

Abstract
The ability to direct the self-assembly of biomolecules on surfaces with true nanoscale control is key for the creation of functional substrates. Herein we report on the fabrication of nanoscale biomolecular arrays, via selective self-assembly on nanopatterned surfaces and minimized non-specific adsorption. We demonstrate that the platform developed allows for the simultaneous screening of specific protein/DNA binding events at the single-molecule level. The strategy here presented is generally applicable, and enables high throughput monitoring of biological activity in real-time and with single-molecule resolution.
Nanoscale control over the organization of biomolecules at solid substrates is a powerful tool for addressing fundamental issues in many areas of biology.1–9 Nanoarrays of biomolecules10–18 can offer unmatched sensitivity, smaller test sample volumes in molecular diagnostics, and high throughput analysis through the ability to monitor (distinct) bio-recognition events in parallel and on the same chip. By approaching the size-scale of individual biomolecules, nanoscale control could conceivably allow us to carry out single-molecule investigations19 (on an array), that in turn enable monitoring of biochemical processes in real time, characterization of transient intermediates, and measurement of the distributions of molecular properties rather than their ensemble averages.20,21 Key issues involved in developing a nanoscale biochip are related to the selectivity (and spot uniformity) of the biomolecular (self)assembly, the consequent minimization of non-specific adsorption of the biomolecules under investigation, and the accessibility of recognition elements within an immobilized biomolecule.22,23 All such issues affect signal-to-noise ratios and prevent proper interpretation of biomolecular binding/recognition events.24,25
Herein we present a strategy that overcomes all the above limitations by controlling the localization of bio-molecules in ordered nanoarrays, allowing for high throughput single-molecule investigations in real time. Specifically, we show how the dimensions and distance of the fabricated arrays’ nanodots allow for both clear addressability and parallel readout of single-molecule events of biological interest via conventional epi-fluorescence microscopy imaging. (As a proof of principle the activity of a DNA-binding enzyme, exemplified here by the restriction endonuclease PvuII, was monitored). This work highlights the clear advantage of true nanoscale confinement in the design of high throughput (and high resolution) heterogeneous assays for biological investigations.
For our studies, we begin by nanopatterning a glass substrate surface via direct electron-beam lithography (EBL) to create 50×50 μm2 arrays of 30 (± 4) nm Au/Pd nanodots spaced 2 μm apart, interspersed with 500 nm registration squares spaced 10 μm apart (see Figure SI-1 and Figure SI-2). Figure 1 shows the approach used to biofunctionalize the nanodots (details given in the Supporting Information). We first form self-assembled monolayers (SAMs) of thiolated alkanes26 exhibiting biotin head-groups. We next passivate the surface against non-specific adsorption of bio-molecules via the formation of a polyethylene glycol-silane (PEG-silane) monolayer on the glass surface. Next, we immobilize streptavidin on the nanodots,27,28 and finally, we tether biotinylated DNA via a second biotin-streptavidin linkage.29,30
Figure 1.
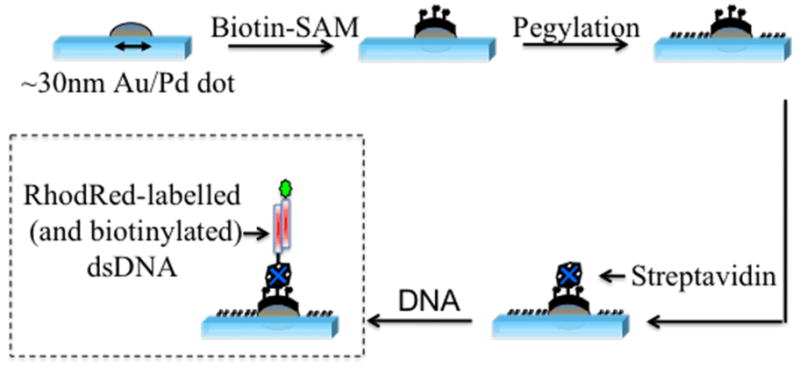
Scheme employed for the chemical functionalization of the nanopatterned substrate
Epi-fluorescence microscopy imaging of the resulting array demonstrates the selectivity of the functionalization at the single-nanodot level. In particular, Figure 2a shows the immobilization of fluorescently labeled streptavidins on every nanodot, while Figure 2b shows the subsequent immobilization of fluorescently labeled double-stranded DNAs. Each employed DNA molecule was labeled with one rhodamine-red fluorophore on the distal end of the duplex, i.e. on the end not attached to the surface-bound streptavidin. Furthermore, in Figures 2a and 2b the uniform passivated regions between the nanodots exhibit a remarkably low fluorescence-background and demonstrate the minimization of non-specific adsorption achieved at the glass substrate. By measuring the average background fluorescence intensity of the glass surface before and after exposure of the substrate to fluorescently labeled DNA (see Supporting Information and figure SI-3), we can determine the physisorbed DNA coverage on the glass surface of our bio-chip to be between 0.1 and 0.5 μm−2 (i.e. less than one DNA every 2 μm2). Noteworthy, because of the size of the nanodots, and therefore the limited number of streptavidins and DNAs attached on each of them (see below for discussion), the ability to resolve single dots requires the ultralow non-specific adsorption which we have achieved.31
Figure 2.

