 10 | 5 | 10p12–13 | 10q21.1 | 10q21.3 | 10q24–25 | 10q26 | ||||
| 11 | 5 | 11p15-ter | 11p13–14 | 11q13 | 11q22 | 11q24 | ||||
| 12 | 6 | 12p13 | 12p11 | 12q12 | 12q15–21 | 12q21–22 | 12q24 | |||
| 13 | 3 | 13q14 | (13q12–13) | 13q21–22 | 13q33 | |||||
| 14 | 3 | 14q12 | 14q24 | 14q32 | ||||||
| 15 | 2 | 15q12–14 | 15q23–24 | |||||||
| 16 | 3 | 16p13 | 16q21 | 16q23–24 | ||||||
| 17 | 4 | 17p12 | 17q12 | 17q22 | 17q24–25 | |||||
| 18 | 4 | 18p11 | 18q12–12 | 18q22 | 18q22–23 | |||||
| 19 | 3 | 19p13 | 19q13.1–13.2 | 19q13.3 | ||||||
| 20 | 2 | 20p13 | 20q13 | |||||||
| 21 | 2 | 21q22 | 21q13 | |||||||
| 22 | 2 | 22q11 | 22q13 | |||||||
| X | 5 | Xp22 | Xp11–21 | Xq13 | Xq25–26 | Xq28 | ||||
| Y | 0 | |||||||||
| Σ = | 109 | |||||||||
The locations of PRINS signals illustrated in Fig. 1C are summarized, as well as the locations of Hsmar2 elements detected by BLASTN searches of the nonredundant nucleotide database. Loci detected by PRINS alone are plain text, by BLASTN are boldface, and by BLASTN alone are in parentheses. The one PRINS signal that was detected by the left but not the right ITR oligonucleotide is underlined.
To demonstrate that the PRINS technique allows the detection of both repeated and unique sequences, control experiments were performed using a telomere-specific oligonucleotide and a chromosome X-specific oligonucleotide. The telomere-specific oligonucleotide detected only the telomeric repeat regions of all human chromosomes (Fig. 2A) whereas the X chromosome-specific oligonucleotide only detected a region on the X chromosome (Fig. 2B).
Figure 2.
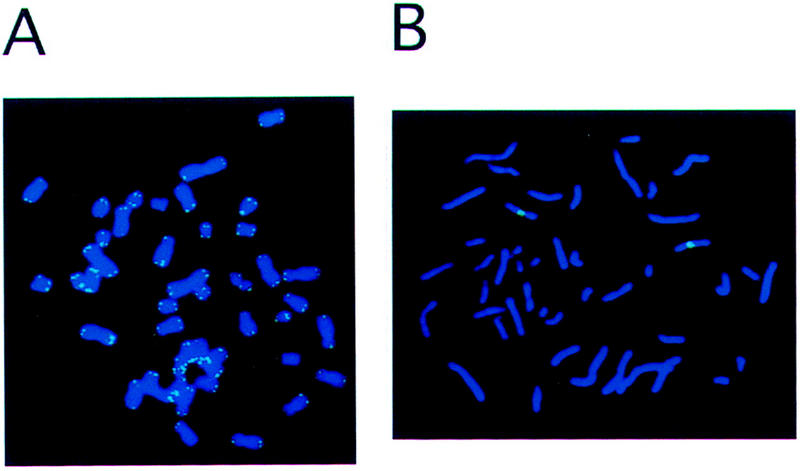
Confirmation of the PRINS technique specificity. (A) Telomere-specific signals. A 50-bp oligonucleotide designed to anneal to the repeated sequences of human telomeres was used for PRINS on a female human metaphase chromosomal spread. The green PRINS signals were detected at the telomeres of all human chromosomes. (B) X chromosome centromere-specific signal. A 50-bp oligonucleotide designed to anneal to a portion of the human X chromosome located at the centromere was used for PRINS on a female human metaphase chromosomal spread. Only two green PRINS signals were detected. These signals were located at the centromeres of the X chromosomes.
