

Abstract
Iron and its homeostasis are intimately tied to the inflammatory response. The adaptation to iron deficiency, which confers resistance to infection and improves the inflammatory condition, underlies what is probably the most obvious link: the anemia of inflammation or chronic disease. A large number of stimulatory inputs must be integrated to tightly control iron homeostasis during the inflammatory response. In order to understand the pathways of iron trafficking and how they are regulated, this chapter will present a brief overview of iron homeostasis. A major focus will be on the regulation of the peptide hormone hepcidin during the inflammatory response and how its function contributes to the process of iron withdrawal. The review will also summarize new and emerging information about other iron metabolic regulators and effectors that contribute to the inflammatory response. Potential benefits of treatment to ameliorate the hypoferremic condition promoted by inflammation will also be considered.
Keywords: hepcidin, anemia of chronic disease, iron withdrawal, iron overload, hemochromatosis, infection
I. Links between iron and inflammation
The rapid evolution of molecular information about iron transport and homeostasis has uncovered a comprehensive understanding of the complex mechanisms involved in this process. It has long been recognized that iron levels must be tightly regulated to provide an essential nutrient that is involved in oxygen delivery, metabolism and redox regulation while guarding against excessive levels of a primary toxicant that can generate reactive oxygen species (ROS) to produce cellular damage and death. Unlike other essential minerals, the delicate balance between iron nutrition and toxicity is maintained by systemic control mechanisms that drive iron conservation and limit uptake until needs are presented – in contrast, homeostasis of many other metals is more simply controlled by eliminating excess. Ultimately, these features of iron and its homeostasis are intimately tied to the response to inflammation and infection and therefore provide major survival mechanisms that are unique in human physiology.
A large body of clinical evidence demonstrates disease susceptibility and the response to infection and inflammation worsen with elevated iron stores. The relationships between iron overload and infectious diseases are particularly well documented. For tuberculosis, it has been demonstrated that parental or oral iron increase mycobacterial growth (66, 80) and that morbidity and mortality increase in patients receiving iron supplementation (91, 120). In fact, dietary iron is associated with occurrence and death from tuberculosis (44). Malaria is a second example of an infectious disease that is strongly influenced by host iron status (62, 68), and the wisdom of iron supplementation and how its judicious use might best be applied in endemic regions is currently debated (118). Iron overload associated with hereditary hemochromatosis has been reported to confer susceptibility to other infectious pathogens, such as Yersinia enterocolitica and Vibrio vulnificus (10, 28, 50). Infections are more frequent in thalassemic patients with iron overload induced by frequent blood transfusions (49, 105). Iron status also appears to strongly influence the course of viral infections (30). In general, iron deficiency confers relative resistance to infection with high iron status promoting the disease state (88). An interesting evolutionary perspective is that iron deficiency may be a protective adaptive response in areas of the world with a higher burden of infectious disease (26).
Inflammatory chronic diseases are also profoundly influenced by iron status. Increased iron stores are correlated with markers of chronic inflammation and other well-established risk factors of diabetes, obesity, and metabolic syndrome (26, 40, 43, 60, 119, 152). Atherosclerosis (171), neurodegeneration (54) and chronic liver disease (158) are frequently associated with iron loading. This growing body of evidence is significant not only due to the prevalence of chronic diseases in modern society, but also because modified dietary iron or manipulation of iron status could represent simple preventive or therapeutic avenues (59, 74). For example, improvement has been found upon reduction of iron stores in diabetics by phlebotomy or iron chelation suggesting new approaches to disease management (8, 35). It is worth noting that iron chelation and deprivation also has been applied in cancer therapy as well (121, 147).
The adaptation to iron deficiency, which confers resistance to infection and improves the inflammatory condition, underlies what is probably the most obvious link between iron and the inflammatory response: the anemia of inflammation (AI), also called the anemia of chronic disease (ACD). Characteristics of the hypoferremic response to inflammation were documented in the 1940s by Cartwright and colleagues (13–15). Today we recognize that the iron regulatory hormone hepcidin is a central player in the coordinated response to reduce systemic iron levels. It is increasingly clear that a large number of stimulatory inputs must be integrated to tightly control hepcidin gene expression during the inflammatory response to modulate AI and make appropriate adjustments to iron homeostasis. In order to understand the pathways of iron trafficking and how they are regulated, this chapter will present a brief overview of iron homeostasis. Its major focus will be on the regulation of hepcidin during the inflammatory response and how its function contributes to the process of iron withdrawal. The review will also summarize new and emerging information about other iron metabolic regulators and effectors that contribute to the inflammatory response. Finally, potential benefits of treatment to ameliorate the hypoferremic condition promoted by inflammation will be considered.
II. Iron homeostasis: a brief overview
The majority of iron necessary to fulfill primary needs of the body is recycled from senescent red blood cells by the reticuloendothelial system (RES). The conservation of iron is offset by additional nutritional and/or environmental demands (e.g., pregnancy, blood loss, hypoxia, etc.) to ultimately dictate the body’s total iron burden. This fine-tuning is primarily adjusted through iron absorption by duodenal enterocytes. In some disease states, e.g. hereditary hemochromatosis, homeostatic regulation is miscued such that excess iron enters the system and balance is shifted towards overload status. Conversely, iron deficiency evolves during the anemia of inflammation or chronic disease by limiting iron absorption and retaining the metal in the RES.
In general, the flux of iron through duodenal enterocytes and macrophages of the RES involves a similar cohort of proteins and functions. The major intestinal iron importer is divalent metal transporter DMT1 (38, 55) which is thought to require an associated ferrireductase activity to reduce Fe(III) to Fe(II). DcytB has been proposed to fulfill this function (84). Transfer of iron across the intestinal mucosa involves basolateral export by ferroportin (29, 85, 170) along with its oxidation by membrane-bound hephaestin (157) or circulating ceruloplasmin (24). Ultimately, absorbed iron is bound by serum transferrin (Tf) for delivery to peripheral tissues by receptor-mediated endocytosis of transferrin receptor-1 (TfR1). Release of iron from the endosome also requires membrane translocation by DMT1 (37) or other transporters (48), along with the action of a ferrireductase called STEAP3 (133). For RES macrophages, iron can be delivered not only by Tf-TfR1, but also through erythrophagocytosis. Despite differences in iron acquisition, macrophages export iron in a manner that is similar to duodenal enterocytes in that ferroportin mediates this transport activity in conjunction with ceruloplasmin.
The regulation of the elements involved in the movement of iron through various compartments is critical to iron homeostasis. A central role is played by ferroportin expression since this transporter mediates entry of iron into circulating pools from both enterocytes (dietary absorption) and macrophages (RES recycling of heme iron obtained from destruction of red blood cells). Flux from the latter mechanism (~ 20 mg/day) is much greater than across the intestine (~ 2 mg/day), and this internal checkpoint provides a means for immediate control over available and circulating serum iron with more modest adjustments made to entry of iron from the diet to match longer term needs. Ferroportin expression is regulated at several levels, including transcriptional and post-transcriptional events (29, 65, 85, 170), but a primary source of regulatory control is exerted by the peptide hormone hepcidin. Hepcidin binds to ferroportin thereby inducing its internalization and degradation (97). The hepcidin-ferroportin axis of control presents a simple and elegant on-off switch for export of iron across the intestinal mucosa and from RES macrophages containing phagocytosed iron from senescent red blood cells. Thus far, the only known consequence of hepcidin’s action is the post-translational down-regulation of ferroportin. Circulating hepcidin levels are generally regulated by total body iron status and respond to physiological demands. Iron deficiency and hypoxia – conditions that place iron demand – are reflected in low hepcidin while iron loading increases its synthesis (41, 99, 101). Importantly, infection and inflammation are also associated with increased hepdicin production and these events ultimately link iron homeostasis to the inflammatory response through the acquired anemia of inflammation or chronic disease.
III. Anemia of inflammation or chronic disease
The upregulation of hepcidin by inflammatory stress response pathways is a major critical event triggering systemic iron withdrawal and sequestration due to its down-regulation of ferroportin. Reduced ferroportin levels limit dietary iron absorption and promote iron retention by the RES (45, 125). Characteristics of AI or ACD include hypoferremia, low serum iron-binding capacity, and normal-to-elevated ferritin levels. Reduced iron availability results in iron-restricted erythropoiesis. Mice that over-express hepcidin recapitulate this response by developing iron deficiency anemia (100, 127). Patients with glycogen storage disease type 1a (GSD1a) who have hepatic adenomas are thought to suffer from anemia due to over-expressed hepcidin. The fact that anemia resolves when tumors are removed supports the physiological importance of hepcidin (164). Despite the large contribution of hepcidin, it should be recognized that other stress response pathways also participate in the inflammatory response, including cytokine-mediated affects elicited by tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) (165). Uncovering the molecular networks involved is critical to understanding the integration of different pathways into the global response of iron status to inflammation (58).
