

Abstract
The acute respiratory distress syndrome (ARDS) causes 40% mortality in approximately 200,000 critically ill patients annually in the United States. ARDS is caused by protein-rich pulmonary edema that causes severe hypoxemia and impaired carbon dioxide excretion. The clinical disorders associated with the development of ARDS include sepsis, pneumonia, aspiration of gastric contents, and major trauma. The lung injury is caused primarily by neutrophil-dependent and platelet-dependent damage to the endothelial and epithelial barriers of the lung. Resolution is delayed because of injury to the lung epithelial barrier, which prevents removal of alveolar edema fluid and deprives the lung of adequate quantities of surfactant. Lymphocytes may play a role in resolution of lung injury. Mortality has been markedly reduced with a lung-protective ventilatory strategy. However, there is no effective pharmacologic therapy, although cell-based therapy and other therapies currently being tested in clinical trials may provide novel treatments for ARDS.
Keywords: acute lung injury, pulmonary edema
INTRODUCTION
The acute respiratory distress syndrome (ARDS) was described originally in 1967 by the late Dr. Thomas L. Petty and coworkers (1). ARDS is a syndrome of acute respiratory failure that presents with progressive arterial hypoxemia, dyspnea, and a marked increase in the work of breathing. Most patients require endotracheal intubation and positive pressure ventilation. There are several clinical disorders associated with the development of ARDS, including sepsis, pneumonia, aspiration of gastric contents, and major trauma (2). The pathology of ARDS in the lung was first described in 1977 by Bachofen & Weibel (3) in a seminal publication. Since the early clinical and pathologic descriptions of ARDS, considerable basic and clinical research has been devoted to understanding the epidemiology, pathogenesis, and determinants of clinical outcomes in ARDS. Also, several clinical trials have been carried out to test new therapies for ARDS.
The primary objective of this review is to briefly discuss the epidemiology, definitions, and clinical disorders associated with ARDS, including the associated nonpulmonary organ failures that frequently accompany this syndrome. The second objective is to consider what is known regarding the pathogenesis of lung injury, primarily mechanisms of injury to the lung endothelium and epithelium that result in the development of acute pulmonary edema. The discussion of pathogenesis also focuses on the contribution of positive pressure ventilation to lung injury in patients with ARDS. The third objective is to consider new insights into the mechanisms responsible for the resolution of ARDS, specifically the mechanisms responsible for the clearance of pulmonary edema and the resolution of acute lung inflammation. The final objective is to briefly review the current treatment modalities, which are primarily focused on improved supportive care. We also discuss promising new therapeutic approaches.
EPIDEMIOLOGY: DEFINITIONS AND ASSOCIATED CLINICAL DISORDERS
In 2005, the incidence of acute lung injury (ALI) and ARDS in adults was estimated to be approximately 200,000 patients annually in the United States, with a mortality of approximately 40% (4). Because of the aging population and the growing incidence of sepsis, this figure probably underestimates the actual incidence. Further, ARDS does occur in children, and several observational studies indicate that it is an important cause of acute respiratory failure in the pediatric population (5, 6). There is new information that African American and Hispanic patients have a higher mortality with ARDS than do Caucasian patients (7); however, the contribution of genetic and/or environmental factors to this racial and ethnic disparity in mortality is not yet well understood.
The standard definition of ALI/ARDS that has been used in most clinical trials as well as for observational clinical research over the past 15 years was recommended by the American/European Consensus Conference in 1994 (8). This definition identifies patients with ALI as those who have bilateral pulmonary infiltrates with arterial hypoxemia using the concentration of arterial oxygen in the blood divided by the inspired fraction of oxygen (i.e., a PaO2/FiO2 ratio of less than 300). If the patient’s PaO2/FiO2 ratio is less than 200, then a diagnosis of ARDS can be made. The arterial hypoxemia is caused by accumulation of edema fluid in the distal air spaces of the lung, resulting in defects in blood gas exchange. Carbon dioxide excretion is also abnormal, which increases the respiratory rate, the minute ventilation, and the work of breathing. To make a diagnosis of ALI or ARDS, the presence of left atrial hypertension should be excluded on the basis of clinical findings, although one clinical trial (9) established that the pulmonary arterial wedge pressure is greater than 18 mm Hg in 29% of patients with ALI/ARDS. Interestingly, most of the patients in that trial had a normal cardiac output. Thus, intravascular volume overload, probably secondary to volume resuscitation that is necessitated by shock from trauma or sepsis, seems to complicate ALI/ARDS in many patients.
Several clinical disorders have been associated with the development of ALI/ARDS, but the majority of patients develop the syndrome in the presence of an established pulmonary or nonpulmonary infection. The most common cause of ALI/ARDS is primary pneumonia, which can be bacterial, viral, or fungal (2, 3). The second most common cause of lung injury is severe sepsis, which may be associated with pneumonia or a nonpulmonary infectious source, such as peritonitis. The other important major causes of ALI/ARDS include aspiration of gastric contents; hemorrhage and shock following major trauma; and several other less common causes such as severe acute pancreatitis, transfusion-associated lung injury, and drug reactions (1, 2, 5, 6, 10).
Importantly, many patients with acute respiratory failure from ARDS also develop non-pulmonary organ failure, such as cardiovascular failure requiring vasopressor support, renal failure that requires dialysis, abnormal liver function, and/or hematologic abnormalities including anemia and thrombocytopenia. Often, the nonpulmonary organ failures are related to severe sepsis, but in other instances they may be associated with the initial insult of shock and transfusion of blood products. One large study (11) reported that the development of acute kidney injury after the development of ALI increased mortality from 28% to 58%.
PATHOLOGY
The characteristic pathological findings in the lungs of patients with ARDS were best described in a classic study in 1977 that included ultrastructural details at different time points in the acute, subacute, and chronic phases (Figure 1) (3). In the acute phase (the first 1–6 days), there is evidence of interstitial and alveolar edema with accumulation of neutrophils, macrophages, and red blood cells in the alveoli (Figure 1a,b). There is also evidence of both endothelial and epithelial injury, often with denuding of the alveolar epithelium (Figure 1c). There are prominent hyaline membranes in the alveoli as well (Figure 1b). In the subacute phase (the next 7–14 days), some of the edema has usually been reabsorbed, and there is evidence of attempts at repair with proliferation of alveolar epithelial type II cells. There may also be infiltration of fibroblasts and some evidence of collagen deposition. In the chronic phase (after 14 days), there is resolution of the acute neutrophilic infiltrate (unless there has been superimposed nosocomial pneumonia) with more mononuclear cells and alveolar macrophages in the alveoli, and often more fibrosis with ongoing evidence of alveolar epithelial repair. In many patients, resolution progresses without fibrosis and simply with gradual resolution of the edema and acute inflammation.
