

Summary
Glucagon-like-peptide-1 receptor (GLP-1R) activation within the nucleus tractus solitarius (NTS) suppresses food intake and body weight (BW), but the intracellular signals mediating these effects are unknown. Here, hindbrain (4th icv) GLP-1R activation by Exendin-4 increased PKA and MAPK activity and decreased phosphorylation of AMPK in NTS. PKA and MAPK signaling contribute to food intake and BW suppression by Exendin-4, as inhibitors RpcAMP and U0126 (4th icv), respectively, attenuated Exendin-4's effects. Hindbrain GLP-1R activation inhibited feeding by reducing meal number, not meal size. This effect was attenuated with stimulation of AMPK activity by AICAR (4th icv). The PKA, MAPK and AMPK signaling responses by Ex-4 were present in immortalized GLP-1R-expressing neurons (GT1-7). In conclusion, hindbrain GLP-1R activation suppresses food intake and BW through coordinated PKA-mediated suppression of AMPK and activation of MAPK. Pharmacotherapies targeting these signaling pathways, which mediate intake-suppressive effects of CNS GLP-1R activation, may prove efficacious in treating obesity.
Introduction
Glucagon-like-peptide-1 (GLP-1), a neuropeptide endogenously produced from “L” cells in the gastrointestinal tract and centrally in neurons of the nucleus tractus solitarius (NTS), plays an important role in glycemic regulation and energy balance [see (Hayes et al., 2010a; Holst, 2007) for review]. Accumulating clinical and basic science data suggests that pharmacotherapies targeting the GLP-1 system are ameliorative not only for Type II Diabetes Mellitus (T2DM), but for obesity as well (Lovshin and Drucker, 2009). Interestingly, exogenous activation of either peripheral or central GLP-1 receptors (GLP-1R) produces a comparable profile of physiological responses that include: enhanced glucose-stimulated insulin secretion (Komatsu et al., 1989), inhibition of gastric emptying (Hayes et al., 2008b, c; Imeryuz et al., 1997; Wettergren et al., 1993), and inhibition of food intake (Chelikani et al., 2005; Gutzwiller et al., 1999; Hayes et al., 2009a; Hayes et al., 2008c; Kinzig et al., 2002; Moran et al., 1996; Turton et al., 1996). Although much is known about the mechanisms by which peripheral GLP-1R activation engages these physiological responses, the relevant central GLP-1R populations and their projections, as well as the intracellular signaling cascades that mediate the food intake inhibitory effects of central GLP-1R-activation have received considerably less attention.
GLP-1Rs are widely expressed throughout the central nervous system (CNS) in many sites relevant to energy balance regulation, including nuclei in the caudal brainstem (NTS; parabrachial nucleus), hypothalamus (lateral hypothalamus, LH; paraventricular hypothalamus, PVH), and basal forebrain (bed nucleus of stria terminalis; central nucleus of the amygdala) (Goke et al., 1995; Larsen et al., 1997; Merchenthaler et al., 1999). Recent reports highlight the importance of hindbrain GLP-1R-expressing nuclei in the control of food intake and body weight (Hayes et al., 2009a; Hayes et al., 2008c; Kinzig et al., 2002). Specifically, the endogenous activation of GLP-1R within the caudal medial-subnucleus of the NTS (mNTS) was shown to contribute to the normal control of food intake by mediating satiation signaling that arises from mechanical distension of the stomach (Hayes et al., 2009a). The intracellular signaling pathways within GLP-1R-expressing NTS neurons that mediate the food intake and body weight suppressive effects of CNS GLP-1 action have not been characterized.
In pancreatic β-cells, GLP-1R activation leads to stimulation of adenylate cyclase, elevated cAMP, and subsequent activation of protein kinase A (PKA) (Gomez et al., 2002; Perfetti and Merkel, 2000). Given that PKA signaling is upstream of many signaling pathways implicated in energy balance regulation, it is likely that the intake suppressive effects of hindbrain GLP-1R activation involve endemic signaling pathways that alter intracellular Ca2+-influx and/or longer-term alterations in gene transcription and protein synthesis to integrate various anorectic signals. Whether the intake suppressive effects of hindbrain GLP-1R activation are indeed dependent upon an increase in PKA activity within GLP-1R-expressing NTS neurons remains to be tested.
In neurons, elevated cAMP levels increase phosphorylation of the p44/42 mitogen-activated protein kinase [p44/42 MAPK; also known as: extracellular signal-regulated kinase (ERK)-1/2] (Dugan et al., 1999; Vossler et al., 1997), presumably through a PKA-dependent pathway (Ma et al., 2007). GLP-1 stimulates p44/42 MAPK phosphorylation in pancreatic β-cells through a mitogen-activated protein kinase kinase (MEK)-dependent, but Raf/Ras-independent pathway that requires PKA activation, an influx of extracellular Ca2+, and calmodulin-dependent protein kinase II (CaMKII) activation (Gomez et al., 2002). Previous reports highlight a role for p44/42 MAPK signaling within the NTS in food intake control (Sutton et al., 2005; Sutton et al., 2004). Yet, direct in vivo and in vitro evaluation of the MAPK pathway mediating the intake suppressive effects of hindbrain GLP-1R activation remains unaddressed.
PKA activation is also known to inhibit calmodulin-dependent protein kinase kinase (CaMKK) (Hurley et al., 2006). CaMKK, together with LKB1, have been identified as the principal upstream kinases which phosphorylate the fuel sensing enzyme AMP-activated protein kinase (AMPK) α catalytic subunit at Thr172 [see (Xue and Kahn, 2006) for review]. Thus, a PKA-induced inhibition of CaMKK activity would lead to decreased AMPK activation (Hurley et al., 2006). AMPK has been implicated in CNS control of energy balance (Minokoshi et al., 2004; Seo et al., 2008; Xue and Kahn, 2006), and its activity is altered by food deprivation (da Silva Xavier et al., 2003; Minokoshi et al., 2004; Xue and Kahn, 2006) and by the effects of leptin, insulin-induced hypoglycemia or 2-DG cytoglucopenia (Han et al., 2005; Hayes et al., 2009b; Hayes et al., 2010b; Kim and Lee, 2005; Kim et al., 2004b; Minokoshi et al., 2004). In addition, increased hypothalamic AMPK mRNA following food deprivation was attenuated by GLP-1R activation (Seo et al., 2008). Recently, the role of AMPK activity in NTS control of energy balance has also been evaluated (Hayes et al., 2008a). Similar to previous reports evaluating AMPK activity in various hypothalamic nuclei (Minokoshi et al., 2004; Xue and Kahn, 2006), it was found that food deprivation increases AMPK activity in NTS lysates and that AMPK activity within the NTS is physiologically relevant to the normal control of food intake (Hayes et al., 2009b; Hayes et al., 2010b). Together, these data support the hypothesis that reduced AMPK activity in the hindbrain may mediate the suppression of intake that follows GLP-1R activation.
Using a combination of in vivo and in vitro techniques we evaluate the cellular signaling pathways that mediate the food intake and body weight suppressive effects of hindbrain GLP-1R activation by the GLP-1R selective agonist, Exendin-4 (Ex-4). We hypothesize that activation of NTS-GLP-1R-expressing neurons suppresses food intake through coordinated PKA-mediated-suppression of AMPK activity and PKA-mediated-activation of p44/42 MAPK.
Results
Hindbrain GLP-1R activation reduces food intake by increasing PKA activity
Hindbrain administration of the GLP-1R agonist Ex-4 (0.3μg; 4th icv), significantly increased PKA activity in the dorsal vagal complex [DVC; which includes NTS, dorsal motor nucleus of the vagus (DMV) and area postrema (AP)] at 10 and 20min-post injection compared to aCSF (Figure 1a). By 30 min, the Ex-4 driven elevation in NTS PKA activity had returned to baseline levels.
Figure 1.

(A) Increased PKA activity (nmol/g/min) in tissue of the caudal DVC following 4th icv Ex-4 (0.3μg) administration. * = P< 0.05 from aCSF. Cumulative chow intake (B) and 24h body weight change (C) following 4th icv delivery of the PKA inhibitor RpcAMP (20μg) or Ex-4 (0.2μg) alone or in combination, counterbalanced with vehicle/vehicle (aCSF/aCSF). Data are mean ± SEM. * = P< 0.05 from vehicle/vehicle. † = P< 0.05 from vehicle/Ex-4.