IV. Hepcidin
Hepcidin was initially identified as liver-derived antimicrobial peptide (LEAP-1) (69, 109, 116) and immediately uncovered as a major product released in response to inflammatory cytokines, in particular interleukin-6 (IL-6) (32, 96, 98, 101). The peptide is also produced by cells of the immune system (e.g., macrophages and neutrophils) during the innate immune response through a toll-like receptor-4 (TLR-4) dependent pathway (112, 140, 144). In vitro studies have shown that other cytokines, including IL-1β, stimulate hepcidin production (75–76, 83), and its up-regulation by activation of TLR-2 has been demonstrated (67). Hepcidin circulates bound to α2-macroglobulin (110) and assays for serum levels of the peptide are available (46). While hepcidin expression responds to inflammatory stimuli, appropriate adjustments in levels of the iron-regulatory hormone must also reflect the regulation of body iron status to direct the correct level of ferroportin iron export activity. The importance of hepcidin in iron homeostasis is underscored by the fact that common genetic defects leading to hereditary hemochromatosis involve disruption of its expression and/or function (77). Thus, the inflammatory response must be coordinated with many other cues that regulate iron balance. The major mechanistic elements integrating the response to inflammation are summarized below.
a. STAT3 regulation
Inflammatory regulation of hepcidin gene expression involves transcriptional control through a signal transducer and activator of transcription (STAT) site on the hepcidin promoter (154, 167). This binding motif appears to be regulated by STAT3. IL-6 is a major hepatic regulator of the acute phase response to inflammatory stimuli. Ligand binding to the IL-6 receptor in turn leads to activation of Janus kinases that phosphorylate STAT3. Translocation of STAT3 to the nucleus results in upregulation of hepcidin gene expression, effectively promoting hypoferremia through down-regulation of ferroportin iron export activity from duodenal enterocytes and RES macrophages. It has been further proposed that STAT3 activation itself, without inflammation, can regulate hepcidin levels. Wrighting and Andrews (167) have suggested GSD1a patient adenomas may alter STAT3 activity to induce hepcidin expression since inflammatory cytokines do not fully explain the associated anemia (164). This concept is supported by RNAi knockdown of STAT3 in hepatoma cells, which alone is sufficient to reduce basal levels of hepcidin expression (154). STAT1 also has been implicated in control of hepcidin synthesis (139, 148).
b. Hemojuvelin (HJV) regulation
An inherited juvenile form of hemochromatosis is produced by mutations in HJV (73, 107, 115). HJV is a coreceptor for bone morphogenic proteins (BMPs) and it has been demonstrated that the HJV/BMP complex regulates hepcidin expression (3). Stimulation of hepcidin by BMPS 2, 4 and 9 has been reported in vitro (149). BMPs are members of the transforming growth factor-β (TGF-β) family and signal through receptor phosphorylation of Smad proteins. Smads translocate to the nucleus and activate gene transcription. The importance of this pathway in iron homeostasis was revealed through studies of liver-specific Smad4 deficient mice, which display iron overload and impaired hepcidin expression (161). Interestingly, IL-6 induction of hepcidin gene expression appears inactive when signaling of hepatic Smad4 is impaired (161), and LPS-induced hepcidin up-regulation is attenuated by deficiency in BMP6 (87), indicating that Smad signaling plays a key role in staging the appropriate response to inflammation. In contrast, experiments using IL-6−/− mice suggest that cytokine signaling is not required for BMP regulation of hepcidin transcription (149). Recent studies of the hepcidin promoter have further established a theoretical framework for transcriptional activation involving crosstalk between the STAT binding site and two BMP-responsive elements (BMP-REs) in proximal (BMP-RE1) and distal (BMP-RE2) regions of the murine promoter (16). Truska et al. (148) propose that physical interactions between these proximal and distal elements in the murine promoter’s organizing structure lead to hierarchical regulation. BMP-RE1 is close to the STAT-binding site and appears to be the critical element defining cross-talk between the HJV/BMP pathway and the inflammatory response (16, 148). One of the conundrums remaining to be resolved is the observation that HJV itself is a target of inflammatory regulation (70); in particular, HJV levels appear to be regulated by the pro-inflammatory cytokine TNF-α (20). Reduced HJV should counteract its signaling influence as a BMP co-receptor during inflammation and thus a complete explanation for the observed response to TNF-α remains to be developed. The recent discovery of matriptase-2/TMRSS6 and its suppressive effects on hepcidin synthesis through HJV cleavage creates additional complexity for understanding how the HJV/BMP signaling mechanism integrates with inflammatory control (31, 36, 137); moreover, how signals from the erythrokines GDF15 and TWSG1 modulate BMP signaling in response to ineffective erythropoiesis must also be integrated to gain a comprehensive understanding of the anemia of inflammation (142–143). Both are members of the TGF-β family and are released under conditions of iron-restricted erythropoiesis.
c. Transferrin (Tf) and HFE
The major form of hereditary hemochromatosis, called Type I hemochromatosis, results from variations in HFE (34). HFE associates with both TfR1 and TfR2, but it is thought that the HFE/TfR2 complex senses Tf saturation to modulate hepcidin transcription (47). Further studies have suggested that BMP signaling is independent of HFE and TfR2, but the precise mechanism of regulation of gene expression for HFE/TfR2 signaling remains unknown (149). Loss of HFE function results in reduced hepcidin levels and increased ferroportin levels, lowering iron levels in monocytes and macrophages (12, 90). In terms of the effects of HFE on the hepcidin inflammatory response, results from studies of Hfe−/− mice have been mixed: one report indicated these mice fail to induce appropriate levels of hepcidin in response to inflammatory stimuli (126), while other groups noted that isolated Hfe−/−mouse hepatocytes did respond to IL-6 in vitro (75) and that knockout mice do develop hypoferremia during acute phase response (42). Recent mechanistic studies of Hfe knockout mice have demonstrated impaired inflammatory responses due to the lower levels of macrophage iron in these animals (160). In response to infection, Hfe−/−mouse macrophages produce less TNF-α and IL-6, important pro-inflammatory cytokines. A similar effect is reported for human patients with HFE-associated hemochromatosis since their monocytes produce less TNF-α in vitro (52). This general defect can be recapitulated in a macrophage model system over-expressing ferroportin, and in wild-type macrophages treated with iron chelators (160). Under these reduced iron conditions, translation of the cytokine mRNAs was blocked, an effect that involves TLR4 signaling (159). In vivo, reduced cytokine production is associated with attenuated inflammatory response to Salmonella infection (160), revealing an underlying cause for susceptibility of patients with Type I hemochromatosis to infections like Salmonella, Vibrio, and Yersinia (10, 28).
d. Hypoxia-inducible factors (HIFs)
HIFs are multi-subunit transcription factors regulated by hydroxylation of an unstable α subunit controlling its degradation. This modification involves prolylhydroxylases requiring both oxygen and iron, thus under hypoxic or low iron conditions, HIFα subunits are stabilized. HIF2α has emerged as an important regulator of dietary iron absorption through the transcriptional control of intestinal DMT1 expression (82, 134). Since hepcidin levels decrease in response to hypoxia (101), HIF regulation is an attractive control mechanism for its function under low oxygen conditions, however, it does not appear to be directly responsible for this effect (156). An alternate hypothesis for indirect effects is HIF induction of the protease furin with subsequent release of a soluble circulating form of HJV predicted to down-regulate hepcidin during hypoxia or exercise (136), but the physiological relevance of this speculative pathway remains unknown. It has been established that HIF1α plays an important role in immunity, and myeloid-specific disruption of its function results in increased susceptibility to infection and impaired cell-mediated inflammation (22, 111). Liver-specific disruption of HIF1α also interferes with hepcidin regulation, further implicating a role for this transcription factor family in the regulation of hepdicin through the inflammatory response (113). HIF1α itself is transcriptionally controlled by the master immunoregulator NF-κB (122). In turn, the HIFs are major regulators of a number of genes involved in iron homeostasis and inflammation, including TfR1, heme oxygenase-1, Nramp1, DMT1, erythropoietin, Dcytb, and ferroportin. Many of these factors directly contribute to mechanisms mediating the iron withdrawal response to inflammation as discussed later in this chapter. Nonetheless, the precise mechanism of HIF regulation of hepcidin levels remains controversial and needs further investigation.