Figure 1.
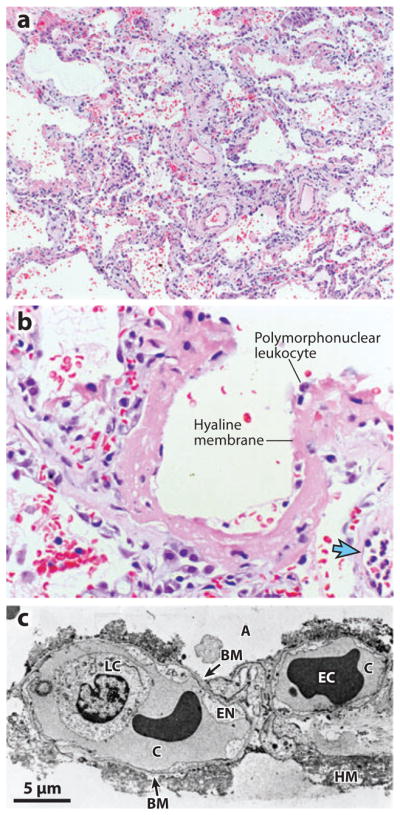
Histologic and ultrastructural analysis of the injured lung has been integral to current concepts of pathogenesis of acute lung injury/acute respiratory distress syndrome (ALI/ARDS). (a) A low-power light micrograph of a lung biopsy specimen collected two days after the onset of ALI/ARDS secondary to gram-negative sepsis demonstrates key features of diffuse alveolar damage, including hyaline membranes, inflammation, intra-alveolar red cells and neutrophils, and thickening of the alveolar-capillary membrane. (b) A higher-power view of a different field illustrates a dense hyaline membrane and diffuse alveolar inflammation. Polymorphonuclear leukocytes are imbedded in the proteinaceous hyaline membrane structure. The blue arrow points to the edge of an adjacent alveolus, which contains myeloid leukocytes. (c) An electron micrograph from a classic analysis of ALI/ARDS showing injury to the capillary endothelium and the alveolar epithelium. Abbreviations: A, alveolar space; BM, exposed basement membrane, where the epithelium has been denuded; C, capillary; EC, erythrocyte; EN, blebbing of the capillary endothelium; LC, leukocyte (neutrophil) within the capillary lumen. The histologic sections in panels a and b are used courtesy of Dr. K. Jones, University of California, San Francisco. Reprinted with permission from the American Thoracic Society.
PATHOGENESIS
The pathogenesis of ALI/ARDS can best be understood by focusing on (a) the factors that are responsible for the accumulation of protein-rich and neutrophilic pulmonary edema in the lung interstitium and in the distal air spaces of the lung and (b) the mechanisms that impair the removal of pulmonary edema fluid and inflammatory cells from the lung. The protein-rich edema fluid in ARDS is associated with large numbers of neutrophils; monocytes; denuded epithelial cells; and proinflammatory markers including cytokines, proteases, oxidants, and procoagulant factors (Figure 2).
Figure 2.

(a) The normal alveolus and (b) the injured alveolus in the acute phase of acute lung injury and the acute respiratory distress syndrome. In the acute phase of the syndrome (b), there is sloughing of both the bronchial and alveolar epithelial cells; protein-rich hyaline membranes form on the denuded basement membrane. Neutrophils adhere to the injured capillary endothelium and marginate through the interstitium into the air space, which is filled with protein-rich edema fluid. In the air space, alveolar macrophages secrete cytokines; interleukin (IL)-1, -6, -8, and -10; and tumor necrosis factor α(TNF-α), which act locally to stimulate chemotaxis and activate neutrophils. IL-1 can also stimulate the production of extracellular matrix by fibroblasts. Neutrophils can release oxidants, proteases, leukotrienes, and other proinflammatory molecules such as platelet-activating factor (PAF). A number of anti-inflammatory mediators also present in the alveolar milieu include IL-1 receptor antagonist, soluble TNF receptor, autoantibodies against IL-8, and cytokines such as IL-10 and -11 (not shown). The influx of protein-rich edema fluid into the alveolus leads to the inactivation of surfactant. Abbreviation: MIF, macrophage-inhibitory factor. Adapted with permission from the Massachusetts Medical Society.
Lung Endothelial Injury
Lung vascular injury is the most important initial cause of ALI/ARDS. There is considerable evidence that an increase in lung vascular permeability occurs primarily at the level of lung microcirculation, which in turn results in the accumulation of protein-rich pulmonary edema, even in the presence of normal lung vascular pressure (12, 13). Injury to the lung endothelium can occur by several mechanisms, although neutrophil-dependent lung injury is probably the best-documented pathway (14, 15). In many experimental models, including acid-induced lung injury (16, 17) and transfusion-associated lung injury (18), neutrophils are a critical final pathway of lung injury. In the setting of both infectious and noninfectious lung injury, neutrophils accumulate in the lung microvasculature and become activated, leading to degranulation and the release of several toxic mediators, including proteases, reactive oxygen species, proinflammatory cytokines, and procoagulant molecules, which result in increased vascular permeability and a sustained loss of normal endothelial barrier function. The concept of neutrophil-dependent lung injury is important, but it also needs to be placed in the context of the vital role that neutrophils play in host defense, particularly against bacterial infection (19). Thus, although neutrophil depletion may attenuate or altogether prevent lung injury in animal models, the lack of functioning neutrophils obviously impairs innate immunity.
There is interesting new evidence that platelets may play an important role in neutrophil-mediated lung injury. Several recent lines of evidence point to an additive and even synergistic effect of platelets in conjunction with neutrophils in causing lung endothelial injury. Platelets can directly interact with neutrophils and monocytes and are themselves a source of proinflammatory cytokines. In recent experimental studies of transfusion-associated lung injury (20) as well as of acid-induced lung injury, platelet depletion markedly reduced lung injury in mouse models. In these mouse models, platelet sequestration in the lung is neutrophil dependent, although neutrophil sequestration is not platelet dependent. There is also evidence that platelets play a role in the thrombotic complications that develop in sickle cell anemia (21). The molecular pathways that link platelet- and neutrophil-mediated injury are incompletely understood (22). Neutrophil recruitment is mediated by step-by-step interactions with the endothelium, usually referred to as rolling adhesion, that are often followed by extravasation. There seem to be several signaling pathways induced during the rolling phase, which result in the transition to leukocyte adhesion; this transition appears to contribute to chemokine-mediated activation. Neutrophils integrate signals received from the endothelium, and there seems to be communication between the vessel wall and platelets—interactions that can cause vascular injury (23). However, platelets can release sphingosine-1-phosphate, which in turn promotes endothelial barrier function (24). As emphasized by some investigators (25), the mechanisms of lung endothelial injury are often distinct from the pathways of injury to the systemic endothelium.