To test the hypothesis that the intake suppressive effects of hindbrain GLP-1R activation requires PKA activity, intake of standard rodent chow was examined following counterbalanced 4th icv administration of Ex-4 (0.2μg) alone or in combination with the widely used PKA-inhibitor, RpcAMP (20μg). Figure 1b shows that cumulative food intake was significantly suppressed at 3h, 6h and 24h following Ex-4 administration. This intake suppression was dependent on an increase in hindbrain PKA activity, as 4th icv pretreatment with the PKA inhibitor RpcAMP reversed the intake suppressive effects of 4th icv Ex-4 at 3h and 6h, and attenuated the intake suppression at 24h. 4th icv Ex-4 administration also significantly suppressed body weight 24h after administration compared to vehicle/vehicle (aCSF/aCSF) treatment (Figure 1c). Pretreatment with RpcAMP significantly attenuated the suppression of body weight by 4th icv Ex-4, whereas RpcAMP alone did not affect body weight.
Hindbrain GLP-1R activation suppresses food intake by increasing phosphorylation of MAPK
Immunoblot analysis revealed that 4th icv Ex-4 (0.3μg) administration increased p44/42 MAPK phosphorylation in DVC tissue compared to vehicle (aCSF) (Figure 2a; data presented as phosphorylated / total MAPK). Specifically, hindbrain Ex-4 significantly increased phosphorylation of p44 MAPK at 10min- and 30min-post administration with no change in total p44 levels. Likewise, 4th icv Ex-4 increased phosphorylation of p42 at 10, 20, and 30min-post administration. For both p44 and p42 MAPK, the greatest increase in phosphorylation was observed at 30min following hindbrain GLP-1R activation.
Figure 2.
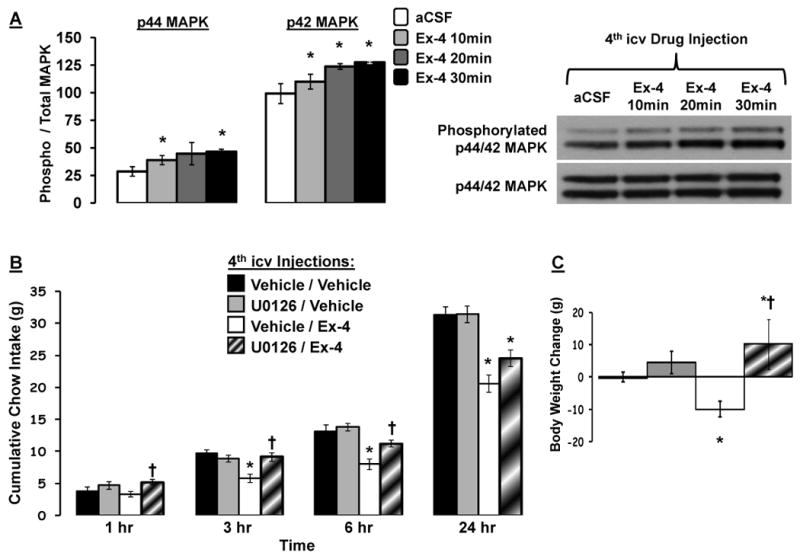
(A) Increased phosphorylation of p44/42-MAPK in caudal DVC tissue lysates following 4th icv Ex-4 (0.3μg) administration. Representative immunoblots for phosphorylated and total p44/42-MAPK are shown. * = P< 0.05 from aCSF. Cumulative chow intake (B) and 24h body weight change (C) following 4th icv delivery of the MEK inhibitor U0126 (2μg) or Ex-4 (0.2μg) alone or in combination, counterbalanced with vehicle/vehicle (DMSO/aCSF). Data are mean ± SEM. * = P< 0.05 from vehicle/vehicle. † = P< 0.05 from vehicle/Ex-4.
Food intake was significantly suppressed following 4th icv Ex-4 (0.2μg) administration in a separate group of rats at 3h, 6h and 24h post-injection compared to vehicle/vehicle (DMSO/aCSF) administration (Figure 2b). Blockade of MAPK signaling by 4th icv administration of the MEK inhibitor, U0126, had no effect on food intake when administered alone. Pretreatment with U0126, however, significantly reversed the inhibition of intake by Ex-4 at 3h and 6h. Body weight was also significantly suppressed 24h after 4th icv Ex-4 administration compared to vehicle/vehicle treatment (Figure 2c). 4th icv administration of U0126, while having no effect on 24h body weight when administered alone, significantly attenuated the suppression of body weight by 4th icv Ex-4.
Hindbrain GLP-1R activation reduces food intake via inhibition of AMPK signaling
Immunoblot analysis revealed that hindbrain GLP-1R activation by 4th icv Ex-4 (0.3μg) significantly reduced phosphorylation of AMPKα2 in DVC tissue at 20min- and 30min-post administration, with no change in total AMPKα levels (Figure 3a; data presented as pAMPKα2 / total AMPKα). Inhibition of NTS AMPK activity following hindbrain GLP-1R activation was greatest at 30min.
Figure 3.
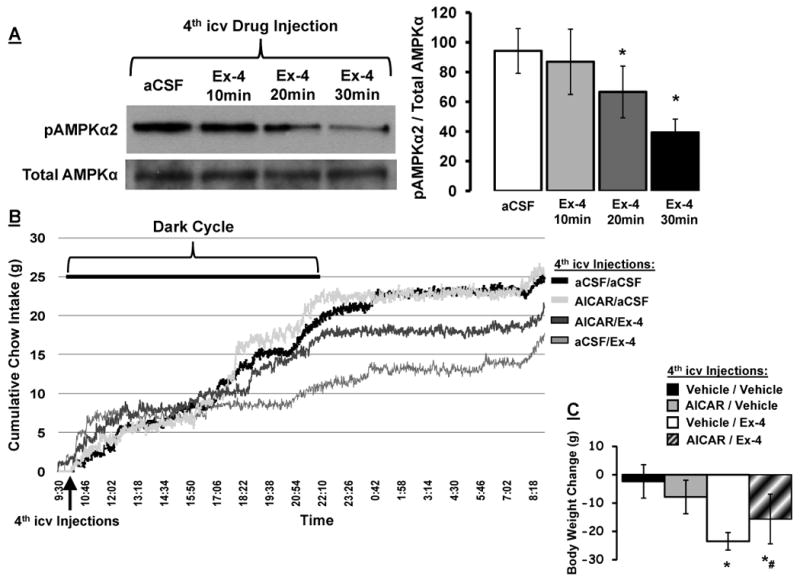
(A) Decreased phosphorylation of AMPKα2 in caudal DVC tissue lysates following 4th icv Ex-4 (0.3μg) administration. Representative immunoblots for pAMPKα2 and total AMPKα are shown. * = P< 0.05 from aCSF. Cumulative chow intake (B) and 24h body weight change (C) following 4th icv delivery of the AMPK activity promoter AICAR (300μg) or Ex-4 (0.1μg) alone or in combination, counterbalanced with vehicle/vehicle (aCSF/aCSF). Chow intake recorded via automated feedometers that continuously record food intake to the nearest ±0.1g/m/24h. Data are mean ± SEM. * = P< 0.05 from vehicle/vehicle. # = P=0.065 from vehicle/Ex-4.
In a separate cohort of rats, the AMPK activity promoter, AICAR, was used to investigate whether inhibition of AMPK activity mediates the suppression of food intake by Ex-4 (measured in automated feedometers that record food intake to the nearest ±0.1g/m/24h every 1min). Hindbrain GLP-1R activation by 4th icv Ex-4 suppressed food intake in the dark cycle by reducing meal number and increasing inter-meal-interval, not by altering meal size (Figure 4a). A maximum 44% intake suppression by Ex-4 occurred 12h-post injection (at the start of light cycle; Figure 3b). 4th icv AICAR attenuated Ex-4's intake suppressive effect by approximately 50% (Figure 3b) and attenuated Ex-4's suppression of meal number and increase in inter-meal-interval (Figure 4a). The change in 24h body weight was significantly suppressed by 4th icv Ex-4 (Figure 3c) compared to body weight change following vehicle. AICAR alone did not alter 24h body weight. Pretreatment with AICAR produced a non-significant trend of an attenuation in the suppression of body weight by 4th icv Ex-4 (p=0.065 for body weight change comparison between vehicle/Ex-4 and AICAR/Ex-4 treatments), whereas AICAR alone did not alter 24h body weight.
Figure 4.