a) Epi-fluorescence microscopy image of the electron-beam written nanoarray functionalized with Alexa488-labeled streptavidins (100 ms exposure time); b) Epi-fluorescence microscopy image of the nanoarray functionalized with Rhodamine Red-labeled dsDNAs (100 ms exposure time); the insets at the top right-hand corners of (a) and (b) show a zoomed fluorescence image of the array.
To demonstrate the general suitability of our platform for monitoring biomolecular interactions, we carried out proof-of-principle restriction enzyme experiments on the functionalized nanoarrays. We anchored to the surface of our nanoarray a 20-basepair DNA labeled with a rohodamine-red fluorophore (one fluorophore per DNA) on the distal end of the duplex, i.e. on the end not attached to the nanodots via the biotin-avidin linkage. The arrays were then incubated with PvuII-HF a well-known and commercially available restriction enzyme with minimal star activity.32 In the presence of the 5′-CAGCTG-3′ PvuII recognition site33 on the employed DNA, we observe a complete loss of fluorescence intensity localized at the individual nanodots, within seconds of addition of the enzyme (Figure 3). This is ascribable to DNA cleavage by the enzyme, and consequent loss of the fluorescently labeled segment of the anchored DNA (see scheme in Figure 3). Notably, no loss of localized fluorescence intensity at the nanodots is observed in the absence of the recognition site, consistent with a lack of DNA cleavage by PvuII (see Figure SI-4). Thus, the interaction of PvuII with the nanodot-immobilized DNA on our nanoarray is highly specific. This observation demonstrates that our platform is generally well suited for the rapid, reliable, and specific real-time monitoring of biomolecular interactions via conventional epi-fluorescence microscopy.
Figure 3.

Scheme and epi-fluorescence microscopy images of the PvuII recognition, and cleavage, of the nanodot-immobilized DNA (200 msec exposure times)
The minimized crowding of the immobilized DNA that arises from the nanoscopic size and microscopic spacing of the nanodots, in combination with the high selectivity and consequently high signal-to-noise ratio achieved, enabled us to obtain single-molecule resolution in monitoring the DNA-PvuII interaction. Each nanodot in our nanoarray is optically resolvable from its neighbors, being spaced 2 μm apart, so we can monitor the loss of fluorescence at the single nanodot level. This results in a loss of fluorescence intensity that occurs in discrete steps, as shown in Figure 4a. By extracting the time delay between when PvuII was first delivered to the nanoarray and when each single-DNA cleavage event was observed, we built a histogram of single-molecule fluorescence extinction as a function of time (Figure 4b).34 It is noteworthy that the histogram is well described by a difference of two exponentials (as shown in Figure SI-6); this implies the existence of at least two rate-determining steps in the PvuII-DNA cleavage reaction, consistent with the existence of a Michaelis-Menten complex.35,36 In addition, our extrapolated value of the overall catalytic rate, or “turnover rate constant”, k, for PvuII (k~1 sec−1) is comparable to previously reported values from ensemble measurements under the same buffer conditions (k~0.3 sec−1).37–39
Figure 4.

a) A plot of the fluorescence intensity versus time of a representative single nanodot containing a single Rhodamine Red-labeled DNA; the single step loss of fluorescence intensity derives from the PvuII cleavage of the DNA (200 ms exposure time). b) Representative histogram of single-molecule DNA cleavage events over an entire nanoarray.