In addition to the effects of neutrophils on the endothelium, some inflammatory mediators act directly on the lung endothelium, resulting in the increased expression of chemokines as well as cell-surface molecules that are important for leukocyte adhesion (23). As a result, leukocyte adhesion to the endothelium and the accumulation of neutrophils in the microcirculation are increased (26, 27), which in turn leads to inflammatory lung injury and further accumulation of other leukocytes in the lung (24). Thus, a key hypothesis in pathogenesis rests on the concept that endothelial activation is central to lung injury (15, 28).
A more detailed review of the molecular mechanisms that account for an increase in lung vascular permeability is beyond the scope of this review. However, there are several publications that describe new insights into the mechanisms of altered lung vascular permeability (15, 29–32).
Alveolar Epithelial Injury
Although lung endothelial injury is a prerequisite for the development of protein-rich pulmonary edema in ARDS, injury to the lung endothelium alone is usually not sufficient to cause the syndrome of ARDS in the absence of some degree of injury to the lung epithelium. For example, there is experimental evidence in large-animal models that moderately severe lung endothelial injury can occur without alveolar epithelial injury (33). In those studies, intravenous and/or intra-alveolar endotoxin produced sustained lung endothelial injury in sheep but did not cause the accumulation of alveolar edema, probably because the alveolar epithelium was morphologically and functionally intact. Alveolar edema developed in this model only when epithelial function was impaired by instillation of live bacteria. In addition to experimental evidence that epithelial injury is required for ARDS, the classic pathologic studies by Bachofen & Weibel (3) in 1977 documented that patients with ARDS demonstrate both lung endothelial and alveolar epithelial injury.
What are the mechanisms responsible for epithelial injury in ALI/ARDS? Although the evidence is incomplete, it appears that neutrophils and their products may be primarily responsible for increased paracellular epithelial permeability in ALI/ARDS. Neutrophils can cross the alveolar epithelium without inducing an increase in lung epithelial permeability (33, 34). However, in pathologic states, the migration of large numbers of neutrophils can result in epithelial injury. In addition, the degree to which neutrophils are activated (primed) by exposure to chemokines and other proinflammatory stimuli seems to play an important role in the effect they may have on the alveolar epithelium as they cross into the distal air spaces.
As summarized in a recent review (35), transepithelial migration of neutrophils into the distal air spaces of the lung involves three sequential steps: adhesion, migration, and post-migration (Figure 3). In the first stage of transepithelial migration in vivo, neutrophils adhere to the basolateral epithelial surface by β2-integrins. It appears that CD11b/CD18 is the primary molecule involved in the initial adhesion of neutrophils to the basolateral surface (36), although there is some evidence for CD18-independent transmigration of neutrophils as well (37). The identity of the epithelial counterreceptor for CD11b/CD18 remained elusive for many years, although a heparan sulfate proteoglycan form of CD44v3 was recently discovered in the gastrointestinal epithelium to bind CD11b/CD18 and facilitate neutrophil transmigration (38).
Figure 3.

Neutrophil migration across epithelia can be considered in three sequential stages: adhesion, migration, and postmigration. The initial stage of neutrophil transepithelial migration is characterized by adhesion of the neutrophils to the basolateral epithelial membrane. Adhesion is mediated by ligation of CD11b/CD18 on the neutrophil surface to several molecules on the epithelial surface, including fucosylated glycoproteins; junctional adhesion molecule C ( JAM-C); and probably other, as-yet-unidentified molecules. After initial adhesion, neutrophils crawl along the epithelial cell membrane via sequential binding to a number of epithelial cell–surface molecules. Both epithelial and neutrophil CD47 molecules are involved during this stage, and CD47 on both cell types may bind to and signal through signal regulatory protein α (SIRPα). In addition, SIRPα probably signals via pathways that are independent of CD47 during neutrophil transepithelial migration. Once neutrophils have completely traversed the epithelial monolayer, they adhere to the apical epithelial surface, where they resist fluid flow and mechanical forces and constitute a defense barrier against invading microorganisms. See Reference 35 for more details. Adapted with permission from the American Thoracic Society.
As the neutrophils migrate along a para-cellular route to the distal air spaces, CD47, a cell-surface molecule expressed on epithelia and neutrophils, may play a role in transepithelial migration. In vitro studies have demonstrated that CD47 blockade impairs neutrophil transepithelial migration (39, 40). Moreover, neutrophil influx into the air spaces and the associated lung permeability caused by inhaled endotoxin are substantially attenuated in CD47-deficient mice (41). Current evidence suggests that once neutrophils have traversed the epithelium and enter the air spaces, they adhere to the apical surface, where they may phagocytize and kill bacteria. Under physiologic conditions, neutrophils can cross the paracellular space and reseal epithelial intercellular junctions, thereby maintaining an intact epithelial barrier and dry air spaces. However, under pathologic conditions, large numbers of activated neutrophils can damage the alveolar epithelium, probably by the release of toxic intracellular molecules that induce dissolution of tight junctions as well as apoptosis and necrosis of alveolar epithelial type I and type II cells (Figure 4). These toxic mediators include proteases such as elastase and matrix metalloproteinases, cationic peptides such as defensins, and reactive oxygen species (35). In addition to creating paracellular gaps, there is considerable evidence that apoptosis of the alveolar epithelium may be important in human lung injury (Figure 5) (42), as well as in experimental studies (43).
Figure 4.