BLAST Searches and Genomic Disorders
As an independent confirmation of the ability to detect complete Hsmar2 elements by the PRINS technique, BLASTN searches were performed using the 50-bp left and right ITR oligonucleotides against the current nonredundant nucleotide database. Thirteen matches were identified in this database by BLASTN (Table 1, bold). These matches contained a left and right ITR and extended at least 22-bp with a perfect match for the last four 3′-end bases. These criteria were presumed to be enough homology to promote priming and extension. Only 2 of the 13 Hsmar2 elements detected by the BLASTN searches were not also detected by PRINS (Table 1, parentheses). Interestingly, within the limits of resolution of the technique (∼400 chromosomal Giemsa bands) the locations of PRINS signals coincide with the chromosomal map locations of twelve known genomic disorders that result from a homologous recombination event within misaligned repeated DNA sequences. The names of these diseases, chromosomal locations, types of rearrangement, and lengths of the repeating unit are summarized in Table 2.
Table 2.
Genomic Disorders That Coincide with the Locations of Hsmar2 Elements Detected by PRINS
| Genomic disease | Type of rearrangement | Length of repeat (bp) | Locus |
|---|---|---|---|
| 21-Hydroxylase deficiency | deletion | (?)a | 6p21.3 |
| Williams–Bueren syndrome | deletion | >30,000 | 7q11.2 |
| Glucocorticoid-remediable aldosteronism | duplication | 10,000 | 8q21 |
| Prader–Willi/Angelman syndrome | deletion | (?) | 15q13 |
| CMT1A/HNPP | duplication/deletion | 24,000 | 17p12 |
| Growth hormone deficiency | deletion | 2,200 | 17q22–24 |
| Debrisoquine sensitivity | deletion | 2,800 | 22q13.1 |
| X-Linked ichthyosis | deletion | 20,000 | Xp22.32 |
| α–Thalessemia | deletion | 4,000 | Xq13 |
| Color blindness | deletion | 39,000 | Xq28 |
| Hunter mucopolysaccharidosis | inversion | 3,000 | Xq28 |
| Hemophila A | inversion | 9,500 | Xq28 |
(?) The length of the repeat involved is unknown.
DISCUSSION
We have demonstrated by PRINS that multiple copies of the mariner element Hsmar2 are spread throughout the human genome. At least 108 of these copies appear to contain both a left and right inverted terminal repeat (Table 1). The genomic locations of these elements were easily identified by PRINS prior to the completion of the human genome project. Although there is still no direct evidence for the involvement of mariner elements in the stimulation of homologous recombination in humans, we provide here some preliminary evidence that the locations of these elements may coincide with at least 12 locations where homologous recombination is the proposed molecular mechanism responsible for the human disease phenotypes (Table 2). It should be noted, however, that at this resolution the correlation between Hsmar2 elements and genomic disorders is not statistically significant. Further studies of the submicroscopic regions containing the elements identified in this study will be necessary to determine if mariner has a role in promoting homologous recombination in humans.
The Hsmar2 element previously called MITE (Reiter et al. 1996) was identified in the CMT1A region (17p12). In addition, an Hsmar2 element was identified in the PWS/AS region (15q13) where large deletions are also a common disease mechanism (Christian et al. 1995; Robinson et al. 1998) and in the WS region (7q11) where flanking repeat sequences have been implicated as a molecular mechanism for disease (Urban et al. 1996; Pérez Jurado et al. 1998). Further studies of these traveling mariner genes in the human genome may not only illuminate our understanding of how molecular rearrangements involving repeated DNA sequences can cause human disease but also may reveal the manner in which the genome itself continues to evolve.