e. Endoplasmic reticulum (ER) stress response
Stress pathways signaling through the cellular ER unfolding protein response (UPR) have also been found to induce hepcidin expression. The UPR has been implicated in the hepatic acute phase response to lipopolysaccharide (LPS), IL-6, and IL-1β (86, 172) suggesting that hepcidin gene expression might be regulated through an additional layer of endogenous control. Two transcription factors have been implicated: cAMP-responsive element binding protein H (CREBH) (153) and CCAAT/enhancer-binding protein (C/EBP) homologousprotein (CHOP), which is a negative regulator of C/EBPα (103). The ER stressors homocysteine and dithiothreitol induce the UPR response, and have been shown to modulate hepcidin expression in a biphasic pattern through changes in the level of CHOP. Ultimately, down-regulation of CHOP relieves inhibition of C/EBPα to increase hepcidin transcription (103). This effect is supported by the independent evidence suggesting that C/EBPα is a transcriptional regulator of hepcidin (21, 56, 83) and that it may serve as a modulator of hepcidin synthesis via erythropoietin signaling of enhanced erythropoiesis (117)
CREBH appears to act in a different pathway induced by ER stress. Activation of CREBH by UPR stress promotes expression of acute phase response genes. Recent studies demonstrate this transcription factor also influences hepcidin expression possibly along with a second ER stress-activated transcription factor, XBP-1 (153). Treatment of CREBH knockout mice with ER stressors fails to induce hepcidin expression to the same level as wild-type mice and although LPS can elicit a hepcidin response in the knockout animals, it is lower than controls (153). The response to ER stress may also reflect the regulation of hepcidin transcription through the STAT pathway, suggesting once again that multiple stimuli coordinate promoter activity.
f. Heme Regulated Inhibitor (HRI)
A generalized response to stress is inhibition of translation initiation through phosphorylation of eukaryotic initiation factor 2a (eIF2a). For example, pathways of ER stress response include the activation of pancreatic ER kinase (PERK) via the UPR. PERK not only causes inhibition of translation, but also promotes expression of genes with upstream open reading frames like activating transcription factor 4 (ATF4), which in turn regulates factors like CHOP (145). Stress associated with heme deficiency activates a similar eIF2a kinase called HRI (heme regulated inhibitor), which is known to block globin synthesis in certain anemias (17). Although HRI is expressed mainly in erythroid precursors, studies of Hri knockout mice reveal that it plays a role in macrophage iron recycling and iron homeostasis (79). Interestingly, loss of HRI activity is associated with reduced inflammatory response and lower levels of hepcidin upon LPS challenge (79). Although the precise molecular mechanism responsible for this effect remains to be fully elucidated, perhaps the loss of HRI may interfere with eIF2a stress regulation to alter CHOP regulation of hepcidin.
g. Summary
Figure 1 depicts the various inputs hypothesized to regulate transcription of hepcidin and their activation by inflammatory signals. Much of the work to date has focused on the murine promoter and reporter analysis has been extensively carried out using liver-derived cell lines or exogenous expression systems (149). There are caveats to such studies since although commonly used liver cell lines like HepG2 cells are IL-6 and BMP-responsive (149), they do not effectively respond to Tf/HFE signaling (47), and may not fully recapitulate features of systemic iron regulation. How metabolic stress response signals are integrated to control hepcidin synthesis must be understood to elucidate the hierarchy regulating its activity.
Figure 1. Regulators of hepcidin synthesis during inflammation.
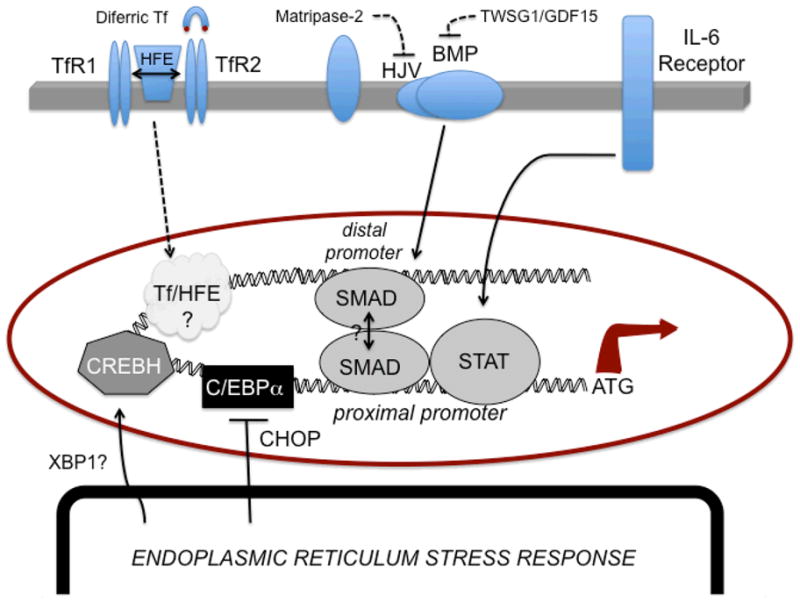
Activation of STAT1/3 by cytokine signaling promotes transcription through a STAT element. IL-6 plays a predominant role in this inflammatory response. This element appears to be regulated through the nearby BMP-RE1 via SMAD activation, which is required for full promoter activity. A distal BMP-RE2 site is thought interact with the SMAD/STAT complex that brings distal and proximal regions of the hepcidin promoter into physical contact. SMAD signaling is activated through the HJV/BMP pathway. HJV is negatively regulated by matripase-2 cleavage; BMP signaling is negatively regulated by GDF15 and TWSG1. HFE interacts with TfR1 and may be released upon binding of diferric Tf. While binding of both diferric Tf and HFE to the TfR2 are known to stimulate hepcidin synthesis, the molecular aspects of this pathway remain unknown. In addition, ER stress response pathways involved in inflammation are known to induce hepcidin through at least two mechanisms: 1) negative regulation of C/EBPα by CHOP and 2) activation of CREBH, possibly in conjunction with activated (spliced) XBP-1.
V. Tumor necrosis factor-α (TNF-α)
TNF–α is a pro-inflammatory cytokine that promotes hypoferremia through mechanisms that are independent of the induction of hepcidin (135). It is clear from studies of human TNF-α polymorphisms that increased levels of the cytokine are associated with systemic iron deficiency (2). Mice treated with TNF-α down-regulate mucosal transfer of iron (71), a response that would be expected to coordinate with iron homeostasis to reduce body iron levels (1). Duodenal ferritin levels are induced in these animals, providing for iron storage during the acute phase response (146). This mechanism would prevent dietary iron exsorption across the mucosa to circulation. However, the profile of TNF-α induced changes in the expression of iron transport proteins is more complicated, and does not completely correlate with the expected iron withdrawal response and retention by macrophages (71, 81, 95, 162). One explanation for the observed effects on macrophage iron levels is that erythrophagocytosis is enhanced by TNF-α (64, 89). It has also been shown that treatment of bronchial epithelial cells with TNF-α promotes up-regulation of DMT1 (162); this effect has been related to a detoxification pathway that would remove iron from the environment to inhibit growth of pathogens in the lungs.
Paradoxically, treatment of macrophages with TNF-α is associated with a transient rise in intracellular free or labile iron that is required for activation of the transcription factor NF-κB (169). Such iron transients have been observed to play an important role in other stress signaling responses (25). NF-κB is an important regulator of inflammatory responses, including the production of TNF-α itself (106). Reduction of intracellular iron by chelation has an inhibitory effect on NF-κB induction of TNF-α and other cytokines (131, 150). Further studies on the mechanism of TNF-α action are needed to more fully understand its reciprocal relationship with iron and control of the NF-κB inflammatory response.
VI. Interferon-γ (IFN-γ)
IFN-γ is a key modulator of macrophage iron status and immune functions. A major mechanism of macrophage-mediated innate immune response is the synthesis of nitric oxide (NO), which is stimulated by IFN-γ. A complex iron-regulated network controls inducible NO synthase (iNOS), the key enzyme in NO production. Through the use of iron chelators and exogenous addition of iron, Weiss and co-workers (166) have demonstrated an inverse relationship between host cell iron status and iNOS expression. Increased iron results in decreased iNOS activity while iron depletion enhances iNOS activity. This inverse effect of iron reflects altered iNOS mRNA levels (166). Iron can also exert inhibitory effects on other IFN-γ responses as well (102). In a reciprocal fashion, IFN-γ can modulate iron status, and has been shown to reduce the availability of iron to S. typhimurium within the macrophage (92). In vivo studies have shown that a high iron diet will reduce IFN-γ (104), implicating its role in the deleterious effects that iron-loading can have on the immune response. Recent in vitro studies further suggest that IFN-γ may modulate hepcidin induction by M. tuberculosis infected macrophages, further confirming the link between iron metabolism and host response to pathogens (139).