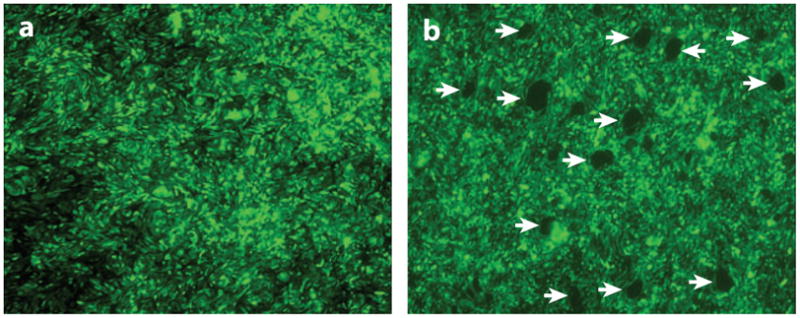
Migration of large numbers of neutrophils results in the death of epithelial cells with the formation of circular areas of denudation at the sites where neutrophils impale the monolayer. Calu-3 lung epithelial cells were grown to confluence on the underside of inverted semipermeable membranes in Transwell inserts. (a) In the control state, the epithelial monolayer is intact. However, when freshly isolated human neutrophils are added to the upper chamber (corresponding to the basolateral surface of epithelial cells) and induced to migrate in a physiological basolateral-to-apical direction across the epithelial monolayer by the addition of a chemoattractant (formyl-methionyl-leucyl-phenylalanine) to the lower chamber (apical surface), individual “scout” neutrophils migrate across the epithelium at specific sites. This initial breach of the epithelium results in a localized high concentration of chemoattractant at these sites. Therefore, as transmigration proceeds, the trailing neutrophils migrate towards the chemotactic gradient and follow in the “tracks” of the leading cells, crossing the monolayer at these sites. (b) The epithelial monolayer after neutrophil migration. The migration of large numbers of neutrophils at specific sites in the epithelial monolayer with the formation of clusters of neutrophils on the apical surface results in disruption of the monolayer and the creation of circular areas of denudation (arrows). The images represent the top-down view of the surface of the epithelial monolayer (not the cross section); the neutrophils have migrated from above the picture plane to below the picture plane.
Figure 5.

Localization of markers of apoptosis in lung tissue sections from patients. (Left) Lung tissue sections from a patient who died with acute lung injury (ALI) or acute respiratory distress syndrome (ARDS). (Right) Lung tissue sections from a patient who died without pulmonary disease. (a–f ) Tissue sections that are counterstained with hematoxylin. ( g–j) Tissue sections that were imaged using differential interference contrast optics, without counterstain, because the epithelial cell immunostain reaction product was subtle. The rows of pictures are matched for one marker of apoptosis: (a,b) TUNEL, (c,d ) caspase-3, (e, f ) Bax, ( g, h) Bcl-2, and (i, j) p53. Cells lining, and in, the alveolar walls demonstrate more TUNEL-labeled nuclei, caspase-3-labeled cytoplasm (arrow), Bax-labeled cytoplasm (arrow), and p53-labeled cytoplasm (arrow) in the tissue sections from the patient who died with ALI or ARDS compared with the patient who died without pulmonary disease. However, Bcl-2-labeled cells lining, and in, the alveolar walls are more prominent in the tissue sections from the patient who died without pulmonary disease compared with the patient who died with ALI or ARDS, as expected. All of the panels are of the same magnification. Reprinted with permission from the American Society of Pathology.
Several neutrophil-derived mediators, including proteases, cationic peptides, reactive oxygen species, and matrix metalloproteinases, appear to be important in causing an increase in epithelial permeability by neutrophils and may be important in alveolar epithelial injury (35). The role of oxidants derived from neutrophils, as well as other cellular sources of oxidants, has been explored in several experimental studies (44). In brief, oxidants have indirect proinflammatory effects and also cause epithelial injury directly via either apoptotic or necrotic pathways. Also, there is evidence from experiments in mice that angiotensin II may be an important mediator of alveolar epithelial apoptosis from hyperoxic-induced lung injury (45).
Ventilator-associated lung injury
A discussion of the pathogenesis of ARDS is not complete without an examination of the contribution of positive pressure ventilation to the lung injury. In the 1970s and 1980s, a few experimental studies (46–48) suggested that the common practice of ventilating patients with a tidal volume of 12 to 15 ml kg−1 and airway pressures greater than 35 to 40 cm H2O may compound ALI. However, definitive proof of this phenomenon came with the results of a single-center study in Brazil in 1998 (49) and from the multicenter National Heart, Lung and Blood Institute–supported ARDS Network clinical trial in 2000 (50). The multicenter ARDS Network trial of 861 patients demonstrated a marked reduction in mortality and in severity of lung injury with a lung-protective ventilatory strategy, and it provided convincing evidence that previously accepted ventilatory strategies using high tidal volumes and high airway pressures amplified lung injury in patients with ARDS. Subsequently, animal studies revealed that a ventilation strategy with lower tidal volumes and lower airway pressures is protective in ALI due to several mechanisms, including reduced lung endothelial injury, reduced lung epithelial injury, reduced lung inflammation, and accelerated resolution of alveolar edema (51, 52). Thus, ventilation with higher tidal volumes and elevated airway pressures causes more lung inflammation and probably results in direct mechanical injury to the lung epithelium and endothelium as well. Follow-up human studies have demonstrated that patients who are ventilated with lower tidal volumes and lower airway pressures had a reduction both (a) in plasma levels of interleukin (IL)-8, IL-6, and soluble TNF receptor 1 (53) and (b) in the number of neutrophils and inflammatory markers in the air spaces of the lung (54). Furthermore, levels of the alveolar epithelial type II cell marker SP-D (55) and the receptor for advanced glycation end products (RAGE) (56) were also reduced in patients who were ventilated with a lower tidal volume. Interestingly, elevated plasma RAGE levels identified those patients who are most likely to benefit from a lower–tidal volume strategy. Thus, unfavorable ventilatory strategies compound the degree of lung injury in patients with ALI/ARDS through a variety of mechanisms.
Genetic factors of the host and the pathogen
There is rapidly growing interest in the potential role of genetic factors in patients who develop ARDS. Genome-wide association screening of patients who either have ARDS or are at risk of developing ARDS is under way. The results of several candidate-gene studies have been summarized in a recent review (57), and the accompanying editorial (58) provided important additional insights. Another likely contributing factor to the development and severity of lung injury is the virulence of the infecting organism, be it bacterial, viral, fungal, or parasitic. Some studies have shown experimentally that the virulence capacity of Pseudomonas aeruginosa is a major determinant of the severity of lung injury (59).
RESOLUTION OF ALVEOLAR EDEMA AND INFLAMMATION
The first step toward resolution of ALI/ARDS is to remove the alveolar edema fluid to the lung interstitium, where net clearance can occur through lung lymphatics, the pulmonary microcirculation, and even bulk flow into the pleural space. Removal of alveolar edema fluid from the air spaces requires vectorial transport of sodium and chloride across the apical and basolateral membranes of alveolar epithelial type I and II cells, which creates a miniosmotic gradient for the reabsorption of water (Figure 6) (60, 61). Thus, sodium is actively absorbed across the apical surface of alveolar epithelial type I and II cells, predominantly by the epithelial sodium channel; the sodium is extruded from the cell by the Na/K-ATPase pumps on the basolateral membrane, thereby creating a miniosmotic gradient for the absorption of water from the alveoli. Chloride is transported by the transcellular and perhaps the paracellular route, although the molecular pathways are not completely understood.
