

Abstract
Although intraocular tumors reside in an immune privileged site, some tumors are rejected nonetheless. For example, intraocular adenovirus-induced (Ad5E1; adenovirus type 5 early region 1) tumors are rejected in syngeneic C57BL/6 mice by one of two pathways. One pathway leads to extensive necrosis of innocent bystander cells and culminates in destruction of the eye, a condition called phthisis. The second pathway is characterized by piecemeal tumor cell death that rids the eye of the tumor while preserving the architecture and function of the eye. To study the mechanisms of phthisical tumor rejection, we isolated a cell clone–designated clone 2.1 that consistently undergoes rejection in a phthisical manner. CD4+ T cells and macrophages were required for phthisical rejection of intraocular clone 2.1 tumors and M1 macrophages were involved in mediating tumor rejection. In vitro and in vivo inhibition of iNOS (inducible nitric oxide synthase) abolished macrophage-mediated killing of tumor cells and rejection of intraocular tumors. A role for M1 macrophages was further supported by investigations showing that intraocular tumors grew progressively in IFN-γ KO (knockout) mice. Studies in mice deficient in TNF-α, TNF receptor-1, or TNF receptor-2 revealed that although TNF-α was not needed for tumor rejection, it was required for the development of necrotizing inflammation and phthisis of tumor-bearing eyes. Together, our findings suggest new strategies to successfully eliminate ocular tumors while preserving the integrity of the eye.
Introduction
Ocular immune privilege is essential for preserving vision and preventing immune-mediated inflammation that destroys normal ocular cells (1, 2). Multiple mechanisms maintain immune privilege in the eye. The absence or reduced expression of MHC class I antigens on the corneal endothelium diminishes the susceptibility of these nonregenerative cells to bystander killing by CD8+ CTL (3). Aqueous humor contains multiple immunosuppressive factors including TGF-β, macrophage migration inhibitory factor (MIF), vasoactive intestinal protein (VIP), somatostatin, and α-melanocyte–stimulating hormone (α-MSH), which inhibit inflammation and down-regulate adaptive immune responses (1, 2). Moreover, antigens introduced into the anterior chamber (AC) induce an antigen-specific downregulation of Th1 and Th2 immune responses by a phenomenon known as anterior chamber–associated immune deviation (ACAID).
Ocular immune privilege would seem to provide an ideal environment for unfettered ocular tumor growth. However, some experimental ocular tumors undergo immune rejection (4–6). Thus, ocular immune privilege can be circumvented, allowing for T-cell–dependent immune rejection of intraocular tumors (6). There are 2 patterns by which intraocular tumors can undergo T-cell–dependent immune rejection. The first pattern is characterized by piecemeal necrosis of intraocular tumor cells and preservation of the architecture of the eye (7, 8). The second pattern involves ischemic necrosis and extensive damage to both the tumor and innocent bystander cells within the eye (9). This pattern results in phthisis or atrophy of the eye (7). Clearly, the immune responses that determine which pattern of rejection occurs have a significant impact on the fate of the eye and the preservation of vision.
In addition to intraocular tumor rejection, some inflammatory ocular diseases, such as sympathetic ophthalmia, can culminate in phthisis and blindness (10). Thus, understanding the cellular mechanisms of intraocular tumor rejection that lead to phthisis will allow a better understanding of tumor immunity in the eye and provide insights into the pathophysiology of phthisis in other inflammatory eye diseases.
To study the immune mechanisms that circumvent immune privilege and result in phthisical rejection of intraocular tumors, we used a murine tumor, adenovirus type 5 early region 1 (Ad5E1), which was generated by transformation of embryonic C57BL/6 mouse cells by transfection with the human Ad5E1 gene (11). Previous results indicated that Ad5E1 tumors undergo spontaneous immune rejection in the AC of syngeneic C57BL/6 mice (12, 13). Rejection of Ad5E1 tumors requires CD4+ T cells, IFN-γ, and macrophages but does not require TNF-α, FasL, TRAIL (TNF-related apoptosis-inducing ligand), perforin, B cells, NK (natural killer) cells, or CD8+ T cells (13–17). During the course of our experiments, we noticed that Ad5E1 tumors occasionally underwent immune rejection that culminated in phthisis, whereas tumor rejection in other mice was pristine and left the eye intact. This prompted us to explore the T-cell–dependent immune mechanisms that lead to phthisical tumor rejection.
Materials and Methods
Animals
C57BL/6 (H-2b) mice, IFN-γ knockout (KO) mice (B6.129S7-Ifnγtm1Ts/J), severe combined immunodeficiency mutation (SCID; B6.CB17-Prkdcscid/SzJ), TNF KO mice (B6.129S6-TNFtm1Gk1/J), and TNF receptor (TNFR) 1 KO mice (B6.129-Tnfsf1atm1Mak/J), and TNFR2 KO (B6.129S2.Tnfr2tm1Mwm/J) were obtained from The Jackson Laboratory. Animals were cared for in accordance with the Association for Research in Vision and Ophthalmology Guidelines regarding the Use of Animals in Ophthalmic and Vision Research.
Tumor cells and ocular cells
Ad5E1 tumor cells were kindly provided by Dr. Rene E.M. Toes (Leiden University Medical Center, the Netherlands) and were cultured as previously described (11). Transfection of Ad5E1 tumor cells with E1A and E1B genes was confirmed by Northern blotting (18). Expression of H-2b class I antigens was reported previously (18) and was confirmed by flow cytometry during the course of this study. A C57BL/6 corneal endothelial cell line was established and immortalized with human papilloma virus genes E6 and E7 using the disabled recombinant retroviral vector pLXSN16E6/E7 and was cultured as previously described (19). Nonimmortalized iris/ciliary body (I/CB) cells were isolated from the eyes of C57BL/6 mice and cultures were established in complete RPMI 1640 medium as described elsewhere (20). All cells were screened for mycoplasma by ELISA and found to be negative.