VII. Effectors of iron withdrawal
While the focus of recent research has been on the regulation of iron metabolism during the inflammatory response by hepcidin, cytokines and other acute phase reactants, the effector molecules that are ultimately responsible for limiting circulating iron or sequestering it from micro-organisms and rapidly growing cancer cells have long been studied. Some of the major mechanistic elements responsible for iron withdrawal during the inflammatory response are highlighted below.
a. Ferroportin
As discussed above, ferroportin is the major target of hepcidin’s control during infection and inflammation. However, inflammatory stimuli can also directly regulate expression of ferroportin to promote macrophage iron retention (81, 170). A central role for this ferroportin response to infection is supported by in vitro studies (18, 93, 108). Mechanisms regulating ferroportin expression include both transcriptional and post-transcriptional events, since ferroportin mRNA harbors a 5′ iron-responsive element (IRE) (85). The presence of the IRE confers regulation of ferroportin translation to iron regulatory proteins (IRPs) (33, 57). IRPs are master regulators of iron metabolism including other factors such asDMT1, ferritin and TfR1. Control of NO synthesis by IFN-γ is thought to modulate IRP2 activity, and thus may contribute to the action of effectors like ferroportin.
b. Ferritin
Ferritin serves as the depot site for iron storage during sequestration responses elicited by inflammatory response. In addition to control by IRP action triggered by iron accrual in liver and other tissues, inflammatory cytokines regulate ferritin translation through an “acute phase box” in the 5′ region of its transcript (124, 146). Transcriptional control mechanisms serve to further up-regulate ferritin expression, and increased serum ferritin levels are well-recognized as part of the acute phase response (146). Combined, cell-mediated down-regulation of ferroportin and up-regulation of ferritin act in concert to induce iron retention and sequestration. Although how TNF-α down-regulates intestinal iron absorption remains to be fully elucidated, it is clear that its induction of ferritin synthesis provides a necessary block on dietary iron entry across the mucosa.
c. Tf/Tf receptor/DMT1
As outlined above, cellular delivery of iron via endocytosis of Tf is a major pathway for iron clearance. In addition, stimulation of non-Tf bound iron uptake by DMT1 by monocytes has been reported in response to cytokines (81). Although inflammation down-regulates Tf receptor levels, possibly through IRP control mechanisms (11, 81), an early transcriptional response to stimuli promoting iron accumulation has been suggested (141). This activation of iron uptake via TfR1 is thought to precede the induction of hepcidin and subsequent down-regulation of ferroportin in macrophage cell lines, however, the temporal nature of changes in iron trafficking remain to be fully elucidated in vivo. In general, iron status is “sensed” through changes in Tf saturation (61, 123) and reflected in HFE functional interactions with Tf receptor-1 and -2 (47, 130). Tight coordination of this network is necessary to maintain the appropriate immune response without impairing iron metabolism. It is clear that Tf and its receptor are key limiting factors in immune cell growth due to their requirement for iron (132) such that iron-deficient mice have reduced T cell proliferation and impaired immunity (104).
d. Nramp1
The phagosomal transporter called natural resistance associated macrophage protein-1 (Nramp1) is thought to play a major role in the inflammatory response since its mutation in mice confers susceptibility to a number of intracellular pathogens (155). Human mutations in the homologous gene are also known to influence tuberculosis infection and rheumatic disease (7). Nramp 1 expression is up-regulated by inflammatory cytokines (53), and it is thought to function by limiting the availability of phagosomal iron needed for growth of invading organisms. It also contributes to production of nitric oxide (NO) and other pro-inflammatory macrophage responses (7, 168). Loss of Nramp1 function in mice impairs iron recycling by macrophages after erythrophagocytosis (138) and reduces the inflammatory response to infection (151). How these functions are regulated by Nramp1 cation transport activity is less clear. It has been proposed that mechanistic effects are exerted through ROS-regulated signaling pathways (72). Recent biochemical studies have suggested that Nramp1 regulates protein tyrosine phosphatase activity, suggesting that the transporter may modulate signal transduction pathways involved in the macrophage inflammatory response (51).
e. Lactoferrin
Lactoferrin is often considered to be another important factor for host inflammatory defense due to its iron-binding ability. Lactoferrin is structurally related to Tf, but unlike its homologue, lactoferrin does not release ferric iron at low pH (5). The mammary gland and epithelial cells produce lactoferrin and it is present in milk and mucosal secretions. Lactoferrin is also produced by neutrophils and is released during inflammation. It is considered to be an iron scavenger at sites of infection and at mucosal surfaces. Although its bacteriocidal actions have been confirmed in many different studies (78), lactoferrin’s true function in immunity and inflammation is less clear. Lactoferrin knockout mice do not show impaired immune response or altered iron homeostasis (163), with only a slight response to staphylococcal infections and no difference to S. aureus or P. aeruoginosa. Lactoferrin may overlap in the function of other iron-binding proteins, or perhaps may even be specific for certain micro-organisms.
f. Lipocalin-2
Lipocalin-2 is an acute phase response protein also referred to as siderocalin or neutrophilgelatinase-associated lipocalin (NGAL). Lipocalin-2 binds siderophores, chelators produced by micro-organisms in the battle to acquire iron from the host (19). Lipocalin-2 knockout mice have decreased survival when infected with E. coli or M. tuberculosis, supporting its importance in the inflammatory defense against invading micro-organisms (6, 39, 128). Importantly, lipocalin-2 production appears to be up-regulated by deficiency in HFE (94). Although a receptor for lipocalin-2 has been identified (27), whether this potential iron transport system has an endogenous “siderophore-like” ligand remains unknown.
VIII. Hypoferremia of inflammation: benefit or liability of treatment?
The anemia acquired in response to inflammation represents a unique facet of human iron metabolism. The evidence that supplemental iron can promote both infectious and chronic inflammatory diseases is clear, but what is the correct balance and should iron treatment or chelation be approached therapeutically? The recent Pemba trial, which led to adverse events in children receiving iron in a malaria-endemic region (129), underscores the need for caution. Nonetheless, it has been argued that negative effects from supplementation of iron-replete individuals may be offset by positive measures in iron-deficient groups such that a rationalized approach would screen iron status and other genetic predispositions when considering various therapies (2, 118). For example, disorders like ferroportin disease, which predisposes individuals to macrophage iron retention (114), may impart particular susceptibility to obligate intracellular pathogens like M. tuberculosis. Ferroportin disease arises from gene variants that lead to loss of iron export function or loss of cell surface expression of ferroportin. The result is an iron-loaded state in macrophages that can promote the growth of bacteria (18, 93). Paradkar et al. (108) have used macrophages from flatiron mice, a model of this form of hemochromatosis, to show that oral iron chelators can limit bacterial growth. This study supports the idea that iron chelation therapy may be useful in certain situations.
Castleman’s disease is an example where interfering with hepcidin production offers an attractive therapeutic avenue for treatment. This inflammatory disorder is associated with increased IL-6 (9), which induces upregulation of hepcidin expression. Treatment with antibodies against the IL-6 receptor has been shown to reduce hepcidin levels in several patients, suggesting an approach to handling the anemia associated with disorder (63). Other new tools that target hepcidin regulation include suppression of BMP signaling. Two compounds, dorsomorphin (4) and LDN-193189 (23) have been shown to decrease to Salmonella-induced inflammation in mice and to reduce intestinal inflammation in a murine model of chronic colitis, respectively (159). Both small molecules reduce BMP signaling and hepcidin production. In addition, hepcidin levels and inflammation were also reduced in the colitis model in response to HJV.Fc, a soluble form of the HJV domain that acts as a co-receptor for BMP signaling (159). Such novel strategies could be future therapeutic options in treating both inflammatory diseases and infection.
Acknowledgments
M.W.-R. is supported by NIH grants R01 DK064750, R01 ES014638, R21 DA025573, RC1 DK086774, and R03 DA027030. The author gratefully acknowledges the help of Dr. Bobby Cherayil for critical reading and comment on the manuscript.