Figure 6.
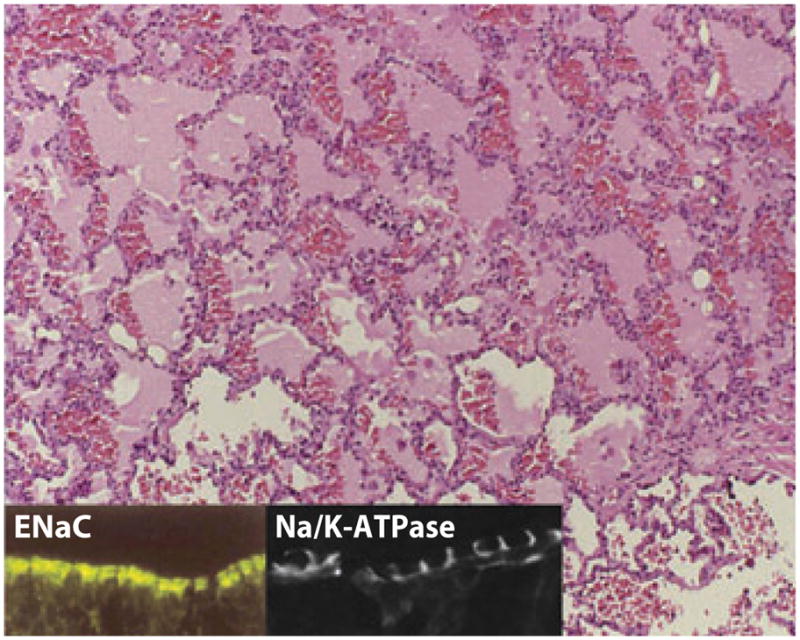
A histologic section of acute respiratory distress syndrome in a human lung sample. Note the fluid-filled alveolar spaces with significant red blood cell infiltration. (Inset) Apical epithelial sodium channel (ENaC) staining and basolateral Na/K-ATPase staining in lung epithelia. Reprinted with permission from the American Physiological Society.
In cardiogenic edema, alveolar edema is readily absorbed across the alveolar epithelium because the epithelium is not injured. In contrast, in patients with ALI, net removal of alveolar edema fluid cannot proceed at a normal rate because the alveolar epithelium is injured due to either (a) frank denuding of the alveolar epithelium (Figure 1c) or (b) inflammation or oxidant- or toxin-mediated injury to the ion-transport machinery in the epithelial cells (60, 61). Several experimental and clinical studies have demonstrated that the capacity of the alveolar epithelium to remove edema fluid is markedly impaired in the presence of ALI (60–64). In contrast, as explained above, the resolution of alveolar edema is normal in most patients with hydrostatic pulmonary edema (65), probably because the alveolar epithelium is not usually injured and can therefore efficiently remove alveolar edema fluid once the elevated pulmonary microvascular pressure normalizes.
Because the recovery of an intact epithelium depends on the cessation of inflammatory injury, resolution of lung inflammation has become an active area of investigation. One group (66, 67) has shown that lipid mediators termed lipoxins and resolvins can control local inflammatory responses, inhibit neutrophil activation, and begin the switch to resolution of lung injury. Analogously, the presence of anti-inflammatory cytokines such as IL-10 may play an important role in the resolution of lung inflammation (68). In addition, recent work based on experimental studies of endotoxin lung injury in mice has highlighted the likely contribution of lymphocytes to the resolution of lung injury (69). In these studies, investigators found that the infusion of CD4+CD25+Foxp3+ T regulatory cells (Tregs) hastened the resolution of endotoxin-mediated lung injury in Rag 1 knockout mice, in part by upregulating transforming growth factor β, and Treg depletion delayed recovery in wild-type mice. Finally, apoptosis and phagocytosis of inflammatory cells are critical for the resolution of inflammation and mitigation of tissue damage (70, 71).
TREATMENT
A large number of pharmacologic therapies have been evaluated in Phase II and Phase III clinical trials for the treatment of ALI/ARDS. These treatments include glucocorticoids, surfactants, inhaled nitric oxide, antioxidants, protease inhibitors, and a variety of other anti-inflammatory treatments. Unfortunately, to date none of these pharmacologic treatments has proven to be effective (72), although some of them may be effective in a subgroup of patients with specific causes of lung injury that might make them more responsive than others. However, despite the lack of a specific pharmacologic treatment, lung-protective ventilation has reduced the mortality of ALI from 40% in 2000 to 25% in 2006 (73). Further, the use of a fluid-conservative strategy after patients with ARDS are no longer in shock has reduced the duration of mechanical ventilation (9).
In terms of promising new therapies, cell-based approaches to treating ALI have been explored in preclinical studies. Recent studies in mice indicated that mesenchymal stem cells (MSCs) may be of value in treating ALI as well as sepsis. In one study from our research group, intratracheal MSCs were effective at reducing pulmonary edema and mortality in mice that had been treated with high-dose intratracheal endotoxin (5 mg kg−1) (74). The benefit of the MSCs in this mouse model appeared to be related to a decrease in proinflammatory cytokines and an increase in anti-inflammatory cytokines, particularly IL-10. Another mouse study reported that intravenous administration of MSCs after the development of peritoneal sepsis reduced mortality and organ injury, and this effect was again mediated primarily by increased IL-10 levels. Interestingly, this study (68) showed that secretion of IL-10 occurred primarily by monocytes and alveolar macrophages in response to prostaglandin E2 released by the MSCs. Also, two recent studies (75–77) reported that administration of MSCs or the cultured medium from MSCs can reverse hyperoxic-induced lung injury in perinatal mouse and rat models. Finally, our own work with a perfused ex vivo human lung preparation indicated that administration of MSCs 1 h after severe endotoxin-induced lung injury was effective in normalizing lung vascular and epithelial permeability to protein as well as reducing pulmonary edema and increasing the rate of alveolar fluid clearance (78). In this study, the cultured media from the MSCs were also effective as treatment. The MSCs and the media from the MSCs were equally effective in reversing the histologic abnormalities in the perfused endotoxin-injured lung (Figure 7). In addition, further studies demonstrated that the release of the paracrine factor, keratinocyte growth factor, was a major mediator of the protective effect. More preclinical studies will be needed to assess the extent to which and the mechanisms by which MSCs are effective in ALI. Clinical trials with allogeneic MSCs may be warranted if preclinical studies continue to show that MSC therapy could be effective in preclinical models.