METHODS
PRINS Labeling and Detection
Primers used for PRINS analysis were designed based on the published consensus sequence for Hsmar2 constructed by Hugh Robertson (accession no. U49974). The left ITR primer begins at the 5′ end of Hsmar2 and ends at base 50 of the consensus sequence whereas the right ITR primer was designed from the complementary strand of Hsmar2 beginning at base 1309 and ending 50 bases internal to the element at base 1279. Primers designed to the open reading frame of the Hsmar2 element were not used because this region is highly variable among the elements examined in the databases. Peripheral blood lymphocytes from a healthy male donor were used for PRINS analysis using the left and right ITR oligonucleotides. Lymphocytes from a healthy female donor were used for the X-chromosome and telomere-specific oligonucleotides. Cells in metaphase were denatured on microscope slides in denaturation solution (80 μl of deionized formamide, 10 μl of ddH20, 10 μl of 20× SSC) covered with a 24 × 50-mm coverslip and heated to 75°C on a heating plate for 5 min. The coverslip was then removed and the slides were subjected to a series of ethanol washes (70%/90%/100%) and air dried. The 50-μl PRINS reaction solution was then added to each slide: 3 μl each of 10 mm dATP, dCTP, and dGTP; 1 μl of 10 mm dTTP; 3 μl of 10 mm biotin-dUTP; 1 μl of 1 pm/μl primer; 1 μl 10× BSA; 2 μl 25 mm MgCl2; 0.5 μl Thermus aquaticus (Taq) polymerase; 10× Taq polymerase buffer; 27.5 μl ddH20. All reagents were from Pharmacia except for biotin-dUTP and BSA (Boehringer Mannheim). Slides were then sealed with a coverslip and rubber cement by warming to 40°C. Incorporation of labeled dUTP was accomplished by initial denaturation of the DNA for 5 min at 94°C followed by a 50-min annealing/extension period at 58°C for the ITR primers, 55°C for the telomere specific primer, and 60°C for the X-chromosome primer. The reaction was stopped by removing the cover slip and soaking the slides at 60°C in 500 ml of stop solution (50 mm EDTA, 5 mm NaCl in ddH20). Detection of the biotin labeled oligonucleotides was performed using the thyramid detection system (TSA-Kit; MEL731A, NEN-Dupont) according to the manufacturer’s instructions.Metaphase spreads were counterstained with DAPI (diamidinophenylindol) solution, 20 μl of the antifade Vectashield (Vectorlabs) was added to the slide, and it was covered with a coverslip. An enhanced DAPI banding pattern was derived by 5-bromo-2-deoxyuridine (BrdU) incorporation into the chromosomes during cultivation of the lymphocytes used for PRINS (Heng and Tsui 1993). The DAPI banding pattern indicated the exact subchromosomal localizations of the probes. Evaluation was done on a Zeiss Axioplan fluorescence microscope and pictures were taken using a CCD-camera and the software of MetaSystems (Altlussheim, Germany).
Acknowledgments
We thank P.J. Hastings and J. Rosen for their critical review of the manuscript. This work was supported in part by grants from the National Institute of Neurological Disorders and Strokes, National Institutes of Health (RO1NS2742) and the Muscular Dystrophy Association (M.D.A.) to J.R.L. L.R. was a fellow of the Boehringer Foundation and the CMT Association (CMTA) during the course of this study. B.R. is supported by grants from the Deutsche Forschungsgemeinschaft (DFG), Deutsche Gesellschaft Muskelkranke (DGM), and Wilhelm-Sander Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jlupski@bcm.tmc.edu; FAX (713) 798-5073.