Acronyms
- AI/ACD
anemia of inflammation/anemia of chronic disease
- ATF4
activating transcription factor 4
- BMP
bone morphogenic protein
- BMP-RE
bone morphogenic protein responsive element
- C/EPBα
CCAAT/enhancer-binding protein-α
- CHOP
CCAAT/enhancer-binding protein homologous protein
- CREBH
cAMP-responsive element binding protein H
- DcytB
duodenal cytochrome B
- DMT1
divalent metal transporter-1
- eIF2α
eukaryotic initiation factor 2α
- ER
endoplasmic reticulum
- GDF15
growth differentiation factor-15
- GSD1a
glycogen storage disease type 1a
- HIF
hypoxia-inducible factor
- HJV
hemojuvelin
- HRI
heme regulated inhibitor
- IL-6
interleukin-6
- INF-γ
interferon-γ
- iNOS
inducible nitric oxide synthase
- IRE
iron-responsive element
- IRP
iron regulatory proteins
- LEAP-1
liver-derived antimicrobial peptide
- LPS
lipopolysaccharide
- NGAL
neutrophilgelatinase-associated lipocalin
- NO
nitric oxide
- Nramp1
natural resistance associated macrophage protein-1
- PERK
pancreatic endoplasmic reticulum kinase
- RES
reticuloendothelial system
- ROS
reactive oxygen species
- STAT
signal transducer and activator of transcription
- Tf/TfR
transferrin/transferrin receptor
- TLR
Toll-like receptor
- TNF-α
tumor necrosis factor-α
- TWSG1
twisted gastrulation
- UPR
unfolded protein response
Literature Cited
- 1.Alvarez-Hernandez X, Liceaga J, McKay IC, Brock JH. Induction of hypoferremia and modulation of macrophage iron metabolism by tumor necrosis factor. Lab Invest. 1989;61:319–22. [PubMed] [Google Scholar]
- 2.Atkinson SH, Rockett KA, Morgan G, Bejon PA, Sirugo G, et al. Tumor necrosis factor SNP haplotypes are associated with iron deficiency anemia in West African children. Blood. 2008;112:4276–83. doi: 10.1182/blood-2008-06-162008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–9. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 4.Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117:1933–9. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker EN, Baker HM. Molecular structure, binding properties and dynamics of lactoferrin. Cell Mol Life Sci. 2005;62:2531–9. doi: 10.1007/s00018-005-5368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:1834–9. doi: 10.1073/pnas.0510847103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackwell JM, Goswami T, Evans CA, Sibthorpe D, Papo N, et al. SLC11A1 (formerly NRAMP1) and disease resistance. Cell Microbiol. 2001;3:773–84. doi: 10.1046/j.1462-5822.2001.00150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bofill C, Joven J, Bages J, Vilella E, Sans T, et al. Response to repeated phlebotomies in patients with non-insulin-dependent diabetes mellitus. Metabolism. 1994;43:614–20. doi: 10.1016/0026-0495(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 9.Brandt SJ, Bodine DM, Dunbar CE, Nienhuis AW. Dysregulated interleukin 6 expression produces a syndrome resembling Castleman’s disease in mice. J Clin Invest. 1990;86:592–9. doi: 10.1172/JCI114749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bullen JJ, Spalding PB, Ward CG, Gutteridge JM. Hemochromatosis, iron and septicemia caused by Vibrio vulnificus. Arch Intern Med. 1991;151:1606–9. [PubMed] [Google Scholar]
- 11.Byrd TF, Horwitz MA. Regulation of transferrin receptor expression and ferritin content in human mononuclear phagocytes. Coordinate upregulation by iron transferrin and downregulation by interferon gamma. J Clin Invest. 1993;91:969–76. doi: 10.1172/JCI116318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cairo G, Recalcati S, Montosi G, Castrusini E, Conte D, Pietrangelo A. Inappropriately high iron regulatory protein activity in monocytes of patients with genetic hemochromatosis. Blood. 1997;89:2546–53. [PubMed] [Google Scholar]
- 13.Cartwright GE, Lauritsen MA, Humphreys S, Jones PJ, Merrill IM, Wintrobe MM. The Anemia Associated With Chronic Infection. Science. 1946;103:72–3. doi: 10.1126/science.103.2664.72. [DOI] [PubMed] [Google Scholar]
- 14.Cartwright GE, Lauritsen MA, Humphreys S, Jones PJ, Merrill IM, Wintrobe MM. The Anemia of Infection. Ii. The Experimental Production of Hypoferremia and Anemia in Dogs. J Clin Invest. 1946;25:81–6. doi: 10.1172/JCI101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cartwright GE, Lauritsen MA, Jones PJ, Merrill IM, Wintrobe MM. The Anemia of Infection. I. Hypoferremia, Hypercupremia, and Alterations in Porphyrin Metabolism in Patients. J Clin Invest. 1946;25:65–80. doi: 10.1172/JCI101690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casanovas G, Mleczko-Sanecka K, Altamura S, Hentze MW, Muckenthaler MU. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J Mol Med. 2009;87:471–80. doi: 10.1007/s00109-009-0447-2. [DOI] [PubMed] [Google Scholar]
- 17.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109:2693–9. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chlosta S, Fishman DS, Harrington L, Johnson EE, Knutson MD, et al. The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect Immun. 2006;74:3065–7. doi: 10.1128/IAI.74.5.3065-3067.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clifton MC, Corrent C, Strong RK. Siderocalins: siderophore-binding proteins of the innate immune system. Biometals. 2009;22:557–64. doi: 10.1007/s10534-009-9207-6. [DOI] [PubMed] [Google Scholar]
- 20.Constante M, Wang D, Raymond VA, Bilodeau M, Santos MM. Repression of repulsive guidance molecule C during inflammation is independent of Hfe and involves tumor necrosis factor-alpha. Am J Pathol. 2007;170:497–504. doi: 10.2353/ajpath.2007.060437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Courselaud B, Pigeon C, Inoue Y, Inoue J, Gonzalez FJ, et al. C/EBPalpha regulates hepatic transcription of hepcidin, an antimicrobial peptide and regulator of iron metabolism. Cross-talk between C/EBP pathway and iron metabolism. J Biol Chem. 2002;277:41163–70. doi: 10.1074/jbc.M202653200. [DOI] [PubMed] [Google Scholar]
- 22.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cuny GD, Yu PB, Laha JK, Xing X, Liu JF, et al. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg Med Chem Lett. 2008;18:4388–92. doi: 10.1016/j.bmcl.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Domenico I, Ward DM, di Patti MC, Jeong SY, David S, et al. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007;26:2823–31. doi: 10.1038/sj.emboj.7601735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deb S, Johnson EE, Robalinho-Teixeira RL, Wessling-Resnick M. Modulation of intracellular iron levels by oxidative stress implicates a novel role for iron in signal transduction. Biometals. 2009;22:855–62. doi: 10.1007/s10534-009-9214-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denic S, Agarwal MM. Nutritional iron deficiency: an evolutionary perspective. Nutrition. 2007;23:603–14. doi: 10.1016/j.nut.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 27.Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123:1293–305. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 28.Doherty CP. Host-pathogen interactions: the role of iron. J Nutr. 2007;137:1341–4. doi: 10.1093/jn/137.5.1341. [DOI] [PubMed] [Google Scholar]
- 29.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–81. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 30.Drakesmith H, Prentice A. Viral infection and iron metabolism. Nat Rev Microbiol. 2008;6:541–52. doi: 10.1038/nrmicro1930. [DOI] [PubMed] [Google Scholar]
- 31.Du X, She E, Gelbart T, Truksa J, Lee P, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science. 2008;320:1088–92. doi: 10.1126/science.1157121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dzikaite V, Holmstrom P, Stal P, Eckes K, Hagen K, et al. Regulatory effects of tumor necrosis factor-alpha and interleukin-6 on HAMP expression in iron loaded rat hepatocytes. J Hepatol. 2006;44:544–51. doi: 10.1016/j.jhep.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 33.Eisenstein RS. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu Rev Nutr. 2000;20:627–62. doi: 10.1146/annurev.nutr.20.1.627. [DOI] [PubMed] [Google Scholar]
- 34.Feder JN. The hereditary hemochromatosis gene (HFE): a MHC class I-like gene that functions in the regulation of iron homeostasis. Immunol Res. 1999;20:175–85. doi: 10.1007/BF02786472. [DOI] [PubMed] [Google Scholar]
- 35.Fernandez-Real JM, Penarroja G, Castro A, Garcia-Bragado F, Hernandez-Aguado I, Ricart W. Blood letting in high-ferritin type 2 diabetes: effects on insulin sensitivity and beta-cell function. Diabetes. 2002;51:1000–4. doi: 10.2337/diabetes.51.4.1000. [DOI] [PubMed] [Google Scholar]