Figure 7.

Histology sections (two examples for each condition) in the ex vivo perfused human lung lobes exposed to endotoxin with and without treatment with either allogeneic human mesenchymal stem cells (MSCs) or their conditioned medium (CM). Human lungs exposed to endotoxin with and without MSCs or their CM were fixed in 10% formalin at 4 h. Sections were stained with hematoxylin and eosin (magnification 10×). The instillation of human allogeneic MSCs (5 × 106 cells) or their CM 1 h after endotoxin injury reduced the degree of edema and cellularity in the endotoxin-injured lung lobe. Abbreviation: LPS, lipopolysaccharide. Reprinted from Reference 79 with permission.
Other clinical trials are in progress to test the potential value of statin therapy for patients with ALI, based on several circumstantial studies suggesting that statin therapy may have value in patients with sepsis or lung injury (67, 79). In addition, studies of sepsis may also have direct relevance to the treatment of lung injury, given that sepsis is the most lethal and important cause of lung injury. Such ongoing clinical trials include the Prowess-Shock Trial, which is retesting the therapeutic value of activated protein C in severe sepsis, and a trial testing a Toll-4 receptor inhibitor in patients with sepsis.
CONCLUSIONS
ALI/ARDS occurs in 200,000 people per year in the United States and has an estimated mortality rate of approximately 40%. There are several clinical disorders associated with the development of ARDS, but the pathogenesis involves inflammatory injury to the lung endothelium and epithelium, which causes a marked increase in lung vascular and epithelial permeability and the passage of protein-rich edema fluid into the air spaces. The initial lung injury can be compounded by ventilator-associated lung injury, particularly when high tidal volumes and inflation pressures are used. Clinical recovery from ALI/ARDS depends on the use of lung-protective ventilation with lower tidal volumes and the reduction of airway pressures, which facilitate the resolution of inflammation, repair of the injured epithelium, and removal of edema fluid from the air spaces.
Although there has been considerable progress in understanding the pathogenesis of ALI/ARDS, there are several important gaps in our knowledge (Table 1). First, the mechanisms that lead to alveolar epithelial injury, including gap formation and apoptosis and necrosis of type I and type II cells, are not well understood. Second, the mechanisms that contribute to restoration of a normal alveolar epithelium require more study. Third, we need to understand the extent to which the proinflammatory and procoagulant pathways are protective in lung injury because they play a necessary role in host defense against invading bacteria, viruses, and fungi. Fourth, the genetic factors of the host (the patient) and the infecting organisms (virulence factors) probably play a major role in both the susceptibility and the severity of ALI/ARDS, but we do not yet have an adequate understanding of how they influence the course of acute respiratory failure in ALI/ARDS. Fifth, there are probably important environmental factors that contribute to the development and severity of ALI/ARDS that require more study, including the potential influence of exposure to cigarette smoke.
Table 1.
Unresolved issues in the pathogenesis and treatment of acute lung injury
| Issue |
|---|
| 1. Specific signaling pathways activated in neutrophils and epithelial cells during transmigration that play a role in lung injury and repair |
| 2. Development of new therapeutic strategies to prevent neutrophil-mediated epithelial injury without impairing critical host defense functions |
| 3. Mechanisms by which platelets, neutrophils, and the endothelium interact to cause pulmonary vascular injury and the role of antiplatelet therapies in clinical lung injury |
| 4. Role of epithelial and endothelial cell apoptosis in acute lung injury and the potential for antiapoptotic therapies for lung injury |
| 5. Role of lymphocytes and resolvins in the resolution of the inflammation, the restoration of the epithelial and endothelial barrier, and the removal of protein-rich edema fluid |
| 6. Genetic contributions to acute lung injury |
| 7. Effect of bacterial and viral virulence factors on clinical outcomes in acute lung injury |
| 8. Contribution of environmental factors to the development and severity of acute lung injury |
| 9. Role of new ventilatory strategies, including noninvasive ventilation, higher positive end–expiratory pressure, and high-frequency ventilation, for the prevention of ventilator-associated lung injury |
| 10. Role of cell-based therapy for treatment of severe lung injury, especially when associated with multiorgan dysfunction |
Although there are no proven specific treatments for ALI/ARDS, there has been considerable progress in managing patients with ARDS by use of lung-protective ventilation strategies as well as conservative fluid management. On the basis of new knowledge of the mechanisms of ALI, in the near future specific pharmacologic or cell-based therapy may prove effective in treating patients with severe ARDS.
Acknowledgments
M.A.M. and R.L.Z. are deeply indebted to Andrew Manies for his invaluable work on this manuscript. M.A.M. receives grant support from NHLBI R37HL51856 and -HL51854 and NIAIAD PO1AI1053194. R.L.Z. receives grant support from the Flight Attendant Medical and Research Institute, the Parker B. Francis Foundation, and National Jewish Health.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2:319–23. doi: 10.1016/s0140-6736(67)90168-7. [DOI] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–49. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 3.Bachofen M, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis. 1977;116:589–615. doi: 10.1164/arrd.1977.116.4.589. [DOI] [PubMed] [Google Scholar]
- 4.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–93. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 5.Flori HR, Glidden DV, Rutherford GW, Matthay MA. Pediatric acute lung injury: prospective evaluation of risk factors associated with mortality. Am J Respir Crit Care Med. 2005;171:995–1001. doi: 10.1164/rccm.200404-544OC. [DOI] [PubMed] [Google Scholar]
- 6.Randolph AG. Management of acute lung injury and acute respiratory distress syndrome in children. Crit Care Med. 2009;37:2448–54. doi: 10.1097/CCM.0b013e3181aee5dd. [DOI] [PubMed] [Google Scholar]
- 7.Erickson SE, Shlipak MG, Martin GS, Wheeler AP, Ancukiewicz M, et al. Racial and ethnic disparities in mortality from acute lung injury. Crit Care Med. 2009;37:1–6. doi: 10.1097/CCM.0b013e31819292ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am J Respir Crit Care Med. 1994;149:818–24. doi: 10.1164/ajrccm.149.3.7509706. [DOI] [PubMed] [Google Scholar]