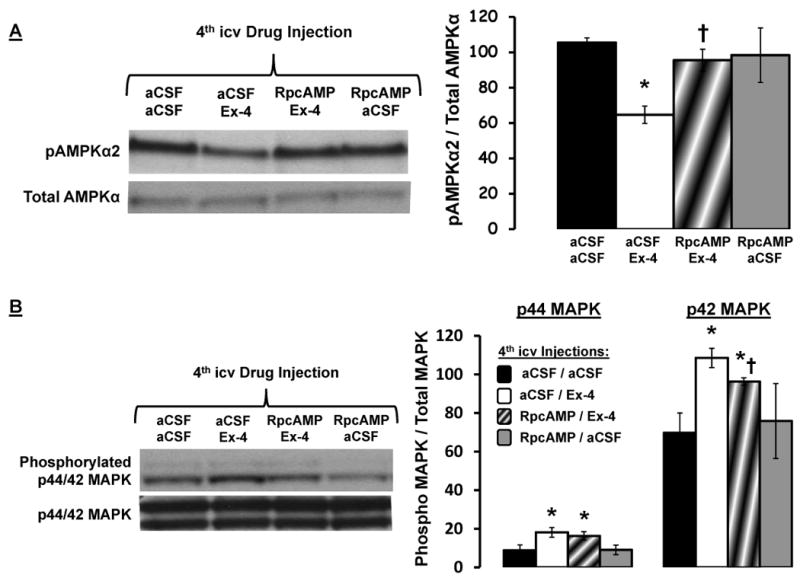
(A) 4th icv Ex-4 (0.3μg)-driven decrease phosphorylation in AMPKα2 in caudal DVC tissue lysates is PKA dependent, as 4th icv administration of the PKA inhibitor RpcAMP (20μg) attenuated the suppression in pAMPKα2 by Ex-4. Representative immunoblots for pAMPKα2 and total AMPKα are shown. * = P< 0.05 from aCSF/aCSF. † = P< 0.05 from aCSF/Ex-4. (B) RpcAMP administration attenuated the increased phosphorylation of p42-MAPK by Ex-4 administration, but did not alter the increased phosphorylation of p44-MAPK by Ex-4. Data are mean ± SEM. * = P< 0.05 from aCSF/aCSF. † = P< 0.05 from aCSF/Ex-4.
Hindbrain GLP-1R activation (4th icv Ex-4) reduces food intake that results from the suppression in meal number, with no alteration in meal size (Supplemental Figures 1a, 1b). In a separate group of rats, IP Ex-4 (3.0μg/kg) administration suppressed food intake by approximately 47% at 5hr-post drug delivery through a reduction in meal size, with no alteration in meal number (Supplemental Figure 1b). Thus, the time point chosen for meal pattern assessment for 4th icv and IP Ex-4 administration in Supplemental Figure 1a and 1b are based on comparable maximum percent suppression of chow intake (∼45% suppression).
The hindbrain GLP-1R-mediated increase in MAPK and reduction in AMPK signaling are PKA-dependent
Immunoblot analysis showed that the increase in MAPK phosphorylation and decrease in pAMPKα2 by 4th icv Ex-4 administration is dependent, in part, on PKA activation. Inhibition of hindbrain PKA activity by 4th icv pretreatment with RpcAMP (20μg) significantly attenuated the Ex-4 (0.3μg; 4th icv)-induced decrease in NTS pAMPKα2 levels (Figure 4a; data presented as pAMPKα2 / total AMPKα). Likewise, RpcAMP significantly attenuated the Ex-4-induced increase in phosphorylated p42-MAPK levels, but not p44-MAPK levels in DVC tissue (Figure 4b; data presented as phosphorylated / total MAPK).
Medial NTS GLP-1R's mediate the intake suppressive effects of hindbrain (4th icv) Ex-4
Ex-4 was administered intraparenchymally to the mNTS, AP and DMV to determine the hindbrain-specific GLP-1R-expressing nucleus within the DVC that mediates the intake suppressive effects of 4th icv Ex-4. First, a sub-threshold dose for intake suppression of Ex-4 was determined for 4th icv delivery, as 4th icv Ex-4 (0.05μg/1μl) significantly suppressed chow intake at 6h- and 24h-post administration, whereas 4th icv Ex-4 (0.025μg/1μl) was without effect (Figure 5a). Unilateral intraparenchymal mNTS administration of Ex-4 at 0.025μg/100nl significantly suppressed chow intake at 3h, 6h, and 24h-post administration; 0.05μg/100nl was also effective (Figure 5b). Conversely, unilateral DMV administration of Ex-4 at 0.05μg/100nl, a dose effective following either 4th icv or mNTS delivery, was without effect on food intake (Figure 5c). A similar absence of an intake-suppressive effect was observed following intraparenchymal Ex-4 administration to the AP at 0.025μg/100nl and 0.05μg/100nl (Figure 5d).
Figure 5.
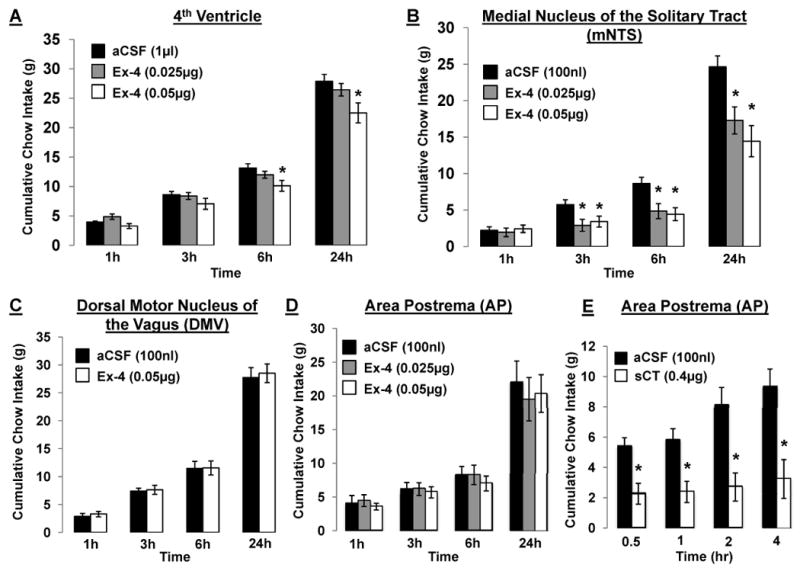
Cumulative chow intake following (A) hindbrain (4th icv) delivery of Ex-4 (0.025μg and 0.05μg in 1μl), (B) unilateral intraparenchymal mNTS delivery of Ex-4 (0.025μg and 0.05μg in 100nl), (C) unilateral intraparenchymal DMV delivery of Ex-4 (0.05μg in 100nl), (D) intraparenchymal AP delivery of Ex-4 (0.025μg and 0.05μg in 100nl), counterbalanced with aCSF. (E) Functional verification of AP injections measured by cumulative 4h chow intake following AP administration of the amylin (calcitonin) receptor agonist, salmon calcitonin (sCT) (0.4μg in 100nl) counterbalanced with aCSF. Data are mean ± SEM. * = P< 0.05 from aCSF.
In addition to post-mortem histological verification of intraparenchymal cannula placement for the mNTS, AP and DMV, functional tests were performed to further confirm placement of mNTS and AP injection sites. The AP has been shown to mediate the intake suppressive effects of the pancreatic hormone amylin and its agonist, salmon calcitonin (sCT) (Mollet et al., 2004; Potes and Lutz, 2010). AP administration of sCT at 0.4μg/100nl, a dose previously established to be just at threshold for intake suppression when administered to the 4th ventricle (Mollet et al., 2004), produced a significant suppression of food intake at all time points measured (Figure 5e), thus providing functional confirmation of our AP site-of-delivery. To confirm mNTS injection sites, the cytoglucopenia-induced sympatho-adrenal mediated glycemic effect resulting from intraparenchymal injection of 24μg of 5-thio-D-glucose in 100nl of aCSF (Hayes et al., 2009b; Hayes et al., 2010b) was performed. A hyperglycemic response (≥100% of basal glycemia) verified cannula placement.
GLP-1R-expressing GT1-7 neurons show Exendin-4-mediated increases in cAMP/PKA and MAPK signaling, and suppression of AMPK signaling
Total cAMP levels were evaluated in GLP-1R-expressing GT1-7 neuronal cells (Beak et al., 1998; Yang et al., 2005) following 15min of Ex-4 treatment (0.0, 0.01, 0.1, 1.0, 10 nmol). Figure 6a shows that GLP-1R activation by Ex-4 dose dependently increased total cAMP levels in GT1-7 neurons, with 0.1, 1.0, and 10.0 nmol of Ex-4 significantly increasing total cAMP levels compared to vehicle (aCSF).
Figure 6.