- 36.Finberg KE, Heeney MM, Campagna DR, Aydinok Y, Pearson HA, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA) Nat Genet. 2008;40:569–71. doi: 10.1038/ng.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A. 1998;95:1148–53. doi: 10.1073/pnas.95.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleming MD, Trenor CC, 3rd, Su MA, Foernzler D, Beier DR, et al. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997;16:383–6. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- 39.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–21. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 40.Ford ES, Cogswell ME. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care. 1999;22:1978–83. doi: 10.2337/diacare.22.12.1978. [DOI] [PubMed] [Google Scholar]
- 41.Frazer DM, Wilkins SJ, Becker EM, Vulpe CD, McKie AT, et al. Hepcidin expression inversely correlates with the expression of duodenal iron transporters and iron absorption in rats. Gastroenterology. 2002;123:835–44. doi: 10.1053/gast.2002.35353. [DOI] [PubMed] [Google Scholar]
- 42.Frazer DM, Wilkins SJ, Millard KN, McKie AT, Vulpe CD, Anderson GJ. Increased hepcidin expression and hypoferraemia associated with an acute phase response are not affected by inactivation of HFE. Br J Haematol. 2004;126:434–6. doi: 10.1111/j.1365-2141.2004.05044.x. [DOI] [PubMed] [Google Scholar]
- 43.Fumeron F, Pean F, Driss F, Balkau B, Tichet J, et al. Ferritin and transferrin are both predictive of the onset of hyperglycemia in men and women over 3 years: the data from an epidemiological study on the Insulin Resistance Syndrome (DESIR) study. Diabetes Care. 2006;29:2090–4. doi: 10.2337/dc06-0093. [DOI] [PubMed] [Google Scholar]
- 44.Gangaidzo IT, Moyo VM, Mvundura E, Aggrey G, Murphree NL, et al. Association of pulmonary tuberculosis with increased dietary iron. J Infect Dis. 2001;184:936–9. doi: 10.1086/323203. [DOI] [PubMed] [Google Scholar]
- 45.Ganz T, Nemeth E. Iron sequestration and anemia of inflammation. Semin Hematol. 2009;46:387–93. doi: 10.1053/j.seminhematol.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganz T, Olbina G, Girelli D, Nemeth E, Westerman M. Immunoassay for human serum hepcidin. Blood. 2008;112:4292–7. doi: 10.1182/blood-2008-02-139915. [DOI] [PubMed] [Google Scholar]
- 47.Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9:217–27. doi: 10.1016/j.cmet.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gao J, Zhao N, Knutson MD, Enns CA. The hereditary hemochromatosis protein, HFE, inhibits iron uptake via down-regulation of Zip14 in HepG2 cells. J Biol Chem. 2008;283:21462–8. doi: 10.1074/jbc.M803150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garcia-Vidal C, Upton A, Kirby KA, Marr KA. Epidemiology of invasive mold infections in allogeneic stem cell transplant recipients: biological risk factors for infection according to time after transplantation. Clin Infect Dis. 2008;47:1041–50. doi: 10.1086/591969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gerhard GS, Levin KA, Price Goldstein J, Wojnar MM, Chorney MJ, Belchis DA. Vibrio vulnificus septicemia in a patient with the hemochromatosis HFE C282Y mutation. Arch Pathol Lab Med. 2001;125:1107–9. doi: 10.5858/2001-125-1107-VVSIAP. [DOI] [PubMed] [Google Scholar]
- 51.Gomez MA, Li S, Tremblay ML, Olivier M. NRAMP-1 expression modulates protein-tyrosine phosphatase activity in macrophages: impact on host cell signaling and functions. J Biol Chem. 2007;282:36190–8. doi: 10.1074/jbc.M703140200. [DOI] [PubMed] [Google Scholar]
- 52.Gordeuk VR, Ballou S, Lozanski G, Brittenham GM. Decreased concentrations of tumor necrosis factor-alpha in supernatants of monocytes from homozygotes for hereditary hemochromatosis. Blood. 1992;79:1855–60. [PubMed] [Google Scholar]
- 53.Govoni G, Gauthier S, Billia F, Iscove NN, Gros P. Cell-specific and inducible Nramp1 gene expression in mouse macrophages in vitro and in vivo. J Leukoc Biol. 1997;62:277–86. doi: 10.1002/jlb.62.2.277. [DOI] [PubMed] [Google Scholar]
- 54.Graham JM, Paley MN, Grunewald RA, Hoggard N, Griffiths PD. Brain iron deposition in Parkinson’s disease imaged using the PRIME magnetic resonance sequence. Brain. 2000;123(Pt 12):2423–31. doi: 10.1093/brain/123.12.2423. [DOI] [PubMed] [Google Scholar]
- 55.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–8. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 56.Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, et al. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006;281:22974–82. doi: 10.1074/jbc.M602098200. [DOI] [PubMed] [Google Scholar]
- 57.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–97. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 58.Hower V, Mendes P, Torti FM, Laubenbacher R, Akman S, et al. A general map of iron metabolism and tissue-specific subnetworks. Mol Biosyst. 2009;5:422–43. doi: 10.1039/b816714c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang R, Ma J, Ascherio A, Stampfer MJ, Willett WC, Hu FB. Dietary iron intake and blood donations in relation to risk of type 2 diabetes in men: a prospective cohort study. Am J Clin Nutr. 2004;79:70–5. doi: 10.1093/ajcn/79.1.70. [DOI] [PubMed] [Google Scholar]
- 60.Jiang R, Manson JE, Meigs JB, Ma J, Rifai N, Hu FB. Body iron stores in relation to risk of type 2 diabetes in apparently healthy women. JAMA. 2004;291:711–7. doi: 10.1001/jama.291.6.711. [DOI] [PubMed] [Google Scholar]
- 61.Johnson MB, Enns CA. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood. 2004;104:4287–93. doi: 10.1182/blood-2004-06-2477. [DOI] [PubMed] [Google Scholar]
- 62.Kabyemela ER, Fried M, Kurtis JD, Mutabingwa TK, Duffy PE. Decreased susceptibility to Plasmodium falciparum infection in pregnant women with iron deficiency. J Infect Dis. 2008;198:163–6. doi: 10.1086/589512. [DOI] [PubMed] [Google Scholar]
- 63.Kawabata H, Tomosugi N, Kanda J, Tanaka Y, Yoshizaki K, Uchiyama T. Anti-interleukin 6 receptor antibody tocilizumab reduces the level of serum hepcidin in patients with multicentric Castleman’s disease. Haematologica. 2007;92:857–8. doi: 10.3324/haematol.10794. [DOI] [PubMed] [Google Scholar]
- 64.Kitagawa S, Yuo A, Yagisawa M, Azuma E, Yoshida M, et al. Activation of human monocyte functions by tumor necrosis factor: rapid priming for enhanced release of superoxide and erythrophagocytosis, but no direct triggering of superoxide release. Exp Hematol. 1996;24:559–67. [PubMed] [Google Scholar]
- 65.Knutson MD, Vafa MR, Haile DJ, Wessling-Resnick M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood. 2003;102:4191–7. doi: 10.1182/blood-2003-04-1250. [DOI] [PubMed] [Google Scholar]
- 66.Kochan I. The role of iron in bacterial infections, with special consideration of host-tubercle bacillus interaction. Curr Top Microbiol Immunol. 1973;60:1–30. doi: 10.1007/978-3-642-65502-9_1. [DOI] [PubMed] [Google Scholar]
- 67.Koening CL, Miller JC, Nelson JM, Ward DM, Kushner JP, et al. Toll-like receptors mediate induction of hepcidin in mice infected with Borrelia burgdorferi. Blood. 2009;114:1913–8. doi: 10.1182/blood-2009-03-209577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koka S, Foller M, Lamprecht G, Boini KM, Lang C, et al. Iron deficiency influences the course of malaria in Plasmodium berghei infected mice. Biochem Biophys Res Commun. 2007;357:608–14. doi: 10.1016/j.bbrc.2007.03.175. [DOI] [PubMed] [Google Scholar]
- 69.Krause A, Neitz S, Magert HJ, Schulz A, Forssmann WG, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480:147–50. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 70.Krijt J, Vokurka M, Chang KT, Necas E. Expression of Rgmc, the murine ortholog of hemojuvelin gene, is modulated by development and inflammation, but not by iron status or erythropoietin. Blood. 2004;104:4308–10. doi: 10.1182/blood-2004-06-2422. [DOI] [PubMed] [Google Scholar]
- 71.Laftah AH, Sharma N, Brookes MJ, McKie AT, Simpson RJ, et al. Tumour necrosis factor alpha causes hypoferraemia and reduced intestinal iron absorption in mice. Biochem J. 2006;397:61–7. doi: 10.1042/BJ20060215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lafuse WP, Alvarez GR, Zwilling BS. Role of MAP kinase activation in Nramp1 mRNA stability in RAW264.7 macrophages expressing Nramp1(Gly169) Cell Immunol. 2002;215:195–206. doi: 10.1016/s0008-8749(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 73.Lanzara C, Roetto A, Daraio F, Rivard S, Ficarella R, et al. Spectrum of hemojuvelin gene mutations in 1q-linked juvenile hemochromatosis. Blood. 2004;103:4317–21. doi: 10.1182/blood-2004-01-0192. [DOI] [PubMed] [Google Scholar]
- 74.Lee DH, Folsom AR, Jacobs DR., Jr Dietary iron intake and Type 2 diabetes incidence in postmenopausal women: the Iowa Women’s Health Study. Diabetologia. 2004;47:185–94. doi: 10.1007/s00125-003-1307-1. [DOI] [PubMed] [Google Scholar]
- 75.Lee P, Peng H, Gelbart T, Beutler E. The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci U S A. 2004;101:9263–5. doi: 10.1073/pnas.0403108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci U S A. 2005;102:1906–10. doi: 10.1073/pnas.0409808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee PL, Beutler E. Regulation of hepcidin and iron-overload disease. Annu Rev Pathol. 2009;4:489–515. doi: 10.1146/annurev.pathol.4.110807.092205. [DOI] [PubMed] [Google Scholar]