- 9.Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, et al. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354:2564–75. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 10.Eisner MD, Thompson T, Hudson LD, Luce JM, Hayden D, et al. Efficacy of low tidal volume ventilation in patients with different clinical risk factors for acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164:231–36. doi: 10.1164/ajrccm.164.2.2011093. [DOI] [PubMed] [Google Scholar]
- 11.Liu KD, Glidden DV, Eisner MD, Parsons PE, Ware LB, et al. Predictive and pathogenetic value of plasma biomarkers for acute kidney injury in patients with acute lung injury. Crit Care Med. 2007;35:2755–61. [PMC free article] [PubMed] [Google Scholar]
- 12.Staub NC. Pulmonary edema. Physiol Rev. 1974;54:678–811. doi: 10.1152/physrev.1974.54.3.678. [DOI] [PubMed] [Google Scholar]
- 13.Staub NC. Pulmonary edema due to increased microvascular permeability. Annu Rev Med. 1981;32:291–312. doi: 10.1146/annurev.me.32.020181.001451. [DOI] [PubMed] [Google Scholar]
- 14.Flick MR, Perel A, Staub NC. Leukocytes are required for increased lung microvascular permeability after microembolization in sheep. Circ Res. 1981;48:344–51. doi: 10.1161/01.res.48.3.344. [DOI] [PubMed] [Google Scholar]
- 15.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–27. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folkesson HG, Matthay MA, Hebert CA, Broaddus VC. Acid aspiration–induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Investig. 1995;96:107–16. doi: 10.1172/JCI118009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiser MR, Pechet TT, Williams JP, Ma M, Frenette PS, et al. Experimental murine acid aspiration injury is mediated by neutrophils and the alternative complement pathway. J Appl Physiol. 1997;83:1090–95. doi: 10.1152/jappl.1997.83.4.1090. [DOI] [PubMed] [Google Scholar]
- 18.Looney MR, Su X, Van Ziffle JA, Lowell CA, Matthay MA. Neutrophils and their Fcγ receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Investig. 2006;116:1615–23. doi: 10.1172/JCI27238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai S, Zemans RL, Young SK, Worthen GS, Jeyaseelan S. Myeloid differentiation protein-2-dependent and -independent neutrophil accumulation during Escherichia coli pneumonia. Am J Respir Cell Mol Biol. 2009;40:701–9. doi: 10.1165/rcmb.2008-0152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Investig. 2009;119:3450–61. doi: 10.1172/JCI38432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009;15:384–91. doi: 10.1038/nm.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Looney MR, Matthay MA. Neutrophil sandwiches injure the microcirculation. Nat Med. 2009;15:364–66. doi: 10.1038/nm0409-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheiermann C, Kunisaki Y, Jang JE, Frenette PS. Neutrophil microdomains: linking heterocellular interactions with vascular injury. Curr Opin Hematol. 2010;17:25–30. doi: 10.1097/MOH.0b013e328333d2a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bozza FA, Shah AM, Weyrich AS, Zimmerman GA. Amicus or adversary: platelets in lung biology, acute injury, and inflammation. Am J Respir Cell Mol Biol. 2009;40:123–34. doi: 10.1165/rcmb.2008-0241TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doyle NA, Bhagwan SD, Meek BB, Kutkoski GJ, Steeber DA, et al. Neutrophil margination, sequestration, and emigration in the lungs of L-selectin-deficient mice. J Clin Investig. 1997;99:526–33. doi: 10.1172/JCI119189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA., Jr Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Investig. 1985;76:2003–11. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmerman GA, McIntyre TM, Prescott SM. Thrombin stimulates the adherence of neutrophils to human endothelial cells in vitro. J Clin Investig. 1985;76:2235–46. doi: 10.1172/JCI112232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmerman GA, Albertine KH, Carveth HJ, Gill EA, Grissom CK, et al. Endothelial activation in ARDS. Chest. 1999;116:18–24S. doi: 10.1378/chest.116.suppl_1.18s. [DOI] [PubMed] [Google Scholar]
- 29.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 30.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–93. doi: 10.1146/annurev-physiol-021909-135833. [DOI] [PubMed] [Google Scholar]
- 31.Parthasarathi K, Ichimura H, Monma E, Lindert J, Quadri S, et al. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J Clin Investig. 2006;116:2193–200. doi: 10.1172/JCI26605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng DS, Han W, Chen SM, Sherrill TP, Chont M, et al. Airway epithelium controls lung inflammation and injury through the NF-κB pathway. J Immunol. 2007;178:6504–13. doi: 10.4049/jimmunol.178.10.6504. [DOI] [PubMed] [Google Scholar]
- 33.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J Clin Investig. 1991;88:864–75. doi: 10.1172/JCI115388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Investig. 1989;84:1609–19. doi: 10.1172/JCI114338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol. 2009;40:519–35. doi: 10.1165/rcmb.2008-0348TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parkos CA, Delp C, Arnaout MA, Madara JL. Neutrophil migration across a cultured intestinal epithelium. Dependence on a CD11b/CD18-mediated event and enhanced efficiency in physiological direction. J Clin Investig. 1991;88:1605–12. doi: 10.1172/JCI115473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doerschuk CM, Winn RK, Coxson HO, Harlan JM. CD18-dependent and -independent mechanisms of neutrophil emigration in the pulmonary and systemic microcirculation of rabbits. J Immunol. 1990;144:2327–33. [PubMed] [Google Scholar]
- 38.Zen K, Liu DQ, Li LM, Chen CX, Guo YL, et al. The heparan sulfate proteoglycan form of epithelial CD44v3 serves as a CD11b/CD18 counter-receptor during polymorphonuclear leukocyte transepithelial migration. J Biol Chem. 2009;284:3768–76. doi: 10.1074/jbc.M807805200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parkos CA, Colgan SP, Liang TW, Nusrat A, Bacarra AE, et al. CD47 mediates postadhesive events required for neutrophil migration across polarized intestinal epithelia. J Cell Biol. 1996;132:437–50. doi: 10.1083/jcb.132.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Merlin D, Burst SL, Pochet M, Madara JL, Parkos CA. The role of CD47 in neutrophil transmigration. Increased rate of migration correlates with increased cell surface expression of CD47. J Biol Chem. 2001;276:40156–66. doi: 10.1074/jbc.M104138200. [DOI] [PubMed] [Google Scholar]
- 41.Su X, Johansen M, Looney MR, Brown EJ, Matthay MA. CD47 deficiency protects mice from lipopolysaccharide-induced acute lung injury and Escherichia coli pneumonia. J Immunol. 2008;180:6947–53. doi: 10.4049/jimmunol.180.10.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albertine KH, Soulier MF, Wang Z, Ishizaka A, Hashimoto S, et al. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol. 2002;161:1783–96. doi: 10.1016/S0002-9440(10)64455-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matute-Bello G, Lee JS, Liles WC, Frevert CW, Mongovin S, et al. Fas-mediated acute lung injury requires fas expression on nonmyeloid cells of the lung. J Immunol. 2005;175:4069–75. doi: 10.4049/jimmunol.175.6.4069. [DOI] [PubMed] [Google Scholar]