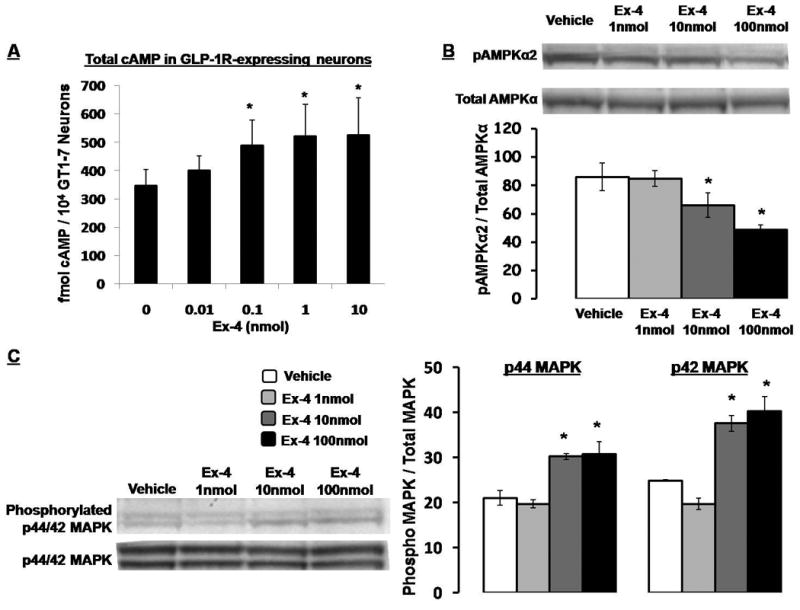
(A) Total cAMP levels in 3hr serum-starved GLP-1R-expressing GT1-7 neuronal cells following 15min treatment with Ex-4 (0.0, 0.01, 0.1, 1.0, 10 nmol). * = P< 0.05 from vehicle (0.0nmol Ex-4). (B) Ex-4 treatment (0.0, 1.0, 10, and 100 nmol; 15min) dose-dependently decreased pAMPKα2 and (C) increased phosphorylated p44/42 MAPK in the GLP-1R-expressing GT1-7 neurons. Data are mean ± SEM. * = P< 0.05 from vehicle (0.0nmol Ex-4).
Immunoblot analysis showed a significant dose-dependent decrease in pAMPKα2 levels in GT1-7 neurons following 15min Ex-4 treatment (0.0, 1.0, 10, and 100 nmol; Figure 6b; data presented as pAMPKα2 / total AMPKα). In addition, Ex-4 treatment dose-dependently increased phosphorylated p44/42 MAPK levels in GT1-7 neurons (Figure 6c; data presented as phosphorylated / total MAPK). Ex-4 treatment did not, however, significantly alter either total AMPKα or total p44/42-MAPK levels.
Discussion
Pharmacological agents targeting the peripheral GLP-1 system are used to treat T2DM, and may also prove to be efficacious in obesity treatment (Hayes et al., 2010a; Holst, 2007; Larsen et al., 2003; Lovshin and Drucker, 2009). A variety of evidence indicates that activation of central GLP-1Rs engages a behavioral and physiological profile of responses that is similar to that observed following activation of peripheral GLP-1Rs and therefore may provide another pharmacological strategy to regulate blood glucose levels and reduce excessive adiposity by decreasing energy intake. Of the various brain regions containing GLP-1R-expressing neurons, previous reports establish those in the NTS as a physiologically relevant population of central GLP-1Rs in food intake control (Hayes et al., 2009a; Hayes et al., 2008c; Kinzig et al., 2002). The intracellular signaling pathways mediating food intake suppression following NTS GLP-1R activation have not been previously established. Here, we show that the suppression of food intake and body weight following activation of hindbrain GLP-1Rs involves a coordinated PKA-mediated suppression in AMPK activity and activation of p44/42-MAPK signaling in the NTS.
Our working model [Figure 7; modified from (Hayes et al., 2010a)] derives from the current data, as well as previous reports examining the intracellular signaling cascades following GLP-1R activation in pancreatic β-cells (Gomez et al., 2002; Perfetti and Merkel, 2000), from known signaling pathways downstream of cAMP/PKA activation in neuronal cells (Dugan et al., 1999; Vossler et al., 1997), and from recent reports showing that increased AMPK mRNA in the hypothalamus following food deprivation is attenuated by GLP-1R activity (Seo et al., 2008). It is likely that the intake suppressive effects following NTS GLP-1R activation, that involve increased PKA and MAPK signaling and decreased AMPK signaling, engage additional intracellular signaling cascades, as well as neuronal projections to other brain regions relevant to energy balance control. In addition to the intake inhibitory responses produced by 4th icv Ex-4 administration, hindbrain GLP-1R signaling also triggers energy expenditure responses including hypothermia and tachycardia (Hayes et al., 2008c). Interestingly, inhibition of hindbrain AMPK activity also increases heart rate and locomotor activity in rats. Therefore, it is plausible that energy expenditure responses produced by hindbrain GLP-1R activation would involve AMPK signaling (Hayes et al., 2009b). However, it remains to be determined whether energetic responses mediated by hindbrain GLP-1R activation involve the same PKA, MAPK, and AMPK intracellular signaling cascades as described here for food intake.
Figure 7.
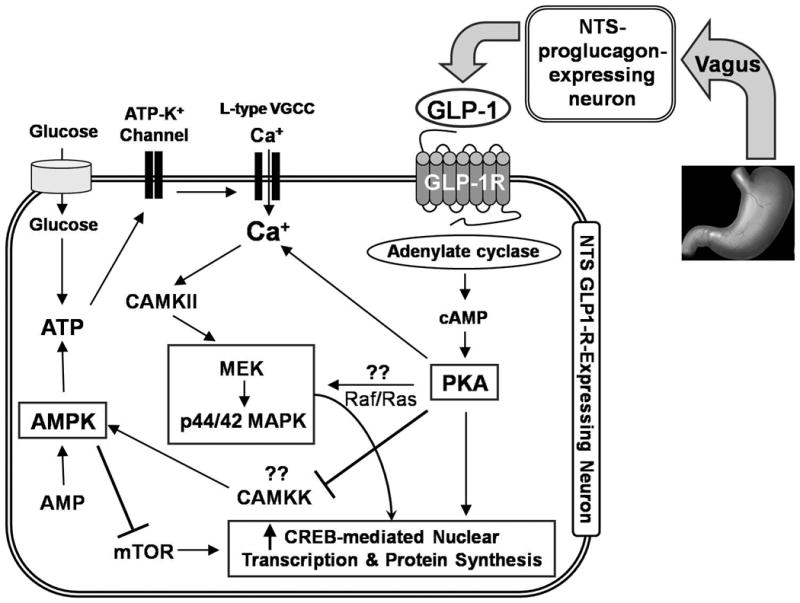
Proposed signaling pathways in NTS-GLP-1R-expressing neurons mediating suppression of intake by central GLP-1. Gastric vagal afferent signaling increases endogenous NTS-derived GLP-1 which activates endemic NTS GLP-1R-expressing neurons (Hayes et al., 2009a) to engage cAMP-dependent increase in PKA activity. Increased PKA activity concomitantly increases phosphorylation of p44/42-MAPK and decreases phosphorylation of AMPK. The combined increase in PKA and p44/42-MAPK activity together with decreased AMPK activity is hypothesized to increase CREB-mediated nuclear transcription and protein synthesis. mTOR = mammalian target of rapamycin; CAMKK = Calmodulin-dependent protein kinase kinase; CAMKII = calcium/calmodulin-dependent protein kinase II; VGCC = voltage gated calcium channel; MEK = mitogen-activated protein kinase kinase.
Previous reports show that the GLP-1R-expressing NTS neurons are the likely mediators of the intake suppressive effects of hindbrain ventricular Ex-4 administration (Hayes et al., 2009a; Hayes et al., 2008c). Direct pharmacological blockade of mNTS GLP-1Rs increases food intake (Hayes et al., 2009a), indicating that endogenous NTS GLP-1R activation contributes to the physiological control of food intake. Vagal afferent signals arising from distension of the stomach appear to engage NTS GLP-1R signaling (Hayes et al., 2009a) following activation of the proglucagon-expressing neurons of the NTS (Vrang et al., 2003) which increases endogenous CNS GLP-1 ligand availability. Data presented in this report show that the food intake suppressive effects of DVC parenchymal application of a GLP-1R ligand is mediated by mNTS neurons, as a significant suppression of intake was observed following Ex-4 delivery to the mNTS at a 4th icv sub-threshold dose (0.025μg); effects not observed following administration of Ex-4 at the same dose or even at a ventricular-effective dose (0.05μg) to other hindbrain GLP-1R-expressing nuclei (i.e. AP or DMV). Moreover, using a combination of in vivo and in vitro techniques with the GLP-1R ligand Ex-4, we demonstrate that the intake suppressive effects of NTS-GLP-1R activation occur through a coordinated PKA-mediated suppression of AMPK activity and activation of p44/42 MAPK signaling. These effects may promote Ca+-dependent depolarization of the GLP-1R expressing neurons and longer-term cAMP response element-binding protein (CREB)-mediated transcriptional effects. The present findings further suggest that the intake suppressive effects of gastric distension may also be mediated in part, by an increase in PKA, MAPK, and possibly CREB signaling, as well as inhibition of AMPK signaling within NTS neurons. It may also be the case that NTS mediation of the intake inhibitory responses of other anorectic signals [e.g. cholecystokinin, melanocortin, leptin] that engage these intracellular signaling pathways (Hayes et al., 2009b; Hayes et al., 2010b; Sutton et al., 2005; Sutton et al., 2004)] could potentially reduce food intake in a synergistic fashion with either CNS GLP-1R ligands and/or gastric distension. Given that PKA inhibition did not completely reverse the 24h intake suppression by 4th icv Ex-4, it is also reasonable to assume that other non-PKA-dependent signaling pathways may mediate the suppression of intake by hindbrain GLP-1R activation.