The discrete step-like drops in fluorescence intensity enabled us to determine the average number of DNA molecules immobilized to single nanodots.40 Our analysis demonstrates that ~60% of the nanodots have one DNA molecule bound per nanodot, ~20% have two DNA molecules bound per nanodot, and ~5% have three DNA molecules bound per nanodot.41 Although each 30 nm nanodot can accommodate up to ~30 streptavidins, with each one anchoring two biotinylated DNAs, we find fewer than four DNAs on ~85% of the nanodots. We postulate that this fortuitously sparse density of the DNA results from a combination of an unfavorable arrangement of the streptavidins at the surface and electrostatic repulsion among DNA molecules during immobilization. In particular the unknown arrangement that biotin-thiols adopt in a mixed SAM on a nanoscale substrate,42 is likely responsible for the limited number of streptavidins, and consequently DNAs, anchored on each nanodot.
In order to demonstrate that the process here presented is scalable, we have monitored distinct bio-recognition events on the same biochip, fabricated employing a low-cost nanopatterning technique. We have co-assembled two different 20-basepair DNA molecules, one endonuclease active, the other not, on a substrate patterned by nanoimprint lithography,43 a lower cost and higher throughput patterning technique. The PvuII-active DNA was labeled with a rhodamine-red fluorophore, while the inactive DNA (i.e. lacking the PvuII recognition site) was labeled with a Cy3 dye; in both cases the fluorophores are localized at the distal end of the duplex. We have co-assembled the DNAs on a 50×50 μm2 array of 10 (± 2) nm Au/Pd nanodots spaced 200 nm apart and fabricated by nanoimprint lithography43 (see Figure SI-7).
Multichannel epi-fluorescence microscopy imaging of the resulting nanoarray enables us to monitor the presence of the two different DNAs co-assembled on the same nanoarray, as shown in Figure 5a and Figure 5b: the rhodamine-red labeled DNA is imaged in the green channel, while the Cy3 labeled DNA in the red channel. The resulting nanoarray was then incubated with PvuII. Within seconds of the addition of the enzyme we observe a loss of fluorescence for the PvuII-active DNA (green channel), as shown in Figure 5c. We attribute this to DNA cleavage by the enzyme, and consequent loss of the fluorescently labeled segment of the anchored DNA, similar to what we showed in figure 3. Notably, no loss of fluorescence intensity is observed on the same nanoarray for the DNA lacking the PvuII recognition site and labeled with the Cy-3 fluorophore (red channel: see Figure 5d). This is consistent with a lack of DNA cleavage by the enzyme, and proves that with our platform we can simultaneously monitor two distinct bio-recognition events on the same biochip (patterned via a low-cost fabrication technique).
Figure 5.

Epi-fluorescence microscopy images of the nanoimprinted nanoarray functionalized with Rhodamine Red-labeled dsDNA, exhibiting the PvuII recognition site (green channel), and Cy3-labelled dsDNA lacking the PvuII recognition site (red channel): 100 ms exposure time; a) and b) show the nanoarray before the adding of the enzyme, while c) and d) show how within seconds of the addition of the enzyme the PvuII active DNA is cleaved, as evidenced by the loss of fluorescence (green channel), while the PvuII inactive DNA is not affected by the presence of the enzyme (red channel).