Identification of tumor clones that undergo phthisical rejection
Monoclonal cell cultures were established from parental Ad5E1 tumor cells by isolating single cells from bulk cultures using a MoFlo XDP cell sorter (Beckman Coulter) and screened for rejection following injection into the AC of normal C57BL/6 mice. Four clones consistently exhibited phthisical rejection in C57BL/6 mice. One of these, designated as Ad5E1 clone 2.1, was used for this study. A clone that underwent nonphthisical rejection was also identified (clone 4.0).
Intraocular tumor cell injections
Tumor cell suspensions were injected into the AC as previously described (21). A 1.0-mL syringe fitted with a glass needle was used to inject 6 μL of a monocellular suspension of Ad5E1 tumor cells (3 × 105 cells/6 μL) into the AC. Eyes were examined 3 times per week and the tumor volume was recorded as the percentage of AC occupied with tumor (21).
Macrophage depletion
Subconjunctival injection of clodronate-containing liposomes (C12MDP-LIP; Sigma) induces the elimination of greater than 95% of the conjunctival macrophages (22) and greater than 99% depletion of F4/80+ macrophages that infiltrate intraocular Ad5E1 tumors (14). Multilamellar liposomes were prepared as described previously (23). Each 100 μL of C12MDP-LIP suspension contained 1 mg of clodronate. The cytotoxicity of C12MDP-LIP and PBS-containing liposomes (PBS-LIP) were tested using the RAW 264.7 macrophage cell line. A liposome suspension (8 μL) was injected into the bulbar conjunctiva using a 30-gauge needle mounted on a 1 mL tuberculin syringe, dispensed into 4 different sites 90 degrees apart. PBS-LIP was used as a negative control. Liposome injections were performed on the day of tumor injection and every 3 to 4 days thereafter (16).
Quantitative real-time PCR
Anterior segments of tumor-bearing eyes and normal eyes were dissected free of the lens and choroid/retina and homogenized and analyzed by real-time quantitative PCR (qPCR). Expression of NOS2, YM1, arginase 1 (Arg1), F4/80, CD11b, and TNF-α mRNA was assessed by real-time qPCR using a MyiQ Single-Color Real-Time PCR Detection system (Bio-Rad). Total RNA was converted into first-strand cDNA using RT2 First Strand Kit (SA Biosciences) and PCR amplification reactions used RT2 qPCR Master Mix (SA Biosciences) according to manufacturer’s instructions. All reactions were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and performed in duplicate.
Isolation of bone marrow–derived macrophages
Bone marrow–derived macrophages (BMDM) were isolated as described previously (24). Bone marrow cells were obtained from the femurs of C57BL/6 mice and cultured with recombinant murine macrophage–colony-stimulating factor (rmM-CSF; 10 ng/mL; R&D Systems) for 7 days.
BMDM-mediated cytotoxicity assay
A macrophage-mediated cytotoxicity assay utilizing BMDMs was performed (25). BMDMs were plated 1 × 105 cells per well in 96-well, flat-bottom plates and incubated with medium alone or medium containing rmIFN-γ (10 U/mL; R&D Systems) plus LPS (lipopolysaccharide; 10 ng/mL; R&D Systems) for 24 hours. Target cells were incubated with 0.2 μCi/mL of 3H-thymidine (MP Biomedicals) for 24 hours and then incubated with 1 × 105 resting or activated macrophages (1:10) for an additional 48 or 72 hours. Target cells were washed twice with PBS and radioactivity was measured in a liquid scintillation counter. Cytotoxicity was calculated using the formula: % cytotoxicity = [(A − B)/A] × 100, in which A = CPM of tumor cells cultured alone and B = CPM in test cultures.
Inhibition of iNOS with L-NAME
L-NG-nitroarginine methyl ester (hydrochloride; L-NAME; Cayman Chemical) was used in both in vitro and in vivo assays to block inducible nitric oxide synthase (iNOS). Final concentrations of 1 and 5 mmol/L L-NAME were used in the in vitro BMDM-mediated tumor killing assays (26, 27). As a control, the biologically inactive isomer D-NG-nitroarginine methyl ester (hydrochloride; D-NAME; Sigma) was used at the same concentrations as L-NAME. To inhibit iNOS in vivo, mice were treated with 50 mg/kg L-NAME daily given i.p. (28, 29). D-NAME was used as a negative control.
TNF-α cytotoxicity assay
TNF-α–induced cytotoxicity was evaluated using a Cyto-Tox96 kit (Promega). Single cell suspensions of clone 2.1 tumor cells (1 × 105 cells/mL), I/CB cells, or corneal endothelial cells were added to 24-well plates (Corning Inc.). Cells were cultured in either medium alone or medium containing various concentrations of murine TNF-α (1, 10, or 100 ng/mL). Cultures were incubated for 48 hours at 37oC, the culture medium was collected, and lactate dehydrogenase (LDH) was measured according to manufacturer’s instructions.
In vitro stimulation of T cells
CD4+ T cells were isolated from draining lymph nodes of tumor rejector mice using CD4 (L3T4) MicroBeads (Miltenyi Biotec) and incubated with either medium alone, tumor antigen–pulsed antigen-presenting cells (APC) or anti-CD3/CD28 beads (25 μL/mL; Invitrogen) for 5 days at 37°C as previously described. Supernatants from T-cell cultures were harvested and the concentration of TNF-α was determined using a mouse TNF-α Quantikine ELISA kit (R&D Systems).
Adoptive transfer experiments
Wild-type (WT) C57BL/6 and TNF-α KO C57BL/6 mice were injected in the AC with tumor cells as described above. Tumors underwent spontaneous rejection within 3 weeks of tumor injection. Following rejection, CD4+ T lymphocytes were isolated from spleen cell suspensions using CD4-specific microbeads and adoptively transferred i.v. into SCID mice at a 1:1 donor:recipient ratio (~5 × 106 cells/mouse). Recipient mice were immediately injected in the AC with clone 2.1 tumor cells.