- 78.Legrand D, Elass E, Carpentier M, Mazurier J. Lactoferrin: a modulator of immune and inflammatory responses. Cell Mol Life Sci. 2005;62:2549–59. doi: 10.1007/s00018-005-5370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu S, Suragani RN, Wang F, Han A, Zhao W, et al. The function of heme-regulated eIF2alpha kinase in murine iron homeostasis and macrophage maturation. J Clin Invest. 2007;117:3296–305. doi: 10.1172/JCI32084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lounis N, Truffot-Pernot C, Grosset J, Gordeuk VR, Boelaert JR. Iron and Mycobacterium tuberculosis infection. J Clin Virol. 2001;20:123–6. doi: 10.1016/s1386-6532(00)00136-0. [DOI] [PubMed] [Google Scholar]
- 81.Ludwiczek S, Aigner E, Theurl I, Weiss G. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood. 2003;101:4148–54. doi: 10.1182/blood-2002-08-2459. [DOI] [PubMed] [Google Scholar]
- 82.Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119:1159–66. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matak P, Chaston TB, Chung B, Srai SK, McKie AT, Sharp PA. Activated macrophages induce hepcidin expression in HuH7 hepatoma cells. Haematologica. 2009;94:773–80. doi: 10.3324/haematol.2008.003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–9. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- 85.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 86.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–26. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 87.Meynard D, Kautz L, Darnaud V, Canonne-Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41:478–81. doi: 10.1038/ng.320. [DOI] [PubMed] [Google Scholar]
- 88.Moalem S, Weinberg ED, Percy ME. Hemochromatosis and the enigma of misplaced iron: implications for infectious disease and survival. Biometals. 2004;17:135–9. doi: 10.1023/b:biom.0000018375.20026.b3. [DOI] [PubMed] [Google Scholar]
- 89.Moldawer LL, Marano MA, Wei H, Fong Y, Silen ML, et al. Cachectin/tumor necrosis factor-alpha alters red blood cell kinetics and induces anemia in vivo. FASEB J. 1989;3:1637–43. doi: 10.1096/fasebj.3.5.2784116. [DOI] [PubMed] [Google Scholar]
- 90.Moura E, Noordermeer MA, Verhoeven N, Verheul AF, Marx JJ. Iron release from human monocytes after erythrophagocytosis in vitro: an investigation in normal subjects and hereditary hemochromatosis patients. Blood. 1998;92:2511–9. [PubMed] [Google Scholar]
- 91.Murray MJ, Murray AB, Murray MB, Murray CJ. The adverse effect of iron repletion on the course of certain infections. Br Med J. 1978;2:1113–5. doi: 10.1136/bmj.2.6145.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nairz M, Fritsche G, Brunner P, Talasz H, Hantke K, Weiss G. Interferon-gamma limits the availability of iron for intramacrophage Salmonella typhimurium. Eur J Immunol. 2008;38:1923–36. doi: 10.1002/eji.200738056. [DOI] [PubMed] [Google Scholar]
- 93.Nairz M, Theurl I, Ludwiczek S, Theurl M, Mair SM, et al. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 2007;9:2126–40. doi: 10.1111/j.1462-5822.2007.00942.x. [DOI] [PubMed] [Google Scholar]
- 94.Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, et al. Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood. 2009;114:3642–51. doi: 10.1182/blood-2009-05-223354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nanami M, Ookawara T, Otaki Y, Ito K, Moriguchi R, et al. Tumor necrosis factor-alpha-induced iron sequestration and oxidative stress in human endothelial cells. Arterioscler Thromb Vasc Biol. 2005;25:2495–501. doi: 10.1161/01.ATV.0000190610.63878.20. [DOI] [PubMed] [Google Scholar]
- 96.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–6. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 98.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461–3. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 99.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98:8780–5. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, et al. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci U S A. 2002;99:4596–601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–44. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Oexle H, Kaser A, Most J, Bellmann-Weiler R, Werner ER, et al. Pathways for the regulation of interferon-gamma-inducible genes by iron in human monocytic cells. J Leukoc Biol. 2003;74:287–94. doi: 10.1189/jlb.0802420. [DOI] [PubMed] [Google Scholar]
- 103.Oliveira SJ, Pinto JP, Picarote G, Costa VM, Carvalho F, et al. ER stress-inducible factor CHOP affects the expression of hepcidin by modulating C/EBPalpha activity. PLoS One. 2009;4:e6618. doi: 10.1371/journal.pone.0006618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Omara FO, Blakley BR. The effects of iron deficiency and iron overload on cell-mediated immunity in the mouse. Br J Nutr. 1994;72:899–909. doi: 10.1079/bjn19940094. [DOI] [PubMed] [Google Scholar]
- 105.Ozkalay N, Anil M, Agus N, Helvaci M, Sirti S. Community-acquired meningitis and sepsis caused by Chryseobacterium meningosepticum in a patient diagnosed with thalassemia major. J Clin Microbiol. 2006;44:3037–9. doi: 10.1128/JCM.00588-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Papa S, Bubici C, Pham CG, Zazzeroni F, Franzoso G. NF-kappaB meets ROS: an ‘iron-ic’ encounter. Cell Death Differ. 2005;12:1259–62. doi: 10.1038/sj.cdd.4401694. [DOI] [PubMed] [Google Scholar]
- 107.Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 108.Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron depletion limits intracellular bacterial growth in macrophages. Blood. 2008;112:866–74. doi: 10.1182/blood-2007-12-126854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–10. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 110.Peslova G, Petrak J, Kuzelova K, Hrdy I, Halada P, et al. Hepcidin, the hormone of iron metabolism, is bound specifically to alpha-2-macroglobulin in blood. Blood. 2009;113:6225–36. doi: 10.1182/blood-2009-01-201590. [DOI] [PubMed] [Google Scholar]
- 111.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–15. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood. 2006;107:3727–32. doi: 10.1182/blood-2005-06-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–32. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32:131–8. doi: 10.1016/j.bcmd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 115.Pietrangelo A. Molecular insights into the pathogenesis of hereditary haemochromatosis. Gut. 2006;55:564–8. doi: 10.1136/gut.2005.078063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–9. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 117.Pinto JP, Ribeiro S, Pontes H, Thowfeequ S, Tosh D, et al. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood. 2008;111:5727–33. doi: 10.1182/blood-2007-08-106195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Prentice AM. Iron metabolism, malaria, and other infections: what is all the fuss about? J Nutr. 2008;138:2537–41. doi: 10.3945/jn.108.098806. [DOI] [PubMed] [Google Scholar]
- 119.Rajpathak SN, Crandall JP, Wylie-Rosett J, Kabat GC, Rohan TE, Hu FB. The role of iron in type 2 diabetes in humans. Biochim Biophys Acta. 2009;1790:671–81. doi: 10.1016/j.bbagen.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 120.Ratledge C. Iron, mycobacteria and tuberculosis. Tuberculosis (Edinb) 2004;84:110–30. doi: 10.1016/j.tube.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 121.Richardson DR, Kalinowski DS, Lau S, Jansson PJ, Lovejoy DB. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim Biophys Acta. 2009;1790:702–17. doi: 10.1016/j.bbagen.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 122.Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Robb A, Wessling-Resnick M. Regulation of transferrin receptor 2 protein levels by transferrin. Blood. 2004;104:4294–9. doi: 10.1182/blood-2004-06-2481. [DOI] [PubMed] [Google Scholar]
- 124.Rogers JT, Bridges KR, Durmowicz GP, Glass J, Auron PE, Munro HN. Translational control during the acute phase response. Ferritin synthesis in response to interleukin-1. J Biol Chem. 1990;265:14572–8. [PubMed] [Google Scholar]
- 125.Roy CN, Andrews NC. Anemia of inflammation: the hepcidin link. Curr Opin Hematol. 2005;12:107–11. doi: 10.1097/00062752-200503000-00001. [DOI] [PubMed] [Google Scholar]
- 126.Roy CN, Custodio AO, de Graaf J, Schneider S, Akpan I, et al. An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide-induced inflammation in mice. Nat Genet. 2004;36:481–5. doi: 10.1038/ng1350. [DOI] [PubMed] [Google Scholar]