In summary, we have demonstrated the ability to control the immobilization of biomolecules at surfaces in arrayed 30nm domains, minimizing non-specific adsorption and allowing for the parallel monitoring of specific protein/DNA binding events at the single molecule level. This also allowed us to determine the average number of DNA molecules immobilized to single 30nm dots: we find fewer than four DNAs on ~85% of the nanodots. Notably, the overall strategy is highly general and can be utilized to immobilize any biotinylated bio-molecule for further studies. By specific design of the biomolecular nanoarray it is possible to record, via conventional epi-fluorescence microscopy imaging, hundreds of single-molecule events of biological interest, simultaneously on a single biochip: to our knowledge this is the first time biological activity is monitored on a nanoarray with such a high density and resolution (i.e. single-molecule investigations carried out in parallel). Furthermore, the fabrication strategy can be easily scaled via nanoimprint lithography, a lower cost and higher throughput patterning technique. In this context, we have also shown that we can fabricate and biofunctionalize arrays of ~10 nm domains, and that we can dynamically monitor distinct bio-recognition events on the same biochip. We envision that the high density and resolution achievable with our platform can find general application in high throughput heterogeneous assays of a wide variety of biomolecular interactions.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the Office of Naval Research under award N00014-09-1-1117, National Institutes of Health through award number PN2EY016586 under the NIH Roadmap for Medical Research and by] the National Science Foundation under award number NSF EF-05-07086. Additional support from the Nanoscale Science and Engineering Initiative of the National Science Foundation under NSF Award Number CHE-0641523 and from the New York State Office of Science, Technology, and Academic Research (NYSTAR) is also gratefully acknowledged. This work was also supported by the National Science Foundation under Award Number CHE-0936923
Footnotes
Supporting Information Available: Experimental procedures, nanofabrication, surface functionalization, fluorescence microscopy, AFM images, scanning electron microscopy images, single-molecule data for physisorbed DNA, control enzyme experiments, bleaching data and single-molecule fitted histogram, as well as the full list of authors for reference 17. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Stephanopoulos N, Solis EOP, Stephanopoulos G. Aiche Journal. 2005;51:1858–1869. [Google Scholar]
- 2.Langer R, Tirrell DA. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 3.Rosi NL, Mirkin CA. Chemical Reviews. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 4.Torres AJ, Wu M, Holowka D, Baird B. Annual Review of Biophysics. 2008;37:265–288. doi: 10.1146/annurev.biophys.36.040306.132651. [DOI] [PubMed] [Google Scholar]
- 5.Wong LS, Khan F, Micklefield J. Chemical Reviews. 2009;109:4025–4053. doi: 10.1021/cr8004668. [DOI] [PubMed] [Google Scholar]
- 6.Nanobiotechnology. Wiley-VCH: Weinheim; 2004. [Google Scholar]
- 7.Aydin D, Schwieder M, Louban I, Knoppe S, Ulmer J, Haas TL, Walczak H, Spatz JP. Small. 2009;5:1014–1018. doi: 10.1002/smll.200801219. [DOI] [PubMed] [Google Scholar]
- 8.Niwa D, Omichi K, Motohashi N, Homma T, Osaka T. Chemistry Letters. 2004;33:176–177. [Google Scholar]
- 9.Zhang GJ, Tanii T, Zako T, Hosaka T, Miyake T, Kanari Y, Funatsu TW, Ohdomari I. Small. 2005;1:833–837. doi: 10.1002/smll.200500091. [DOI] [PubMed] [Google Scholar]
- 10.Nicolau DV, Demers L, Ginger DS. In: Microarray Technology and Its Applications. Nicolau DV, Müller UR, editors. Springer; 2005. pp. 89–118. [Google Scholar]
- 11.Nanobiotechnology II. Wiley-VCH; Weinheim: 2007. [Google Scholar]
- 12.Bulyk ML, Gentalen E, Lockhart DJ, Church GM. Nature Biotechnology. 1999;17:573–577. doi: 10.1038/9878. [DOI] [PubMed] [Google Scholar]
- 13.Demers LM, Ginger DS, Park SJ, Li Z, Chung SW, Mirkin CA. Science. 2002;296:1836–1838. doi: 10.1126/science.1071480. [DOI] [PubMed] [Google Scholar]
- 14.Bruckbauer A, Ying LM, Rothery AM, Zhou DJ, Shevchuk AI, Abell C, Korchev YE, Klenerman D. Journal of the American Chemical Society. 2002;124:8810–8811. doi: 10.1021/ja026816c. [DOI] [PubMed] [Google Scholar]