- 44.Cochrane CG, Spragg R, Revak SD. Pathogenesis of the adult respiratory distress syndrome. Evidence of oxidant activity in bronchoalveolar lavage fluid. J Clin Investig. 1983;71:754–61. doi: 10.1172/JCI110823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, et al. Hyperoxia causes angiopoietin 2–mediated acute lung injury and necrotic cell death. Nat Med. 2006;12:1286–93. doi: 10.1038/nm1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis. 1974;110:556–65. doi: 10.1164/arrd.1974.110.5.556. [DOI] [PubMed] [Google Scholar]
- 47.Parker JC, Townsley MI, Rippe B, Taylor AE, Thigpen J. Increased microvascular permeability in dog lungs due to high peak airway pressures. J Appl Physiol. 1984;57:1809–16. doi: 10.1152/jappl.1984.57.6.1809. [DOI] [PubMed] [Google Scholar]
- 48.Dreyfuss D, Saumon G. Role of tidal volume, FRC, and end-inspiratory volume in the development of pulmonary edema following mechanical ventilation. Am Rev Respir Dis. 1993;148:1194–203. doi: 10.1164/ajrccm/148.5.1194. [DOI] [PubMed] [Google Scholar]
- 49.Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338:347–54. doi: 10.1056/NEJM199802053380602. [DOI] [PubMed] [Google Scholar]
- 50.Acute Resp. Distress Syndr. Netw. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–8. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 51.Frank JA, Gutierrez JA, Jones KD, Allen L, Dobbs L, Matthay MA. Low tidal volume reduces epithelial and endothelial injury in acid-injured rat lungs. Am J Respir Crit Care Med. 2002;165:242–49. doi: 10.1164/ajrccm.165.2.2108087. [DOI] [PubMed] [Google Scholar]
- 52.Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos mRNA expression in an isolated rat lung model. J Clin Investig. 1997;99:944–52. doi: 10.1172/JCI119259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, et al. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med. 2005;33:1–6. 230–32. doi: 10.1097/01.ccm.0000149854.61192.dc. [DOI] [PubMed] [Google Scholar]
- 54.Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, et al. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. J Am Med Assoc. 1999;282:54–61. doi: 10.1001/jama.282.1.54. [DOI] [PubMed] [Google Scholar]
- 55.Eisner MD, Parsons P, Matthay MA, Ware L, Greene K Acute Resp. Distress Syndr. Netw. Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax. 2003;58:983–88. doi: 10.1136/thorax.58.11.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Calfee CS, Ware LB, Eisner MD, Parsons PE, Thompson BT, et al. Plasma receptor for advanced glycation end products and clinical outcomes in acute lung injury. Thorax. 2008;63:1083–89. doi: 10.1136/thx.2008.095588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao L, Barnes KC. Recent advances in genetic predisposition to clinical acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;296:L713–25. doi: 10.1152/ajplung.90269.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong MN. Gene association studies in acute lung injury: replication and future direction. Am J Physiol Lung Cell Mol Physiol. 2009;296:L711–12. doi: 10.1152/ajplung.00080.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moskowitz SM, Wiener-Kronish JP. Mechanisms of bacterial virulence in pulmonary infections. Curr Opin Crit Care. 2010;16:8–12. doi: 10.1097/MCC.0b013e3283354710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 61.Folkesson HG, Matthay MA. Alveolar epithelial ion and fluid transport: recent progress. Am J Respir Cell Mol Biol. 2006;35:10–19. doi: 10.1165/rcmb.2006-0080SF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis. 1990;142:1250–57. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- 63.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–83. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 64.Zemans RL, Matthay MA. Bench-to-bedside review: the role of the alveolar epithelium in the resolution of pulmonary edema in acute lung injury. Crit Care. 2004;8:469–77. doi: 10.1186/cc2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verghese GM, Ware LB, Matthay BA, Matthay MA. Alveolar epithelial fluid transport and the resolution of clinically severe hydrostatic pulmonary edema. J Appl Physiol. 1999;87:1301–12. doi: 10.1152/jappl.1999.87.4.1301. [DOI] [PubMed] [Google Scholar]
- 66.Bonnans C, Levy BD. Lipid mediators as agonists for the resolution of acute lung inflammation and injury. Am J Respir Cell Mol Biol. 2007;36:201–5. doi: 10.1165/rcmb.2006-0269TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Planaguma A, Pfeffer MA, Rubin G, Croze R, Uddin M, et al. Lovastatin decreases acute mucosal inflammation via 15-epi-lipoxin A4. Mucosal Immunol. 2010;3:270–79. doi: 10.1038/mi.2009.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E2–dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15:42–49. doi: 10.1038/nm.1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Investig. 2009;119:2898–913. doi: 10.1172/JCI36498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–64. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- 71.Savill JS, Henson PM, Haslett C. Phagocytosis of aged human neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J Clin Investig. 1989;84:1518–27. doi: 10.1172/JCI114328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cepkova M, Matthay MA. Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. J Intensive Care Med. 2006;21:119–43. doi: 10.1177/0885066606287045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wheeler AP, Bernard GR, Thompson BT, Schoenfeld D, Wiedemann HP, et al. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. N Engl J Med. 2006;354:2213–24. doi: 10.1056/NEJMoa061895. [DOI] [PubMed] [Google Scholar]
- 74.Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow–derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;179:1855–63. doi: 10.4049/jimmunol.179.3.1855. [DOI] [PubMed] [Google Scholar]
- 75.van Haaften T, Byrne R, Bonnet S, Rochefort GY, Akabutu J, et al. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med. 2009;180:1131–42. doi: 10.1164/rccm.200902-0179OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, et al. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med. 2009;180:1122–30. doi: 10.1164/rccm.200902-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abman SH, Matthay MA. Mesenchymal stem cells for the prevention of bronchopulmonary dysplasia: delivering the secretome. Am J Respir Crit Care Med. 2009;180:1039–41. doi: 10.1164/rccm.200909-1330ED. [DOI] [PubMed] [Google Scholar]
- 78.Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin–induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci USA. 2009;106:16357–62. doi: 10.1073/pnas.0907996106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fessler MB, Young SK, Jeyaseelan S, Lieber JG, Arndt PG, et al. A role for hydroxy-methylglutaryl coenzyme a reductase in pulmonary inflammation and host defense. Am J Respir Crit Care Med. 2005;171:606–15. doi: 10.1164/rccm.200406-729OC. [DOI] [PubMed] [Google Scholar]