While caudal brainstem processing (in the absence of forebrain communication) is sufficient to mediate the food intake suppressive effects following 4th icv Ex-4 administration (Hayes et al., 2008c), the NTS is not the only CNS GLP1-R-expressing nucleus relevant to energy balance control (e.g. PVH; central nucleus of the amygdala, CeA; dorsal medial hypothalamus, DMH). Indeed, parenchymal application of GLP-1(7-36) in the PVH suppresses food intake in rats (McMahon and Wellman, 1997, 1998). Whether the intracellular signaling cascades for PVH or other GLP-1R-expressing neurons are identical to those reported here for the DVC GLP-1R remains to be determined. It may be possible that in GLP-1R-expressing nuclei outside the NTS, different intracellular signaling pathways may mediate different behavioral/physiological responses produced by GLP-1R activation. Such a case has been made previously for the angiotensin receptor (Daniels et al., 2005). Nonetheless, it is reasonable to assume that the signaling cascades for GLP-1R-expressing neurons are similar throughout the brain, as the ex vivo signaling responses reported here for the DVC tissue lysates were also found in vitro in immortalized hypothalamic GLP-1R-expressing GT1-7 cells. It is virtually impossible to rule out the possibility that intracellular signaling inhibitors/activators (e.g., U0126/AICAR) attenuate the behavioral intake suppression of 4th icv Ex-4 by acting on both GLP-1R- and non-GLP-1R-expressing neurons.
Hindbrain Ex-4 administration rapidly increases NTS PKA activity, with a peak in activity occurring 10min-post administration and a return to baseline (vehicle) levels seen at 30min-post administration. Yet, behavioral analyses show that the suppression of food intake by 4th icv Ex-4 is observed by the 3rd hour post-administration, an effect dependent upon an increase in PKA and MAPK activity, as well as a decrease in AMPK activity. This distinction in the time-course effects for the intracellular signaling cascades from tissue lysates and those for the food intake response have been reported previously for other anorectic ligands and intracellular signals acting within the brain (Morton et al., 2009; Niswender et al., 2001; Sutton et al., 2005). These observations can be interpreted to suggest that although PKA-, MAPK-, and AMPK-signaling are required to mediate the suppression of intake by NTS GLP-1R activation, other downstream signaling cascades and recruitment of neurons of other CNS nuclei are also required to mediate the intake-suppressive action of hindbrain Ex-4. These additional signaling cascades and neuronal projections arising from NTS GLP-1R-expressing neurons remain to be identified.
That suppression in meal number accounts for the reduced food intake resulting from hindbrain Ex-4 supports the notion that the central GLP-1 system is different, but complementary to the peripheral GLP-1 system. This conclusion is based in part on data presented here, as well as previous observations (Ruttimann et al., 2009; Scott and Moran, 2007) that show that the short term intake inhibitory effects of peripheral GLP-1R activation result from a reduction in meal size, with minimal effect on meal number. It may be possible that peripheral administration of Ex-4 or other DPP-IV-resistant GLP-1R ligands with longer half-lives than GLP-1(7-36) (e.g., liraglutide) may reduce food intake across a longer period of time by simultaneous activation of both peripheral and central GLP-1 systems.
In conclusion, current findings demonstrate that the food intake- and body weight-suppressive effects of hindbrain GLP-1R activation are dependent upon an increase in PKA activity and subsequent simultaneous phosphorylation of p44/42-MAPK and inhibition of AMPK activity in NTS neurons. Ex-4 activation of the same signaling cascades in GT1-7 neuronal cells and in NTS lysates supports the view that these pathways occur in GLP-1R-expressing neurons. Together with our previous reports (Hayes et al., 2009a; Hayes et al., 2008c), current data demonstrate that NTS GLP-1R-expressing neurons play a physiological role in control of food intake and energy balance regulation. These data provide clues to understanding how NTS neurons mediate anorectic actions of other stimuli involved in meal-to-meal food intake control. Future pharmacotherapies targeting not only NTS GLP-1R, but the CNS GLP-1 system as a whole, may prove to be efficacious in ameliorating chronic hyperphagia associated with obesity.
Experimental Procedures
Subjects and Materials
Individually housed, adult male Sprague Dawley rats (300-350g; Charles River), maintained at 23 °C with 12h:12h light/dark cycle (10:00h lights off), had ad libitum access to chow (Purina Rodent Chow 5001) and water except as noted. All procedures conformed to the institutional standards of the animal care and use committee (University of Pennsylvania).
The following compounds were utilized to examine hindbrain GLP-1R-mediated control of food intake: The DPP-IV resistant, GLP-1R agonist, Exendin-4 (Ex-4; American Peptide Co.) (Hayes et al., 2008c); Adenosine-3′,5′-cyclic monophosphorothioate Rp-isomer (RpcAMP; Sigma), a widely used inhibitor of cAMP-dependent activation of PKA (Dostmann et al., 1990; Kelly et al., 2007; Sheriff et al., 2003); The AMP-mimicking promoter of AMPK activity, 5-Aminoimidazole-4-carboxamide-1-β-D-riboside (AICAR; Fisher Scientific) (Hayes et al., 2009b; Kim et al., 2004a); and the selective MEK inhibitor U0126 (Tocris Bioscience), which blocks p44/42 MAPK phosphorylation (Daniels et al., 2003; Sutton et al., 2005; Sutton et al., 2004). All drugs were administered in unrestrained animals in a counterbalanced fashion, with all drug combinations for a given experiment being administered on any given experimental day. For behavioral testing, each experimental trial was separated by a minimum of 48h.
4th intracerebroventricular / mNTS / AP / DMV cannulation surgery
Under ketamine (90 mg/kg), xylazine (2.7 mg/kg) and acepromazine (0.64 mg/kg) anesthesia and analgesia (Metacam 2mg/kg), guide cannulas (Plastics One; 26-gauge) were implanted at the following coordinates: 4th icv: 2.0 mm above 4th ventricle, 2.5 mm anterior to occipital suture, 4.5 mm ventral to dura, and on midline; bilateral caudal mNTS: 2.0 mm above mNTS, 1.0 mm posterior to occipital crest, ± 0.7 mm lateral to midline, 6.7 mm ventral from skull surface; AP: 2.0 mm above AP, 1.0 mm posterior to occipital crest, on midline, 6.4 mm ventral from skull surface; bilateral DMV: 2.0 mm above DMV, 1.0 mm posterior to occipital crest, ± 0.7 mm lateral to midline, 6.9 mm ventral from skull surface (Hayes and Covasa, 2006;Hayes et al., 2009b; Hayes et al., 2010b; Paxinos, 1998). Intended anatomical positions of 4th icv/mNTS injection sites were evaluated 1 wk post-surgery as described (Hayes et al., 2009b; Hayes et al., 2010b). Behavioral testing began ∼12 days following surgery. All DVC intraparenchymal injection sites were anatomically confirmed via postmortem verification of the position of 100nl pontamine sky blue injections. Only animals passing both functional and histological verifications were included in final statistical analyses.
Signaling Analysis
ex vivo tissue collection
Tissue of the caudal DVC of ad libitum chow fed rats, were prepared as described (Hayes et al., 2009b; Minokoshi et al., 2004). Briefly, at dark cycle onset, rats received a 4th icv injection of Ex-4 (0.3μg) or aCSF vehicle and were sacrificed 10, 20, or 30min later by decapitation. Brains were rapidly removed; tissue lysates were collected from the DVC (Hayes et al., 2009b) and samples flash frozen in isopentane and stored at -80°C. To collect caudal DVC tissue, brains were positioned with dorsal surface up and the cerebellum was removed. The calamus scriptorius (most caudal portion of the 4th ventricle) provides a landmark for isolating caudal dorsomedial DVC tissue. Coronal sections were made 1mm anterior and 2mm posterior to the calamus scriptorius. Bilateral sagittal cuts were made ∼2mm lateral to the midline. A horizontal cut was then made 0.5 mm ventral to the central canal (Hayes et al., 2009b; Huo et al., 2006).