- 15.Rodolfa KT, Bruckbauer A, Zhou DJ, Korchev YE, Klenerman D. Angewandte Chemie-International Edition. 2005;44:6854–6859. doi: 10.1002/anie.200502338. [DOI] [PubMed] [Google Scholar]
- 16.Huang SX, Schopf E, Chen Y. Nano Letters. 2007;7:3116–3121. doi: 10.1021/nl071643g. [DOI] [PubMed] [Google Scholar]
- 17.Akbulut O, Jung JM, Bennett RD, Hu Y, Jung HT, Cohen RE, Mayes AM, Stellacci F. Nano Letters. 2007;7:3493–3498. doi: 10.1021/nl0720758. [DOI] [PubMed] [Google Scholar]
- 18.Drmanac R, et al. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 19.Ishijima A, Yanagida T. Trends in Biochemical Sciences. 2001;26:438–444. doi: 10.1016/s0968-0004(01)01860-6. [DOI] [PubMed] [Google Scholar]
- 20.Ritort F. Journal of Physics-Condensed Matter. 2006;18:R531–R583. doi: 10.1088/0953-8984/18/32/R01. [DOI] [PubMed] [Google Scholar]
- 21.Smiley RD, Hammes GG. Chemical Reviews. 2006;106:3080–3094. doi: 10.1021/cr0502955. [DOI] [PubMed] [Google Scholar]
- 22.Houseman BT, Mrkisch M. Angew Chem Int Ed. Vol. 38. 1999. pp. 782–785. [DOI] [PubMed] [Google Scholar]
- 23.Castronovo M, Radovic S, Grunwald C, Casalis L, Morgante M, Scoles G. Nano Letters. 2008;8:4140–4145. doi: 10.1021/nl802370g. [DOI] [PubMed] [Google Scholar]
- 24.Bulyk ML. Analytics of Protein-DNA Interactions. 2007;104:65–85. doi: 10.1007/10_025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Field S, Udalova I, Ragoussis J. Analytics of Protein-DNA Interactions. 2007;104:87–110. doi: 10.1007/10_2006_035. [DOI] [PubMed] [Google Scholar]
- 26.Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chemical Reviews. 2005;105:1103–1169. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 27.Cherniavskaya O, Chen CJ, Heller E, Sun E, Provezano J, Kam L, Hone J, Sheetz MP, Wind SJ. Journal of Vacuum Science & Technology B. 2005;23:2972–2978. [Google Scholar]
- 28.Methods in Enzymology. Academic press; 1990. p. 184. [Google Scholar]
- 29.Ladd J, Boozer C, Yu QM, Chen SF, Homola J, Jiang S. Langmuir. 2004;20:8090–8095. doi: 10.1021/la049867r. [DOI] [PubMed] [Google Scholar]
- 30.Smith CL, Milea JS, Nguyen GH. Top Curr Chem. 2005;261:63–90. [Google Scholar]
- 31.Indeed, if the metal thiolation step in the functionalization process is performed after the PEG-silane step, we have observed that the biotin-thiols will physisorb on the PEG surrounding the nanodots and function as anchoring points for streptavidin and, consequently, for the DNAs, thus increasing the background fluorescence and preventing the ability to resolve single nanodots via epi-fluorescence microscopy imaging.
- 32.New England Biolabs. http://www.neb.com/nebecomm/products/productR3151.asp.
- 33.Athanasiadis A, Vlassi M, Kotsifaki D, Tucker PA, Wilson KS, Kokkinidis M. Nature Structural Biology. 1994;1:469–475. doi: 10.1038/nsb0794-469. [DOI] [PubMed] [Google Scholar]
- 34.Photobleaching occurs at a rate that is more than one order of magnitude slower than the rate of PvuII cleavage, as shown in figure SI-5: this allows us to comfortably dismiss photobleaching in our single-molecule enzyme data.
- 35.Lu HP, Xun LY, Xie XS. Science. 1998;282:1877–1882. doi: 10.1126/science.282.5395.1877. [DOI] [PubMed] [Google Scholar]
- 36.Xie SN. Single Molecules. 2001;2:229–236. [Google Scholar]
- 37.Xie FQ, Dupureur CM. Archives of Biochemistry and Biophysics. 2009;483:1–9. doi: 10.1016/j.abb.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie FQ, Qureshi SH, Papadakos GA, Dupureur CM. Biochemistry. 2008;47:12540–12550. doi: 10.1021/bi801027k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simoncsits A, Tjornhammar ML, Rasko T, Kiss A, Pongor S. Journal of Molecular Biology. 2001;309:89–97. doi: 10.1006/jmbi.2001.4651. [DOI] [PubMed] [Google Scholar]
- 40.This was done by counting the number of discrete step-like drops in the fluorescence intensity versus time trajectories arising from both PvuII activity (on PvuII-treated nanoarrays) and photobleaching (on untreated nanoarrays), .
- 41.The remaining ~15% of nanodots lack discrete, step-like drops in fluorescence intensity, instead exhibiting a relatively continuous decay that likely arises from a large density of immobilized DNAs, thus preventing single-molecule resolution. .
- 42.Jackson AM, Myerson JW, Stellacci F. Nature Materials. 2004;3:330–336. doi: 10.1038/nmat1116. [DOI] [PubMed] [Google Scholar]
- 43.Schvartzman M, Wind SJ. Nano Letters. 2009;9:3629–3634. doi: 10.1021/nl9018512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.