Statistics
Student’s t test was used to assess the statistical significance between experimental and control groups. A value of P < 0.05 was considered significant.
Results
Phthisical rejection of Ad5E1 tumors
The Ad5E1 clone 2.1 tumor cell line was selected because of its consistent phthisical form of immune rejection (Fig. 1A). Intraocular clone 2.1 tumors that underwent phthisical rejection had extensive necrosis that culminated in atrophy of the eye. The AC was compressed, the cornea was opaque and vascularized, and only the lens remained intact (Fig. 1B, bottom), which was in stark contrast to the architecture of an eye following nonphthisical rejection of clone 4.0 tumor (Fig. 1B, top). To confirm that rejection was immune mediated, clone 2.1 cells were injected into the eyes of SCID mice where tumors grew progressively (Fig. 1A). Previous results indicated that rejection of Ad5E1 tumors required IFN-γ (13). To establish the requirement of IFN-γ for rejection, clone 2.1 tumor cells were injected into the AC of IFN-γ KO, which subsequently developed progressive intraocular tumors (Fig. 1A).
Figure 1.

Intraocular Ad5E1 clone 2.1 tumor growth in C57BL/6, IFN-γ KO, and SCID mice. A, clone 2.1 tumor cells were injected into the AC of C57BL/6 WT, C57BL/6 IFN-γ KO, and C57BL/6 SCID mice. Two independent experiments were performed (n = 5). B, photographs of phthisical rejection of clone 2.1 tumor (bottom) and nonphthisical rejection of clone 4.0 tumor (top) in WT C57BL/6 mice.
Phthisical intraocular Ad5E1 tumor rejection requires macrophages
Although a recent emphasis has been placed on macrophages that promote tumor growth, the role of macrophages in tumor rejection has been firmly established (30). We and others have previously reported that rejection of Ad5E1 intraocular tumors requires macrophages (14, 16); however, the role of macrophages in phthisical intraocular tumor rejection has not been examined. Previous studies have shown that phthisical rejection occurs via a CD4+ T-cell–mediated mechanism resulting in destruction of the tumor and normal ocular cells (7). To determine whether macrophages were required for phthisical rejection of clone 2.1 tumors, macrophages were depleted locally through the subconjunctival injections of clodronate liposomes prior to AC tumor injection. Depletion of macrophages in eyes injected with clone 2.1 tumor cells prevented tumor rejection, indicating that macrophages were associated with phthisical rejection of clone 2.1 tumors (Fig. 2A). In contrast, tumor rejection proceeded unabatedly in mice treated with PBS-LIP.
Figure 2.

Cytotoxic M1 macrophages are necessary for rejection of intraocular Ad5E1 clone 2.1 tumors. A, clone 2.1 tumor cells were injected into the AC of WT mice followed by subconjunctival injections of clodronate-containing or PBS-LIP, respectively. Two independent experiments were performed (n = 5). Clone 2.1 tumor cells were injected into the AC of WT mice. Tumor-bearing (TB) eyes were enucleated 14 days after tumor injection and expression of NOS2 (B), Arg1 (C), and YM1 (D) was assessed by real-time qPCR. Results shown are typical of 3 independent experiments. E, BMDMs were activated with rmIFN-γ plus LPS for 24 hours and used as effector cells in cytotoxicity assays. F, corneal endothelial cells and I/CB cells were used as target cells in cytotoxicity assays with BMDM effector cells.
Macrophages in Ad5E1 tumors express characteristics of classically activated (M1) macrophages
The observation that clone 2.1 tumor rejection requires macrophages led us to hypothesize that the predominant population of macrophages involved in the rejection was the M1 phenotype. Intraocular tumors were removed from the AC on day 14 (i.e., the peak time of intraocular tumor growth in WT C57BL/6 mice) and homogenized. RNA was immediately isolated and qPCR was performed to determine the expression of NOS2 (M1 marker), Arg1, and YM1 (M2 markers). Expression of 2 macrophage markers, F4/80 and CD11b, was determined by qPCR to confirm that the isolated cells were macrophages. As a control, cells from the RAW 264.7 macrophage cell line were polarized into either an M1 or M2 phenotype by culturing with either IFN-γ/LPS or IL-4/IL-10/IL-13, respectively. Samples were compared with naive eyes and normalized to GAPDH expression.
The dominant population of macrophages in the intraocular tumors was M1 as shown by the 50-fold increase in the expression of the M1-associated NOS2 gene (Fig. 2B). In contrast, there were very few M2 macrophages as noted by the baseline expression of the Arg1 (Fig. 2C) and YM1 genes (Fig. 2D), both of which are classical markers for M2 macrophages.
Ad5E1 tumors are susceptible to macrophage-mediated killing
Direct in vitro killing of tumor cells by M1 macrophages has been reported by other investigators (31). To determine whether Ad5E1 tumor cells were susceptible to direct macrophage-mediated killing, in vitro cytotoxicity assays were performed. BMDMs were untreated or treated with IFN-γ and activated with LPS. BMDMs were then cocultured with 3H-thymidine–labeled Ad5E1 tumor or B16F10 melanoma cells at 10:1 E:T ratios for 48 or 72 hours. Activated BMDMs killed Ad5E1 clone 2.1 tumor cells (75%–80% cytotoxicity) at both 48 and 72 hour time points (Fig. 2E and data not shown), indicating that clone 2.1 tumor cells are highly susceptible to macrophage-mediated killing.