To determine whether the decrease in pAMPKα2 levels and increase in phosphorylated p44/42-MAPK levels by hindbrain GLP-1R activation are dependent on PKA activation, a separate group of ad libitum fed rats (n=3/treatment) received a 4th icv injection of aCSF vehicle (1μl) counterbalanced with RpcAMP (20μg) at dark cycle onset. Twenty min later, rats received a 4th icv injection of Ex-4 (0.3μg) counterbalanced with aCSF. Rats were sacrificed 20min following the second set of injections and brains were rapidly removed and tissue of the caudal DVC was collected.
in vitro GLP-1R-expressing immortalized GT1-7 neuronal cells
GT1-7 hypothalamic neuroblastoma cells (generous gift from Dr. Sangwon Kim, University of Pennsylvania), that highly express the GLP-1R (Beak et al., 1998; Yang et al., 2005), were maintained at 37°C in 5% CO2 and were cultured in 1× DMEM with 4.5 mg/ml glucose (GIBCO/Invitrogen), 10% (v/v) fetal bovine serum (Thermo Fisher Scientific Inc.), and 2% penicillin (10,000 I.U./ml)-streptomycin (10,000 ug/ml) solution (Mediatech, Inc.). Three hour serum-starved GT1-7 neurons were treated with Ex-4 (0.0, 1.0, 10, 100nmol) for 15 min. At the end of the incubation, medium was removed, cells were washed in ice-cold 0.1M PBS, and lysates were prepared by scraping cells into 500 μl of 1% NP-40 lysis buffer containing fresh protease and phosphatase inhibitors and repetitively passing the lysates through a 26-gauge needle followed by 10min centrifugation (12,000 rpm) at 4 °C to pellet out insoluble material.
Biochemistry
Caudal DVC tissue and GT1-7 neuronal cell-lysates were subjected to SDS-PAGE and transferred to PVDF membranes for immunoblot analysis (Hayes et al., 2009b; Hayes et al., 2010b). pAMPKα-Thr 172 rabbit monoclonal antibody (pAMPKα2; Cell Signaling; Cat. #: 2535S) was used to evaluate AMPK activity and was normalized to total AMPKα (reactive against α1 and α2 subunits; Cell Signaling). Phospho-p44/42 MAPK polyclonal antibody (Thr202/Tyr204; Cell Signaling; Cat. #: 9101S) was used to assess MAPK signaling and was normalized to total p44/42 MAPK (Cell Signaling; Cat. #: 9102). Blots were quantified using NIH Image software. Relative PKA activity of tissue and cell lysates was determined by ELISA (Stressgen Assay Designs; Cat # EKS-390A).
Determination of cAMP levels
Measurement of total cAMP was conducted using the Biotrak cAMP enzyme immunoassay system (GE Healthcare / Amersham Biosciences, USA). Three hour serum-starved GT1-7 neurons, cultured in a 96-well culture plate (6×104 cells/well), were treated with Ex-4 (0.0, 0.01, 0.1, 1.0, 10nmol) for 15 min. cAMP levels were calculated using a standard curve of fmol of cAMP / 104 GT1-7 neurons.
Feeding Behavior
Hindbrain PKA activity
One hour prior to the dark cycle, ad libitum fed rats (n=7) received a 4th icv injection of aCSF vehicle (1μl) counterbalanced with RpcAMP (20μg) [dose selection: (Kelly et al., 2007)] 45min prior to a 4th icv injection of Ex-4 (0.2μg) [dose subthreshold for intake suppression by IP administration from pilot work (data not shown) and (Mack et al., 2006)] counterbalanced with aCSF. Five minutes following the second drug administration, immediately prior to dark onset, rats were presented with pre-weighed maintenance rat chow and intakes were recorded to the nearest 0.1g at 1, 3, 6, and 24h-post food presentation.
Hindbrain MAPK signaling
One hour prior to the dark cycle, a separate group of ad libitum fed rats (n=8) received a 4th icv injection of vehicle [dimethylsulfoxide (DMSO); 1μl] counterbalanced with U0126 (2μg dissolved in DMSO) 45min prior to a 4th icv injection of Ex-4 (0.2μg) counterbalanced with aCSF [U0126 dose and injection time-course chosen from pilot dose response study (not shown) and (Sutton et al., 2005)]. Cumulative chow intake was recorded at 1, 3, 6, and 24h.
Hindbrain AMPK signaling
In a separate group of ad libitum fed rats (n=6), the role of hindbrain AMPK signaling in mediating the intake suppressive effects of 4th icv Ex-4 was evaluated using automated feedometers designed in our lab. Briefly, feedometers consisted of a standard Nalgene rodent bin (18 × 9 × 8 inches for length, width, depth respectively) with permanent, externally mounted food hoppers. Rats had ad libitum access to pelleted chow through a 3.5 × 2.5 inch square hole in the chamber. The externally mounted food hoppers were housed in and rested on a custom-built reinforced acrylic cup (to catch spillage) attached to a leveled load-cell (Omega Engineering, Inc; LCAE-1KG). The weight of the chow + spillage was recorded to the nearest 0.1g/min and relayed to computer via process meter (Omega Engineering, Inc; DP25B). Data were processed by computer software (National Instruments; LabVIEW; Penn 8 Channel Feed Monitor 1.0.0) to determine food ingestion each 1min and a total food intake curve was cumulated for 24h. Two hours prior to the dark cycle, automated food intake readings began. One hour later, rats received 4th icv injection of aCSF (3μl) counterbalanced with AICAR (300μg dissolved in aCSF), followed 45min later by a second 4th icv injection (1μl) of Ex-4 (0.1μg) counterbalanced with aCSF (Hayes et al., 2009b). A meal was defined as a bout of eating at least 0.25mg of food followed by 10min of no eating. These meal parameters accounted for >95% of the 24h cumulative food intake.
Meal pattern analysis of the intake suppressive effects following peripheral GLP-1R activation
In a separate group of ad libitum chow fed rats (n=7), the meal pattern effects mediating the intake suppression following peripheral (IP) Ex-4 (3.0μg/kg) administration were evaluated to allow for comparisons with the aforementioned meal pattern effects following 4th icv Ex-4. As detailed above, data were processed to determine the continuous cumulative intake for 24h, as well as analyzed for the meal-pattern effects of the drugs injected. Two hours prior to the dark cycle, automated food intake readings began. Fifteen minutes prior to the onset of the dark cycle, rats received an IP injection of Ex-4 counterbalanced with 0.9% saline vehicle (1ml/kg). Dose and time-course selection of drug delivery was chosen from (Hayes et al., 2009b) and pilot studies to yield a 40-50% maximum suppression in food intake.
Intraparenchymal mNTS / DMV / AP Ex-4 administration
In separate groups of rats fed chow ad libitum, individual nuclei within the DVC were targeted to determine which site[s] contributed to the mediation of the intake suppressive effects of hindbrain (4th icv) Ex-4 delivery was determined. First, to establish a 4th icv dose of Ex-4 that is without effect on intake we administered Ex-4 4th icv at 0.025μg and 0.05μg counterbalanced with 1μl aCSF, five minutes prior to dark onset. Rats (n=7) intake of chow was recorded at 1, 3, 6, and 24h.
Having established that the 0.025μg dose of Ex-4 is sub-threshold for intake suppression when administered to the 4th ventricle, the relevant GLP-1R-expressing nucleus within the DVC mediating the intake suppressive effects of 4th icv Ex-4 was determined by intraparenchymal administration of Ex-4 at a 100nl volume. A separate group of rats (n=12), implanted with bilateral mNTS cannula (at the level of the AP), received counterbalanced unilateral intraparenchymal mNTS injections of Ex-4 (0.025μg and 0.05μg in 100nl) or aCSF five minutes prior to dark onset. Likewise, rats (n=5) implanted with DMV cannula received counterbalanced injections of Ex-4 (0.05μg in 100nl) or aCSF five minutes prior to dark onset. Cumulative chow intake was recorded at 1, 3, 6, and 24h.
A separate set of rats (n=7) received counterbalanced intraparenchymal injections of Ex-4 (0.025μg and 0.05μg in 100nl) or aCSF into the AP five minutes prior to dark onset. Again, chow intake was recorded at 1, 3, 6, and 24h. To functionally verify the AP-site of injections, at the completion of the AP-Ex-4 experiment, the same rats received counterbalanced AP administration of the amylin agonist, salmon calcitonin (sCT) at 0.4μg/100nl or aCSF five minutes prior to dark onset. Food intake was recorded at 0.5, 1, 2, and 4h-post food presentation. Dose and time-course of food intake recording for the AP sCT experiment was conducted as reported (Mollet et al., 2004).