- 127.Roy CN, Mak HH, Akpan I, Losyev G, Zurakowski D, Andrews NC. Hepcidin antimicrobial peptide transgenic mice exhibit features of the anemia of inflammation. Blood. 2007;109:4038–44. doi: 10.1182/blood-2006-10-051755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Saiga H, Nishimura J, Kuwata H, Okuyama M, Matsumoto S, et al. Lipocalin 2-dependent inhibition of mycobacterial growth in alveolar epithelium. J Immunol. 2008;181:8521–7. doi: 10.4049/jimmunol.181.12.8521. [DOI] [PubMed] [Google Scholar]
- 129.Sazawal S, Black RE, Ramsan M, Chwaya HM, Stoltzfus RJ, et al. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet. 2006;367:133–43. doi: 10.1016/S0140-6736(06)67962-2. [DOI] [PubMed] [Google Scholar]
- 130.Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7:205–14. doi: 10.1016/j.cmet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schwacha MG, Gudewicz PW, Snyder JA, Loegering DJ. Depression of macrophage respiratory burst capacity and arachidonic acid release after Fc receptor-mediated phagocytosis. J Immunol. 1993;150:236–45. [PubMed] [Google Scholar]
- 132.Seligman PA, Kovar J, Gelfand EW. Lymphocyte proliferation is controlled by both iron availability and regulation of iron uptake pathways. Pathobiology. 1992;60:19–26. doi: 10.1159/000163692. [DOI] [PubMed] [Google Scholar]
- 133.Sendamarai AK, Ohgami RS, Fleming MD, Lawrence CM. Structure of the membrane proximal oxidoreductase domain of human Steap3, the dominant ferrireductase of the erythroid transferrin cycle. Proc Natl Acad Sci U S A. 2008;105:7410–5. doi: 10.1073/pnas.0801318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009;9:152–64. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sharma N, Laftah AH, Brookes MJ, Cooper B, Iqbal T, Tselepis C. A role for tumour necrosis factor alpha in human small bowel iron transport. Biochem J. 2005;390:437–46. doi: 10.1042/BJ20050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. 2008;111:924–31. doi: 10.1182/blood-2007-07-100677. [DOI] [PubMed] [Google Scholar]
- 137.Silvestri L, Pagani A, Nai A, De Domenico I, Kaplan J, Camaschella C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8:502–11. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Soe-Lin S, Apte SS, Andriopoulos B, Jr, Andrews MC, Schranzhofer M, et al. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc Natl Acad Sci U S A. 2009;106:5960–5. doi: 10.1073/pnas.0900808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sow FB, Alvarez GR, Gross RP, Satoskar AR, Schlesinger LS, et al. Role of STAT1, NF-kappaB, and C/EBPbeta in the macrophage transcriptional regulation of hepcidin by mycobacterial infection and IFN-gamma. J Leukoc Biol. 2009;86:1247–58. doi: 10.1189/jlb.1208719. [DOI] [PubMed] [Google Scholar]
- 140.Sow FB, Florence WC, Satoskar AR, Schlesinger LS, Zwilling BS, Lafuse WP. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J Leukoc Biol. 2007;82:934–45. doi: 10.1189/jlb.0407216. [DOI] [PubMed] [Google Scholar]
- 141.Tacchini L, Gammella E, De Ponti C, Recalcati S, Cairo G. Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J Biol Chem. 2008;283:20674–86. doi: 10.1074/jbc.M800365200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 143.Tanno T, Porayette P, Sripichai O, Noh SJ, Byrnes C, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009;114:181–6. doi: 10.1182/blood-2008-12-195503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Theurl I, Theurl M, Seifert M, Mair S, Nairz M, et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood. 2008;111:2392–9. doi: 10.1182/blood-2007-05-090019. [DOI] [PubMed] [Google Scholar]
- 145.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–74. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 146.Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99:3505–16. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- 147.Toyokuni S. Role of iron in carcinogenesis: cancer as a ferrotoxic disease. Cancer Sci. 2009;100:9–16. doi: 10.1111/j.1349-7006.2008.01001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Truksa J, Lee P, Beutler E. Two BMP responsive elements, STAT, and bZIP/HNF4/COUP motifs of the hepcidin promoter are critical for BMP, SMAD1, and HJV responsiveness. Blood. 2009;113:688–95. doi: 10.1182/blood-2008-05-160184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Truksa J, Peng H, Lee P, Beutler E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci U S A. 2006;103:10289–93. doi: 10.1073/pnas.0603124103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tsukamoto H. Iron regulation of hepatic macrophage TNFalpha expression. Free Radic Biol Med. 2002;32:309–13. doi: 10.1016/s0891-5849(01)00772-9. [DOI] [PubMed] [Google Scholar]
- 151.Valdez Y, Grassl GA, Guttman JA, Coburn B, Gros P, et al. Nramp1 drives an accelerated inflammatory response during Salmonella-induced colitis in mice. Cell Microbiol. 2009;11:351–62. doi: 10.1111/j.1462-5822.2008.01258.x. [DOI] [PubMed] [Google Scholar]
- 152.Vari IS, Balkau B, Kettaneh A, Andre P, Tichet J, et al. Ferritin and transferrin are associated with metabolic syndrome abnormalities and their change over time in a general population: Data from an Epidemiological Study on the Insulin Resistance Syndrome (DESIR) Diabetes Care. 2007;30:1795–801. doi: 10.2337/dc06-2312. [DOI] [PubMed] [Google Scholar]
- 153.Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, et al. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325:877–80. doi: 10.1126/science.1176639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109:353–8. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 155.Vidal SM, Malo D, Vogan K, Skamene E, Gros P. Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell. 1993;73:469–85. doi: 10.1016/0092-8674(93)90135-d. [DOI] [PubMed] [Google Scholar]
- 156.Volke M, Gale DP, Maegdefrau U, Schley G, Klanke B, et al. Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. PLoS One. 2009;4:e7875. doi: 10.1371/journal.pone.0007875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–9. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 158.Wallace DF, Subramaniam VN. Co-factors in liver disease: the role of HFE-related hereditary hemochromatosis and iron. Biochim Biophys Acta. 2009;1790:663–70. doi: 10.1016/j.bbagen.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 159.Wang L, Harrington L, Trebicka E, Shi HN, Kagan JC, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest. 2009;119:3322–8. doi: 10.1172/JCI39939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wang L, Johnson EE, Shi HN, Walker WA, Wessling-Resnick M, Cherayil BJ. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J Immunol. 2008;181:2723–31. doi: 10.4049/jimmunol.181.4.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang RH, Li C, Xu X, Zheng Y, Xiao C, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 162.Wang X, Garrick MD, Yang F, Dailey LA, Piantadosi CA, Ghio AJ. TNF, IFN-gamma, and endotoxin increase expression of DMT1 in bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L24–33. doi: 10.1152/ajplung.00428.2003. [DOI] [PubMed] [Google Scholar]
- 163.Ward PP, Mendoza-Meneses M, Cunningham GA, Conneely OM. Iron status in mice carrying a targeted disruption of lactoferrin. Mol Cell Biol. 2003;23:178–85. doi: 10.1128/MCB.23.1.178-185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood. 2002;100:3776–81. doi: 10.1182/blood-2002-04-1260. [DOI] [PubMed] [Google Scholar]
- 165.Weiss G. Iron metabolism in the anemia of chronic disease. Biochim Biophys Acta. 2009;1790:682–93. doi: 10.1016/j.bbagen.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 166.Weiss G, Werner-Felmayer G, Werner ER, Grunewald K, Wachter H, Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med. 1994;180:969–76. doi: 10.1084/jem.180.3.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204–9. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wyllie S, Seu P, Goss JA. The natural resistance-associated macrophage protein 1 Slc11a1 (formerly Nramp1) and iron metabolism in macrophages. Microbes Infect. 2002;4:351–9. doi: 10.1016/s1286-4579(02)01548-4. [DOI] [PubMed] [Google Scholar]
- 169.Xiong S, She H, Takeuchi H, Han B, Engelhardt JF, et al. Signaling role of intracellular iron in NF-kappaB activation. J Biol Chem. 2003;278:17646–54. doi: 10.1074/jbc.M210905200. [DOI] [PubMed] [Google Scholar]
- 170.Yang F, Liu XB, Quinones M, Melby PC, Ghio A, Haile DJ. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J Biol Chem. 2002;277:39786–91. doi: 10.1074/jbc.M201485200. [DOI] [PubMed] [Google Scholar]
- 171.Yuan XM. Apoptotic macrophage-derived foam cells of human atheromas are rich in iron and ferritin, suggesting iron-catalysed reactions to be involved in apoptosis. Free Radic Res. 1999;30:221–31. doi: 10.1080/10715769900300241. [DOI] [PubMed] [Google Scholar]
- 172.Zhang K, Shen X, Wu J, Sakaki K, Saunders T, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124:587–99. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]