Normal ocular cells are susceptible to macrophage-mediated killing
M1 macrophages may kill normal ocular cells in addition to killing tumor cells, which may account for the destruction of innocent bystander cells and ischemic necrosis of the eye. To determine whether activated M1 macrophages contribute to the phthisical rejection of intraocular clone 2.1 tumors by bystander killing of normal ocular cells, corneal endothelial and I/CB cells were tested for their susceptibility to in vitro killing by activated macrophages. As shown in Figure 2F, activated macrophages killed 40% to 50% of I/CB cells and 30% to 40% of corneal endothelial cells, indicating that M1 macrophages are able to kill normal ocular cells in vitro and, in addition to rejecting intraocular tumors, M1 macrophages might contribute to the phthisis that occurs in the rejection of clone 2.1 tumors.
Macrophage-mediated killing of Ad5E1 clone 2.1 tumor cells is iNOS dependent
Nitric oxide is a major molecule employed by M1 macrophages to mediate tumor cytotoxicity (32–34). Assessment of NO production indicated that BMDMs activated with IFN-γ and LPS produced NO, could be blocked by the specific NOS inhibitor L-NAME (data not shown). To determine whether BMDMs use NO to kill Ad5E1 2.1 tumor cells, in vitro cytotoxicity assays were performed in either the presence or absence of the iNOS inhibitor L-NAME. As a control, the biologically inactive isomer of L-NAME, D-NAME, was also used. Inhibition of iNOS by L-NAME significantly reduced the ability of BMDMs to kill Ad5E1 tumor cells (Fig. 3A). However, D-NAME did not affect macrophage-mediated killing, indicating that the inhibition of BMDM-mediated killing was NO specific (Fig. 3A). Neither L-NAME nor D-NAME alone was toxic to the tumor cells at the doses used in the in vitro assays (data not shown). These results support the hypothesis that M1 macrophages are the dominant macrophage population present in intraocular tumors undergoing phthisical rejection.
Figure 3.

iNOS is required for rejection of intraocular Ad5E1 clone 2.1 tumors. A, BMDMs were activated with rmIFN-γ plus LPS and used as effector cells in vitro. Cultures were incubated for 48 hours and then harvested. iNOS was inhibited by the addition of 1 or 5 mmol/L L-NAME to activated BMDMs. D-NAME, a biologically inactive isomer of L-NAME, was used as a control. B, WT mice were injected i.p. with L-NAME daily. Controls consisted of C57BL/6 mice injected i.p. with D-NAME daily or left untreated. All C57BL/6 mice were injected in the AC with clone 2.1 tumor cells.
Phthisical intraocular tumor rejection is iNOS dependent
To determine the role of iNOS in phthisical rejection in vivo, NO synthesis was blocked by injecting L-NAME (50 mg/kg/d) prior to AC tumor injection. As controls, untreated mice and a group of mice treated with D-NAME were also challenged in the AC with clone 2.1 tumor cells. Inhibition of NO synthesis prevented phthisical tumor rejection (Fig. 3B). In contrast, naive mice and mice treated with D-NAME rejected intraocular tumors in a phthisical manner that was indistinguishable from untreated controls (Fig. 3B). These results indicate that the phthisical rejection of Ad5E1 clone 2.1 tumors is NO dependent.
IFN-γ is required for phthisical rejection of Ad5E1 clone 2.1 intraocular tumors
The recruitment of M1 macrophages into inflammatory sites often requires IFN-γ, IL-12, and TNF-α (31, 35). Rejection of clone 2.1 tumors required IFN-γ and macrophages (Figs. 1A and 2A), suggesting that IFN-γ might function to recruit and activate M1 macrophages. To address this, tumor cells were injected into the AC of SCID, IFN-γ KO, and WT C57BL/6 mice. Intraocular tumors were collected at day 14, and expression of NOS2, CD11b, and F4/80 was examined by qPCR. Tumors isolated from IFN-γ–deficient mice contained significantly fewer M1 macrophages than WT mice as noted by decreased expression of CD11b and F4/80 genes and the reduction in NOS2 expression (Fig. 4). Tumor-bearing eyes from SCID mice also had fewer M1 macrophages suggesting that T cells were the source of IFN-γ involved in generating M1 macrophages in WT mice (Fig. 4). These results imply that IFN-γ produced by CD4+ T cells is essential for macrophage-dependent rejection of Ad5E1 clone 2.1 intraocular tumors.
Figure 4.
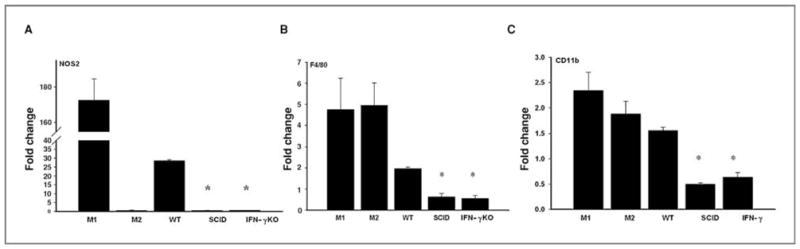
IFN-γ is required for M1 macrophage–mediated tumor rejection. Tumor-bearing eyes from C57BL/6, SCID, and IFN-γ KO mice were collected at day 14 and real-time qPCR was used to detect NOS2 (A), F4/80 (B), and CD11b (C). All samples were normalized to GAPDH and compared with non–tumor-bearing eyes in 2 independent experiments. M1- and M2-polarized RAW 264.7 cells served as controls. Mean ± SD. *, P ≤ 0.05.