Data and Statistical Analyses
Data for each respective study were analyzed separately and expressed as mean ± SEM. For all experiments, comparisons between treatment means were analyzed by one- or two-way ANOVAs and, if appropriate, post-hoc, pair-wise comparisons were made using Tukey's honestly significant difference test with P<0.05 considered statistically significant. Analyses were made using PC-SAS (version 8.02, SAS Institute) mixed procedure.
Supplementary Material
(A) Hindbrain GLP-1R activation by 4th icv Ex-4 (0.1μg) reduced food intake in the 12h dark cycle by suppressing meal number and increasing inter-meal-interval, not by altering meal size. 4th icv delivery of the AMPK activity promoter AICAR (300μg) attenuated both suppression of meal number and increase in inter-meal-interval by 4th icv Ex-4. * = P< 0.05 from vehicle/vehicle. † = P< 0.05 from vehicle/Ex-4. (B) Peripheral GLP-1R activation by IP administration of Ex-4 (3.0μg/kg) significantly suppressed 5h dark cycle chow intake compared to vehicle (saline) by reducing meal size, not by altering meal number. Time points of meal pattern assessment for 4th icv and IP Ex-4 administration are chosen based on comparable maximum percent suppression of chow intake (∼45% suppression). Data are presented as mean ± SEM. * = P< 0.05 from vehicle.
Acknowledgments
We thank: Dr. Karolina Skibicka, Amber Alhadeff, Samantha Fortin, Ryan Tsou, and Dr. Michael May for technical assistance; John Andrews-Labenski, Seth Martin and Raymond Lee for their aid in constructing our automated feedometers; Dr. Sangwon Kim for generously providing us with the GT1-7 neuronal cell line; and Dr. Ted Abel for constructive suggestions and experimental advice. Supported by: NIH Grants DK077484, DK085435 (M.R.H.), DK21397 (H.J.G.), and DK082417 (K.K.B.), by the Institute of Diabetes, Obesity, and Metabolism at The University of Pennsylvania (5P30DK019525), as well as The Obesity Society 2008 New Investigator Grant (M.R.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beak SA, Heath MM, Small CJ, Morgan DG, Ghatei MA, Taylor AD, Buckingham JC, Bloom SR, Smith DM. Glucagon-like peptide-1 stimulates luteinizing hormone-releasing hormone secretion in a rodent hypothalamic neuronal cell line. J Clin Invest. 1998;101:1334–1341. doi: 10.1172/JCI610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelikani PK, Haver AC, Reidelberger RD. Intravenous infusion of glucagon-like peptide-1 potently inhibits food intake, sham feeding, and gastric emptying in rats. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1695–1706. doi: 10.1152/ajpregu.00870.2004. [DOI] [PubMed] [Google Scholar]
- da Silva Xavier G, Leclerc I, Varadi A, Tsuboi T, Moule SK, Rutter GA. Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem J. 2003;371:761–774. doi: 10.1042/BJ20021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D, Patten CS, Roth JD, Yee DK, Fluharty SJ. Melanocortin receptor signaling through mitogen-activated protein kinase in vitro and in rat hypothalamus. Brain Res. 2003;986:1–11. doi: 10.1016/s0006-8993(03)03162-7. [DOI] [PubMed] [Google Scholar]
- Daniels D, Yee DK, Faulconbridge LF, Fluharty SJ. Divergent behavioral roles of angiotensin receptor intracellular signaling cascades. Endocrinology. 2005;146:5552–5560. doi: 10.1210/en.2005-0774. [DOI] [PubMed] [Google Scholar]
- Dostmann WR, Taylor SS, Genieser HG, Jastorff B, Doskeland SO, Ogreid D. Probing the cyclic nucleotide binding sites of cAMP-dependent protein kinases I and II with analogs of adenosine 3′,5′-cyclic phosphorothioates. J Biol Chem. 1990;265:10484–10491. [PubMed] [Google Scholar]
- Dugan LL, Kim JS, Zhang Y, Bart RD, Sun Y, Holtzman DM, Gutmann DH. Differential effects of cAMP in neurons and astrocytes. Role of B-raf. J Biol Chem. 1999;274:25842–25848. doi: 10.1074/jbc.274.36.25842. [DOI] [PubMed] [Google Scholar]
- Goke R, Larsen PJ, Mikkelsen JD, Sheikh SP. Distribution of GLP-1 binding sites in the rat brain: evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur J Neurosci. 1995;7:2294–2300. doi: 10.1111/j.1460-9568.1995.tb00650.x. [DOI] [PubMed] [Google Scholar]
- Gomez E, Pritchard C, Herbert TP. cAMP-dependent protein kinase and Ca2+ influx through L-type voltage-gated calcium channels mediate Raf-independent activation of extracellular regulated kinase in response to glucagon-like peptide-1 in pancreatic beta-cells. J Biol Chem. 2002;277:48146–48151. doi: 10.1074/jbc.M209165200. [DOI] [PubMed] [Google Scholar]
- Gutzwiller JP, Goke B, Drewe J, Hildebrand P, Ketterer S, Handschin D, Winterhalder R, Conen D, Beglinger C. Glucagon-like peptide-1: a potent regulator of food intake in humans. Gut. 1999;44:81–86. doi: 10.1136/gut.44.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SM, Namkoong C, Jang PG, Park IS, Hong SW, Katakami H, Chun S, Kim SW, Park JY, Lee KU, et al. Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia. 2005;48:2170–2178. doi: 10.1007/s00125-005-1913-1. [DOI] [PubMed] [Google Scholar]
- Hayes MR, Bradley L, Grill HJ. Endogenous hindbrain glucagon-like peptide-1 receptor activation contributes to the control of food intake by mediating gastric satiation signaling. Endocrinology. 2009a;150:2654–2659. doi: 10.1210/en.2008-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MR, Covasa M. Dorsal hindbrain 5-HT3 receptors participate in control of meal size and mediate CCK-induced satiation. Brain Res. 2006;1103:99–107. doi: 10.1016/j.brainres.2006.05.058. [DOI] [PubMed] [Google Scholar]
- Hayes MR, De Jonghe BC, Kanoski SE. Role of the glucagon-like-peptide-1 receptor in the control of energy balance. Physiol Behav. 2010a;100:503–510. doi: 10.1016/j.physbeh.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Bence KK, Grill HJ. Dorsal hindbrain AMP-Kinase as an intracellular mediator of energy balance. Endocrinology. 2008a doi: 10.1210/en.2008-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Bence KK, Grill HJ. Dorsal hindbrain 5′-adenosine monophosphate-activated protein kinase as an intracellular mediator of energy balance. Endocrinology. 2009b;150:2175–2182. doi: 10.1210/en.2008-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Grill HJ. Caudal brainstem processing is sufficient for behavioral, sympathetic and parasympathetic responses driven by peripheral and hindbrain glucagon-like-peptide-1 receptor stimulation. Endocrinology. 2008b doi: 10.1210/en.2007-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Grill HJ. Caudal brainstem processing is sufficient for behavioral, sympathetic, and parasympathetic responses driven by peripheral and hindbrain glucagon-like-peptide-1 receptor stimulation. Endocrinology. 2008c;149:4059–4068. doi: 10.1210/en.2007-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Leichner TM, Guarnieri DJ, DiLeone RJ, Bence KK, Grill HJ. Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab. 2010b;11:77–83. doi: 10.1016/j.cmet.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- Huo L, Grill HJ, Bjorbaek C. Divergent regulation of proopiomelanocortin neurons by leptin in the nucleus of the solitary tract and in the arcuate hypothalamic nucleus. Diabetes. 2006;55:567–573. doi: 10.2337/diabetes.55.03.06.db05-1143. [DOI] [PubMed] [Google Scholar]
- Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem. 2006;281:36662–36672. doi: 10.1074/jbc.M606676200. [DOI] [PubMed] [Google Scholar]
- Imeryuz N, Yegen BC, Bozkurt A, Coskun T, Villanueva-Penacarrillo ML, Ulusoy NB. Glucagon-like peptide-1 inhibits gastric emptying via vagal afferent-mediated central mechanisms. Am J Physiol. 1997;273:G920–927. doi: 10.1152/ajpgi.1997.273.4.G920. [DOI] [PubMed] [Google Scholar]