REFERENCES
- Chance PF, Abbas N, Lensch MW, Pentao L, Roa BB, Patel PI, Lupski JR. Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet. 1994;3:223–228. doi: 10.1093/hmg/3.2.223. [DOI] [PubMed] [Google Scholar]
- Chen K-S, Manian P, Koeuth T, Potocki L, Zhao Q, Chinault AC, Lee CC, Lupski JR. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- Christian SL, Robinson WP, Huang B, Mutirangura A, Line MR, Nakao M, Surti U, Chakravarti A, Ledbetter DH. Molecular characterization of two proximal deletion breakpoint regions in both Prader-Willi and Angelman syndrome patients. Am J Hum Genet. 1995;57:40–48. [PMC free article] [PubMed] [Google Scholar]
- Eichler EE, Budarf ML, Rocchi M, Deaven LL, Doggett NA, Baldini A, Nelson DL, Mohrenweiser HW. Interchromosomal duplications of the adrenoleukodystrophy locus: A phenomenon of pericentromeric plasticity. Hum Mol Genet. 1997;6:991–1002. doi: 10.1093/hmg/6.7.991. [DOI] [PubMed] [Google Scholar]
- Heng HHQ, Tsui L-C. Modes of DAPI banding and simultaneous in situ hybridization. Chromosoma. 1993;102:325–332. doi: 10.1007/BF00661275. [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Moran JV. The impact of L1 retrotransposons on the human genome. Nat Genet. 1998;19:19–24. doi: 10.1038/ng0598-19. [DOI] [PubMed] [Google Scholar]
- Kiyosawa H, Chance PF. Primate origin of the CMT1A-REP repeat and analysis of a putative transposon-associated recombinational hotspot. Hum Mol Genet. 1996;5:745–753. doi: 10.1093/hmg/5.6.745. [DOI] [PubMed] [Google Scholar]
- Luke S, Shepelsky M. FISH: Recent advances and diagnostic aspects. Cell Vision. 1998;5:49–53. [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Oosumi T, Belknap WR, Garlick B. Mariner transposons in humans. Nature. 1995;378:672. doi: 10.1038/378672a0. [DOI] [PubMed] [Google Scholar]
- Pentao L, Wise CA, Chinault AC, Patel PI, Lupski JR. Charcot-Marie-Tooth type 1A duplication appears to arise from recombination at repeat sequences flanking the 1.5 Mb monomer unit. Nat Genet. 1992;2:292–300. doi: 10.1038/ng1292-292. [DOI] [PubMed] [Google Scholar]
- Pérez Jurado LA, Wang YK, Peoples R, Coloma A, Cruces J, Francke U. A duplicated gene in the breakpoint regions of the 7q11.23 Williams- Beuren syndrome deletion encodes the initiator binding protein TFII-I and BAP-135, a phosphorylation target of BTK. Hum Mol Genet. 1998;7:325–334. doi: 10.1093/hmg/7.3.325. [DOI] [PubMed] [Google Scholar]
- Reiter LT, Murakami T, Koeuth T, Pentao L, Muzny DM, Gibbs RA, Lupski JR. A recombination hotspot responsible for two inherited peripheral neuropathies is located near a mariner transposon-like element. Nat Genet. 1996;12:288–297. doi: 10.1038/ng0396-288. [DOI] [PubMed] [Google Scholar]
- Reiter LT, Murakami T, Koeuth T, Gibbs RA, Lupski JR. The human COX10 gene is disrupted during homologous recombination between the 24 Kb proximal and distal CMT1A-REPs. Hum Mol Genet. 1997;6:1595–1603. doi: 10.1093/hmg/6.9.1595. [DOI] [PubMed] [Google Scholar]
- Reiter L, Hastings PJ, Nelis E, De Jonhge P, Van Broekhoven C, Lupski JR. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am J Hum Genet. 1998;62:1023–1033. doi: 10.1086/301827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM, Martos R. Molecular evolution of the second ancient human mariner transposon, Hsmar2, illustrates patterns of neutral evolution in the human genome lineage. Gene. 1997;205:219–228. doi: 10.1016/s0378-1119(97)00471-x. [DOI] [PubMed] [Google Scholar]
- Robinson WP, Dutly F, Nicholls RD, Bernasconi F, Penaherrera M, Michaelis RC, Abeliovich D, Schinzel AA. The mechanisms involved in formation of deletions and duplications of 15q11-q13. J Med Genet. 1998;35:130–136. doi: 10.1136/jmg.35.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban Z, Helms C, Fekete G, Csiszsár K, Bonnet D, Munnich A, Donis-Keller H, Boyd CD. 7q11.23 deletions in Williams syndrome arise as a consequence of unequal meiotic crossover. Am J Hum Genet. 1996;59:958–962. [PMC free article] [PubMed] [Google Scholar]