Phthisical destruction of the tumor-containing eye requires TNF-α, although tumor rejection is TNF-α independent
We considered the hypothesis that TNF-α elaborated by T cells during the rejection of intraocular clone 2.1 tumors contributed to phthisis, even though intraocular tumor rejection was not TNF-α dependent. In vitro assays addressed whether TNF-α contributed to phthisis by killing normal ocular cells that line the AC of the eye. Corneal endothelial cells and I/CB cells were incubated with TNF-α and cell death was assessed. As expected, clone 2.1 tumor cells were susceptible to TNF-α–induced apoptosis. Normal ocular cells were also susceptible to killing by TNF-α, albeit less than tumor cells (Fig. 5A). Accordingly, clone 2.1 tumor cells were injected into the AC of TNF-α KO and WT mice and the eyes were observed for tumor growth and resolution. Tumors underwent rejection in TNF-α KO mice and WT mice (Fig. 5B). However, tumor rejection in the TNF-α KO mice did not culminate in phthisis (Table 1). To address the hypothesis that the effect of TNF-α on the development of phthisis was through toxicity of TNF-α on the host’s cells, tumor cells were injected into the AC of TNFR1 KO and TNFR2 KO mice. Contrary to our hypothesis, phthisical tumor rejection occurred in TNFR1 KO, TNFR2 KO, and WT mice (Fig. 5C and D and Table 1). Thus, the rejection phenotype is determined by the tumor and not the host cells.
Figure 5.
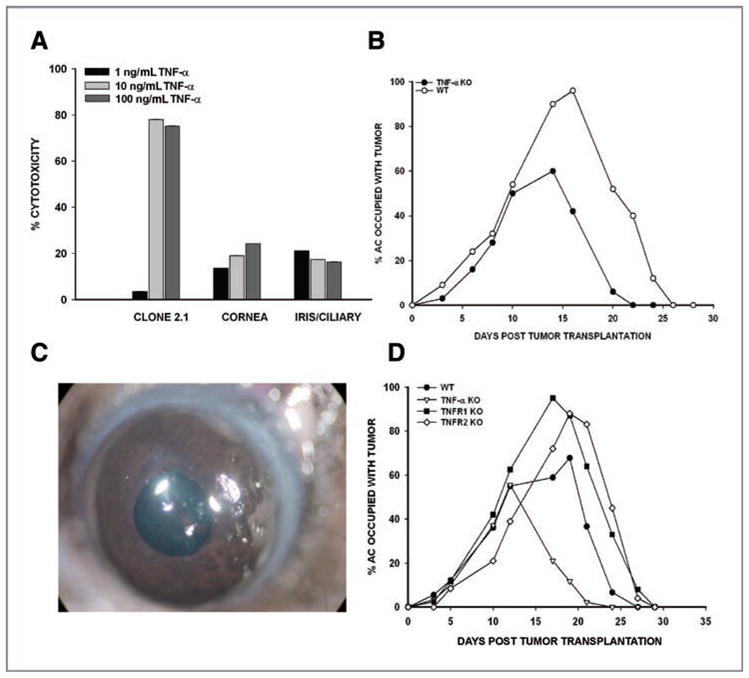
TNF-α is necessary for phthisis but not rejection of intraocular Ad5E1 clone 2.1 tumors. A, clone 2.1 tumor cells, I/CB cells, and corneal endothelial cells were incubated in medium alone or medium containing TNF-α for 48 hours. Cytotoxicity was determined by LDH release. B, Ad5E1 clone 2.1 tumor cells were injected into the AC of TNF-α KO mice or WT C57BL/6 mice. C, photograph of nonphthisical rejection of Ad5E1 clone 2.1. D, AC injection of clone 2.1 tumor cells in WT, TNF-α KO, TNFR1 KO, and TNFR2 KO mice. Experiments were performed twice with similar results. n = 5 mice per group.
Table 1.
Phthisical rejection of intraocular clone 2.1 Ad5E1 tumors requires TNF-α
| Host | Nonphthisical, % | Phthisical, % |
|---|---|---|
| C57BL/6 (n = 34) | 21 | 79 |
| TNF-α KO (n = 33) | 88 | 12 |
| TNFR1 KO(n = 10) | 10 | 90 |
| TNFR2 KO (n = 10) | 0 | 100 |
Phthisical tumor rejection requires T-cell–produced TNF-α
Tumor cells were injected into the AC of SCID, IFN-γ KO, and WT C57BL/6 mice and the expression of TNF-α was examined by qPCR. Intraocular tumors from mice deficient in either T cells or IFN-γ expressed no TNF-α (compared with WT mice) thereby suggesting that T cells are a major source of TNF-α (Fig. 6A). To further examine the hypothesis that T cells produce TNF-α in response to intraocular tumors, CD4+ T cells were isolated from draining lymph nodes of mice that had rejected intraocular clone 2.1 tumors and stimulated in vitro with APCs pulsed with clone 2.1 tumor antigen or stimulated with anti-CD3/CD28 beads. Rejector T cells secreted TNF-α in response to either tumor antigen or anti-CD3/CD28 stimulation (Fig. 6B). Although T cells are a major source of TNF-α, M1 macrophages can also produce this cytokine. To demonstrate that T cells were the source of TNF-α, purified T cells from rejector WT and TNF-α KO mice were transferred to naive SCID mice. Mice were immediately challenged with clone 2.1 tumor cells. As expected, SCID mice that received T cells from rejector mice eliminated intraocular tumors in a phthisical manner. In contrast, SCID mice that received T cells from TNF-α KO mice failed to develop phthisical rejection (Fig. 6C and D). As a control, C57BL/6 mice were injected with clone 2.1 cells and as expected, the tumors underwent phthisical rejection. To demonstrate histologic differences in ocular architecture between phthisical and nonphthisical tumor rejection, tissue sections were prepared and stained with H&E. (hematoxylin and eosin; Fig. 6E and F). The architecture of eyes that underwent nonphthisical rejection was pristine compared with the gross destruction of the ocular architecture that occurred in phthisical rejection.
Discussion
Immune-mediated rejection of intraocular tumors can follow 2 different pathways (7). The first pathway involves piecemeal necrosis and eradication of the tumor without damage to innocent bystander ocular cells. The second pathway involves rejection of intraocular tumors and culminates in extensive damage to innocent bystander cells and phthisis of the eye (7, 8). To our knowledge, this is the first report that has examined the immune mechanisms that mediate phthisical rejection of intraocular tumors.