- Kelly MP, Isiegas C, Cheung YF, Tokarczyk J, Yang X, Esposito MF, Rapoport DA, Fabian SA, Siegel SJ, Wand G, et al. Constitutive activation of Galphas within forebrain neurons causes deficits in sensorimotor gating because of PKA-dependent decreases in cAMP. Neuropsychopharmacology. 2007;32:577–588. doi: 10.1038/sj.npp.1301099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Miller I, Aja S, Landree LE, Pinn M, McFadden J, Kuhajda FP, Moran TH, Ronnett GV. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J Biol Chem. 2004a;279:19970–19976. doi: 10.1074/jbc.M402165200. [DOI] [PubMed] [Google Scholar]
- Kim MS, Lee KU. Role of hypothalamic 5′-AMP-activated protein kinase in the regulation of food intake and energy homeostasis. J Mol Med. 2005;83:514–520. doi: 10.1007/s00109-005-0659-z. [DOI] [PubMed] [Google Scholar]
- Kim MS, Park JY, Namkoong C, Jang PG, Ryu JW, Song HS, Yun JY, Namgoong IS, Ha J, Park IS, et al. Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat Med. 2004b;10:727–733. doi: 10.1038/nm1061. [DOI] [PubMed] [Google Scholar]
- Kinzig KP, D'Alessio DA, Seeley RJ. The diverse roles of specific GLP-1 receptors in the control of food intake and the response to visceral illness. J Neurosci. 2002;22:10470–10476. doi: 10.1523/JNEUROSCI.22-23-10470.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu R, Matsuyama T, Namba M, Watanabe N, Itoh H, Kono N, Tarui S. Glucagonostatic and insulinotropic action of glucagonlike peptide I-(7-36)-amide. Diabetes. 1989;38:902–905. doi: 10.2337/diab.38.7.902. [DOI] [PubMed] [Google Scholar]
- Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience. 1997;77:257–270. doi: 10.1016/s0306-4522(96)00434-4. [DOI] [PubMed] [Google Scholar]
- Larsen PJ, Vrang N, Tang-Christensen M. Central pre-proglucagon derived peptides: opportunities for treatment of obesity. Curr Pharm Des. 2003;9:1373–1382. doi: 10.2174/1381612033454775. [DOI] [PubMed] [Google Scholar]
- Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:262–269. doi: 10.1038/nrendo.2009.48. [DOI] [PubMed] [Google Scholar]
- Ma X, Bruning J, Ashcroft FM. Glucagon-like peptide 1 stimulates hypothalamic proopiomelanocortin neurons. J Neurosci. 2007;27:7125–7129. doi: 10.1523/JNEUROSCI.1025-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mack CM, Moore CX, Jodka CM, Bhavsar S, Wilson JK, Hoyt JA, Roan JL, Vu C, Laugero KD, Parkes DG, et al. Antiobesity action of peripheral exenatide (exendin-4) in rodents: effects on food intake, body weight, metabolic status and side-effect measures. Int J Obes (Lond) 2006;30:1332–1340. doi: 10.1038/sj.ijo.0803284. [DOI] [PubMed] [Google Scholar]
- McMahon LR, Wellman PJ. Decreased intake of a liquid diet in nonfood-deprived rats following intra-PVN injections of GLP-1 (7-36) amide. Pharmacol Biochem Behav. 1997;58:673–677. doi: 10.1016/s0091-3057(97)90017-4. [DOI] [PubMed] [Google Scholar]
- McMahon LR, Wellman PJ. PVN infusion of GLP-1-(7-36) amide suppresses feeding but does not induce aversion or alter locomotion in rats. Am J Physiol. 1998;274:R23–29. doi: 10.1152/ajpregu.1998.274.1.R23. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I, Lane M, Shughrue P. Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J Comp Neurol. 1999;403:261–280. doi: 10.1002/(sici)1096-9861(19990111)403:2<261::aid-cne8>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- Mollet A, Gilg S, Riediger T, Lutz TA. Infusion of the amylin antagonist AC 187 into the area postrema increases food intake in rats. Physiol Behav. 2004;81:149–155. doi: 10.1016/j.physbeh.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Moran TH, Wohn A, Schwartz GJ, Ladenheim EE. Fourth ventricular administration of glucagon-like peptide (7-36) amide (GLP-1) inhibits food intake in rats. Society for Neuroscience. 1996;22:15. [Google Scholar]
- Morton GJ, Blevins JE, Kim F, Matsen M, Figlewicz DP. The action of leptin in the ventral tegmental area to decrease food intake is dependent on Jak-2 signaling. Am J Physiol Endocrinol Metab. 2009;297:E202–210. doi: 10.1152/ajpendo.90865.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Perfetti R, Merkel P. Glucagon-like peptide-1: a major regulator of pancreatic beta-cell function. Eur J Endocrinol. 2000;143:717–725. doi: 10.1530/eje.0.1430717. [DOI] [PubMed] [Google Scholar]
- Potes CS, Lutz TA. Brainstem mechanisms of amylin-induced anorexia. Physiol Behav. 2010;100:511–518. doi: 10.1016/j.physbeh.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Ruttimann EB, Arnold M, Hillebrand JJ, Geary N, Langhans W. Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology. 2009;150:1174–1181. doi: 10.1210/en.2008-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott KA, Moran TH. The GLP-1 agonist exendin-4 reduces food intake in nonhuman primates through changes in meal size. Am J Physiol Regul Integr Comp Physiol. 2007;293:R983–987. doi: 10.1152/ajpregu.00323.2007. [DOI] [PubMed] [Google Scholar]
- Seo S, Ju S, Chung H, Lee D, Park S. Acute effects of glucagon-like peptide-1 on hypothalamic neuropeptide and AMP activated kinase expression in fasted rats. Endocr J. 2008;55:867–874. doi: 10.1507/endocrj.k08e-091. [DOI] [PubMed] [Google Scholar]
- Sheriff S, Chance WT, Iqbal S, Rizvi TA, Xiao C, Kasckow JW, Balasubramaniam A. Hypothalamic administration of cAMP agonist/PKA activator inhibits both schedule feeding and NPY-induced feeding in rats. Peptides. 2003;24:245–254. doi: 10.1016/s0196-9781(03)00037-8. [DOI] [PubMed] [Google Scholar]
- Sutton GM, Duos B, Patterson LM, Berthoud HR. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology. 2005;146:3739–3747. doi: 10.1210/en.2005-0562. [DOI] [PubMed] [Google Scholar]
- Sutton GM, Patterson LM, Berthoud HR. Extracellular signal-regulated kinase 1/2 signaling pathway in solitary nucleus mediates cholecystokinin-induced suppression of food intake in rats. J Neurosci. 2004;24:10240–10247. doi: 10.1523/JNEUROSCI.2764-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, Choi SJ, Taylor GM, Heath MM, Lambert PD, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. 1996;379:69–72. doi: 10.1038/379069a0. [DOI] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- Vrang N, Phifer CB, Corkern MM, Berthoud HR. Gastric distension induces c-Fos in medullary GLP-1/2-containing neurons. Am J Physiol Regul Integr Comp Physiol. 2003;285:R470–478. doi: 10.1152/ajpregu.00732.2002. [DOI] [PubMed] [Google Scholar]
- Wettergren A, Schjoldager B, Mortensen PE, Myhre J, Christiansen J, Holst JJ. Truncated GLP-1 (proglucagon 78-107-amide) inhibits gastric and pancreatic functions in man. Dig Dis Sci. 1993;38:665–673. doi: 10.1007/BF01316798. [DOI] [PubMed] [Google Scholar]
- Xue B, Kahn BB. AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. J Physiol. 2006;574:73–83. doi: 10.1113/jphysiol.2006.113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhou LB, Liu SQ, Tang JF, Li FY, Li RY, Song HD, Chen MD. Expression of feeding-related peptide receptors mRNA in GT1-7 cell line and roles of leptin and orexins in control of GnRH secretion. Acta Pharmacol Sin. 2005;26:976–981. doi: 10.1111/j.1745-7254.2005.00118.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Hindbrain GLP-1R activation by 4th icv Ex-4 (0.1μg) reduced food intake in the 12h dark cycle by suppressing meal number and increasing inter-meal-interval, not by altering meal size. 4th icv delivery of the AMPK activity promoter AICAR (300μg) attenuated both suppression of meal number and increase in inter-meal-interval by 4th icv Ex-4. * = P< 0.05 from vehicle/vehicle. † = P< 0.05 from vehicle/Ex-4. (B) Peripheral GLP-1R activation by IP administration of Ex-4 (3.0μg/kg) significantly suppressed 5h dark cycle chow intake compared to vehicle (saline) by reducing meal size, not by altering meal number. Time points of meal pattern assessment for 4th icv and IP Ex-4 administration are chosen based on comparable maximum percent suppression of chow intake (∼45% suppression). Data are presented as mean ± SEM. * = P< 0.05 from vehicle.