We have previously shown that rejection of intraocular Ad5E1 tumors requires IFN-γ, which can act directly on tumor cells by (a) inhibiting tumor cell proliferation, (b) inducing tumor cell apoptosis, and (c) simultaneously downregulating proangiogenic genes and upregulating antiangiogenic genes in the tumors (16, 36, 37). IFN-γ is important for activation of macrophages that kill microbial pathogens and tumor cells (33, 38, 39). We found evidence that IFN-γ is required for the recruitment and activation of macrophages that promote a phthisical form of tumor rejection that rids the host of an ocular tumor at the expense of the eye.
The present findings indicate that macrophages are crucial for the rejection of intraocular clone 2.1 tumors. We are attracted to the hypothesis that macrophages are also intimately involved in the necrosis that culminates in phthisis. There are 2 basic subsets of macrophages: (a) M1 macrophages, polarized by IFN-γ and (b) M2 macrophages, polarized by IL-4, IL-10, and IL-13. M2 macrophages are typically associated with Th2 responses and tend to reduce tissue destruction through their elaboration of immunosuppressive and antiinflammatory cytokines such as TGF-β and IL-10. Although we found evidence of M2 macrophage–related molecules in the tumor-bearing eyes, M2 cytokine expression was only 2-fold above background levels. In contrast, the same tumor-bearing eyes expressed a 300-fold increase in M1 macrophage–associated NOS2. This supports the conclusion that M1 macrophages were involved in the pathologic sequelae leading to phthisis. M1 macrophages produce large amounts of proinflammatory and Th1-polarizing cytokines including IL-12, IL-6, IL-23 and TNF-α. M1 macrophages also contribute to tissue damage through their production of toxic intermediates such as NO and reactive oxygen intermediates (ROI). Our findings indicate that macrophages can directly kill clone 2.1 tumor cells in vitro, supporting the hypothesis that tumor infiltrating macrophages in Ad5E1 intraocular tumors are of the M1 subset that can directly mediate tumor rejection via secretion of TNF-α and reactive oxygen species (40–42). The present findings demonstrate that Ad5E1 tumor clone 2.1 is highly susceptible to macrophage-mediated killing in vitro and presumably in vivo, based on the progressive intraocular tumor growth in mice whose periocular macrophage population has been deleted by subconjunctival injection of clodronate-containing liposomes. This conclusion is further supported by the observation that inhibition of the M1 macrophage mediator, NO, prevents tumor rejection.
Nitric oxide was one of the first macrophage killing mechanisms that was identified and has pleiotropic tumorigenic properties from promotion of angiogenesis to enhancement of immunosuppression by tumors (43, 44). Paradoxically, the most recognized effector molecule employed by M1 macrophages to kill tumors is NO and it has been well documented that NO produced by M1 macrophages effectively kills tumor cells (45–47). The present results show that inhibition of iNOS reduces macrophage-mediated cytotoxicity in vitro and prevents tumor rejection. In addition to direct killing of tumor cells, a decrease in iNOS could reduce the angiogenic and immunosuppressive properties of tumor cells that are resistant to cytotoxicity, thus further reducing tumor growth.
IFN-γ also plays a pivotal role in recruitment of M1 macrophages into the intraocular tumor microenvironment in addition to its capacity to inhibit angiogenesis and induce tumor cell apoptosis in the intraocular Ad5E1 tumors (13). Progressive growth of clone 2.1 tumors in SCID mice and the failure of these hosts to recruit macrophages into the intraocular tumors suggest that T cells produce IFN-γ that is required for recruitment and activation of M1 macrophages.
The present results and previous reports indicate that Ad5E1 tumor cells circumvent immune privilege and induce the generation of CD4+ T cells that produce IFN-γ and CD8+ T cells that generate TNF-α following injection into the AC (13, 16). The production of IFN-γ polarizes macrophages to an M1 phenotype. M1 macrophages produce NO that effectively destroys the tumor, but also causes bystander damage to normal ocular cells. Although the generation of TNF-α is not required for tumor rejection, it is essential for the development of phthisis. Like NO, TNF-α kills normal ocular cells in vitro and plays a crucial role in the extensive injury to innocent bystander cells in the eye, which culminates in phthisis. Thus, our model demonstrates that it is possible to modify the host’s immune response against tumors such that the immune system eliminates the intraocular tumor while preserving the integrity of the eye. Immune-mediated phthisis underscores the importance of immune privilege in restraining intraocular inflammation and preserving the integrity of ocular tissues, many of which are incapable of regeneration. Understanding the mechanisms that circumvent immune privilege and culminate in phthisis may facilitate the development of immunotherapy that promotes tumor rejection while preserving vision. This may shape the nature of therapies invoked for the treatment of other inflammatory eye diseases such as sympathetic ophthalmia and uveitis.
Figure 6.

T cells are the source of TNF-α. A, expression of TNF-α mRNA from tumor-bearing WT and IFN-γ KO eyes was determined by qPCR and compared with a non-tumor-bearing eye. B, T cells harvested from tumor rejector mice were unstimulated or were stimulated with tumor antigen (TA)-pulsed APCs or with anti-CD3/CD28 beads. Secretion of TNF-α was determined by ELISA. C, T cells from C57BL/6 and TNF-α KO tumor rejector mice were adoptively transferred to SCID or IFN-γ KO mice and recipient mice were injected with clone 2.1 tumor cells in the AC. n = 9 or 10 mice in each group. D, percentage of animals that underwent nonphthisical intraocular tumor rejection in C. Histologic sections of eyes that underwent nonphthisical (E) or phthisical (F) tumor rejection.
Acknowledgments
The authors thank Khrishen Cunnusamy for his technical assistance.
Grant Support
This work was supported by NIH grants EY005631, EY016664, and EY020799 and an unrestricted grant from Research to Prevent Blindness.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Niederkorn JY. Immune privilege in the anterior chamber of the eye. Crit Rev Immunol. 2002;22:13–46. [PubMed] [Google Scholar]
- 2.Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. 2006;7:354–9. doi: 10.1038/ni1328. [DOI] [PubMed] [Google Scholar]
- 3.Whitsett CF, Stulting RD. The distribution of HLA antigens on human corneal tissue. Invest Ophthalmol Vis Sci. 1984;25:519–24. [PubMed] [Google Scholar]
- 4.Niederkorn JY. The immunopathology of intraocular tumour rejection. Eye. 1991;5:186–92. doi: 10.1038/eye.1991.33. [DOI] [PubMed] [Google Scholar]
- 5.Niederkorn JY. Immunoregulation of intraocular tumours. Eye. 1997;11:249–54. doi: 10.1038/eye.1997.60. [DOI] [PubMed] [Google Scholar]
- 6.Niederkorn JY, Wang S. Immunology of intraocular tumors. Ocul Immunol Inflamm. 2005;13:105–10. doi: 10.1080/09273940490518586. [DOI] [PubMed] [Google Scholar]
- 7.Knisely TL, Luckenbach MW, Fischer BJ, Niederkorn JY. Destructive and nondestructive patterns of immune rejection of syngeneic intraocular tumors. J Immunol. 1987;138:4515–23. [PubMed] [Google Scholar]
- 8.Niederkorn JY, Knisely TL. Immunological analysis of a destructive pattern of intraocular tumor resolution. Curr Eye Res. 1988;7:515–26. doi: 10.3109/02713688809031806. [DOI] [PubMed] [Google Scholar]
- 9.Niederkorn JY, Meunier PC. Spontaneous immune rejection of intraocular tumors in mice. Invest Ophthalmol Vis Sci. 1985;26:877–84. [PubMed] [Google Scholar]
- 10.Damico FM, Kiss S, Young LH. Sympathetic ophthalmia. Semin Ophthalmol. 2005;20:191–7. doi: 10.1080/08820530500232100. [DOI] [PubMed] [Google Scholar]
- 11.Toes RE, Offringa R, Blom RJ, Brandt RM, Van Der Eb AJ, Melief CJ, et al. An adenovirus type 5 early region 1B-encoded CTL epitope-mediating tumor eradication by CTL clones is down-modulated by an activated ras oncogene. J Immunol. 1995;154:3396–405. [PubMed] [Google Scholar]
- 12.Schurmans LR, den Boer AT, Diehl L, Van Der Voort EI, Kast WM, Melief CJ, et al. Successful immunotherapy of an intraocular tumor in mice. Cancer Res. 1999;59:5250–4. [PubMed] [Google Scholar]
- 13.Wang S, Boonman ZF, Li HC, He Y, Jager MJ, Toes RE, et al. Role of TRAIL and IFN-gamma in CD4+ T cell-dependent tumor rejection in the anterior chamber of the eye. J Immunol. 2003;171:2789–96. doi: 10.4049/jimmunol.171.6.2789. [DOI] [PubMed] [Google Scholar]
- 14.Boonman ZF, Schurmans LR, van Rooijen N, Melief CJ, Toes RE, Jager MJ. Macrophages are vital in spontaneous intraocular tumor eradication. Invest Ophthalmol Vis Sci. 2006;47:2959–65. doi: 10.1167/iovs.05-1427. [DOI] [PubMed] [Google Scholar]
- 15.Dace DS, Chen PW, Alizadeh H, Niederkorn JY. Ocular immune privilege is circumvented by CD4+ T cells, leading to the rejection of intraocular tumors in an IFN-γ-dependent manner. J Leukoc Biol. 2007;81:421–9. doi: 10.1189/jlb.0806489. [DOI] [PubMed] [Google Scholar]
- 16.Dace DS, Chen PW, Niederkorn JY. CD4+ T-cell-dependent tumour rejection in an immune-privileged environment requires macrophages. Immunology. 2008;123:367–77. doi: 10.1111/j.1365-2567.2007.02700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schurmans LR, Diehl L, den Boer AT, Sutmuller RP, Boonman ZF, Medema JP, et al. Rejection of intraocular tumors by CD4+ T cells without induction of phthisis. J Immunol. 2001;167:5832–7. doi: 10.4049/jimmunol.167.10.5832. [DOI] [PubMed] [Google Scholar]
- 18.Kast WM, Offringa R, Peters PJ, Voordouw AC, Meloen RH, Van Der Eb AJ, et al. Eradication of adenovirus E1-induced tumors by E1A-specific cytotoxic T lymphocytes. Cell. 1989;59:603–14. doi: 10.1016/0092-8674(89)90006-8. [DOI] [PubMed] [Google Scholar]
- 19.Niederkorn JY, Chiang EY, Ungchusri T, Stroynowski I. Expression of a nonclassical MHC class Ib molecule in the eye. Transplantation. 1999;68:1790–9. doi: 10.1097/00007890-199912150-00025. [DOI] [PubMed] [Google Scholar]
- 20.Streilein JW, Bradley D. Analysis of immunosuppressive properties of iris and ciliary body cells and their secretory products. Invest Ophthalmol Vis Sci. 1991;32:2700–10. [PubMed] [Google Scholar]
- 21.Niederkorn J, Streilein JW, Shadduck JA. Deviant immune responses to allogeneic tumors injected intracamerally and subcutaneously in mice. Invest Ophthalmol Vis Sci. 1981;20:355–63. [PubMed] [Google Scholar]
- 22.van Klink F, Taylor WM, Alizadeh H, Jager MJ, van Rooijen N, Niederkorn JY. The role of macrophages in Acanthamoeba keratitis. Invest Ophthalmol Vis Sci. 1996;37:1271–81. [PubMed] [Google Scholar]
- 23.Van Rooijen N. The liposome-mediated macrophage ‘suicide’ technique. J Immunol Methods. 1989;124:1–6. doi: 10.1016/0022-1759(89)90178-6. [DOI] [PubMed] [Google Scholar]
- 24.Spath GF, Schlesinger P, Schreiber R, Beverley SM. A novel role for Stat1 in phagosome acidification and natural host resistance to intracellular infection by Leishmania major. PLoS Pathog. 2009;5:e1000381. doi: 10.1371/journal.ppat.1000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shinohara H, Yano S, Bucana CD, Fidler IJ. Induction of chemokine secretion and enhancement of contact-dependent macrophage cytotoxicity by engineered expression of granulocyte-macrophage colony-stimulating factor in human colon cancer cells. J Immunol. 2000;164:2728–37. doi: 10.4049/jimmunol.164.5.2728. [DOI] [PubMed] [Google Scholar]
- 26.Nascimento FR, Ribeiro-Dias F, Russo M. Cytotoxic activity of BCG-activated macrophages against L929 tumor cells is nitric oxide-dependent. Braz J Med Biol Res. 1998;31:1593–6. doi: 10.1590/s0100-879x1998001200012. [DOI] [PubMed] [Google Scholar]
- 27.Yamaguchi H, Kidachi Y, Umetsu H, Ryoyama K. L-NAME inhibits tumor cell progression and pulmonary metastasis of r/m HM-SFME-1 cells by decreasing NO from tumor cells and TNF-alpha from macrophages. Mol Cell Biochem. 2008;312:103–12. doi: 10.1007/s11010-008-9725-5. [DOI] [PubMed] [Google Scholar]
- 28.She H, Nakazawa T, Matsubara A, Hisatomi T, Young TA, Michaud N, et al. Reduced photoreceptor damage after photodynamic therapy through blockade of nitric oxide synthase in a model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2007;48:2268–77. doi: 10.1167/iovs.06-0979. [DOI] [PubMed] [Google Scholar]
- 29.Zhao X, Mohaupt M, Jiang J, Liu S, Li B, Qin Z. Tumor necrosis factor receptor 2-mediated tumor suppression is nitric oxide dependent and involves angiostasis. Cancer Res. 2007;67:4443–50. doi: 10.1158/0008-5472.CAN-07-0185. [DOI] [PubMed] [Google Scholar]
- 30.Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–61. doi: 10.1111/j.1600-065X.2008.00607.x. [DOI] [PubMed] [Google Scholar]
- 31.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 32.Bogdan C. Nitric oxide and the regulation of gene expression. Trends Cell Biol. 2001;11:66–75. doi: 10.1016/s0962-8924(00)01900-0. [DOI] [PubMed] [Google Scholar]
- 33.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A. 1997;94:5243–8. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–26. doi: 10.1172/JCI112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. 2004;4:432–44. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- 36.Dace DS, Chen PW, Alizadeh H, Niederkorn JY. Ocular immune privilege is circumvented by CD4+ T cells, leading to the rejection of intraocular tumors in an IFN-{gamma}-dependent manner. J Leukoc Biol. 2007;81:421–9. doi: 10.1189/jlb.0806489. [DOI] [PubMed] [Google Scholar]
- 37.Dace DS, Chen PW, Niederkorn JY. CD8+ T cells circumvent immune privilege in the eye and mediate intraocular tumor rejection by a TNF-alpha-dependent mechanism. J Immunol. 2007;178:6115–22. doi: 10.4049/jimmunol.178.10.6115. [DOI] [PubMed] [Google Scholar]
- 38.Bloom BR, Bennett B. Macrophages and delayed-type hypersensitivity. Semin Hematol. 1970;7:215–24. [PubMed] [Google Scholar]
- 39.David JR. Lymphocyte mediators and cellular hypersensitivity. N Engl J Med. 1973;288:143–9. doi: 10.1056/NEJM197301182880311. [DOI] [PubMed] [Google Scholar]
- 40.Bonnotte B, Larmonier N, Favre N, Fromentin A, Moutet M, Martin M, et al. Identification of tumor-infiltrating macrophages as the killers of tumor cells after immunization in a rat model system. J Immunol. 2001;167:5077–83. doi: 10.4049/jimmunol.167.9.5077. [DOI] [PubMed] [Google Scholar]
- 41.Mytar B, Siedlar M, Woloszyn M, Ruggiero I, Pryjma J, Zembala M. Induction of reactive oxygen intermediates in human monocytes by tumour cells and their role in spontaneous monocyte cytotoxicity. Br J Cancer. 1999;79:737–43. doi: 10.1038/sj.bjc.6690118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urban JL, Shepard HM, Rothstein JL, Sugarman BJ, Schreiber H. Tumor necrosis factor: a potent effector molecule for tumor cell killing by activated macrophages. Proc Natl Acad Sci U S A. 1986;83:5233–7. doi: 10.1073/pnas.83.14.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hibbs JB, Jr, Taintor RR, Vavrin Z. Macrophage cytotoxicity: role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science. 1987;235:473–6. doi: 10.1126/science.2432665. [DOI] [PubMed] [Google Scholar]
- 44.Weigert A, Brune B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide. 2008;19:95–102. doi: 10.1016/j.niox.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 45.Lechner M, Lirk P, Rieder J. Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Semin Cancer Biol. 2005;15:277–89. doi: 10.1016/j.semcancer.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Li LM, Kilbourn RG, Adams J, Fidler IJ. Role of nitric oxide in lysis of tumor cells by cytokine-activated endothelial cells. Cancer Res. 1991;51:2531–5. [PubMed] [Google Scholar]
- 47.Stuehr DJ, Nathan CF. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. 1989;169:1543–55. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]