

Abstract
Background. Several studies in patients with primary aldosteronism (PA) have suggested that aldosterone (ALD) is directly contributing to albuminuria. However, there are limited data pertaining to the direct role of ALD in in vivo models in regard to the induction of renal injury and the involved mechanisms. In the present study, we established a high-dose ALD-infused rat model to evaluate urinary albumin excretion rate (UAER) and podocyte damage. Moreover, we studied the effect of eplerenone (EPL), telmisartan (TEL) and amlodipine (AML) on ALD-induced renal structural and functional changes.
Methods. Immunohistochemical and real-time PCR analyses, and TUNEL assays were performed to evaluate nephrin expression and podocyte injury.
Results. ALD-receiving rats (ARR) showed a progressive increase in BP, UAER and proteinuria when compared with control rats (CR). Conversely, BP was significantly reduced in ALD + EPL (A/ERR)-, ALD + AML (A/ARR)- and ALD + TEL (A/TRR)-treated rats. However, UAER and proteinuria were decreased only in A/ERR and A/TRR, but not in A/ARR. Only EPL administration provided protection against ALD-induced podocyte apoptosis. Renal tissue of ARR revealed enhanced expression of nephrin protein and mRNA. This effect of ALD was inhibited by EPL, but not by TEL or AML.
Conclusions. ALD induces direct glomerular injury independent of its haemodynamic effects; this effect of ALD is, at least in part, mediated through activation of the mineralocorticoid receptor.
Keywords: aldosterone, amlodipine, eplerenone, podocyte, telmisartan
Introduction
Aldosterone, traditionally viewed as a key regulator of extracellular fluid volume and electrolyte homeostasis, contributes to the progression of cardiovascular and renal injury via activation of the mineralocorticoid receptor (MR) [1–5]. However, the available data elucidating the pathogenic role of aldosterone are indirect, and mainly based on the interruption with non-specific MR blockers in clinical studies and aldosterone-non-specific animal models [6–10]. Patients with human primary aldosteronism (PA), characterized by elevated plasma aldosterone concentration, show higher albuminuria when compared with patients with essential hypertension with similar blood pressure [11,12]. Also, uninephrectomized rats treated with exogenous aldosterone infusion and a high salt diet exhibited glomerular and tubulointerstitial lesions and proteinuria [7]. Since both renal ablation [13,14] and high salt intake [15,16] have the potential to induce proteinuria, it was difficult to determine the contribution of aldosterone in the induction of proteinuria in this model. At the same time, there are limited data on the occurrence of renal lesions in PA. Therefore, we used an aldosterone-specific animal model to mimic the state of PA and to investigate the involved molecular mechanisms of aldosterone-induced proteinuria.
It is noteworthy that podocytes play a pivotal role in constituting the major size-selective permeability barrier and provide a defence against urinary protein loss [17]. Accumulating evidence has implicated that podocyte injury plays a critical role in the initiation and progression of proteinuria and glomerulosclerosis [18,19]. Furthermore, adjacent foot processes of podocytes are connected with the slit diaphragm, a unique apparatus consisting of multiple interacting proteins. Interestingly, nephrin was the first identified protein on the slit diaphragm and found to be crucial for the integrity of the slit diaphragm and podocyte function [20,21]. Moreover, alterations in the slit diaphragm components have been demonstrated in a variety of proteinuric diseases. Recently, Shibata et al. demonstrated that podocyte injury and reduced expression of nephrin are associated with oxidative stress and Sgk1 upregulation in aldosterone/salt-receiving proteinuric animals [9].
We previously demonstrated that angiotensin II (Ang II)-receiving rats developed proteinuria and also showed podocyte apoptosis and alteration in glomerular nephrin expression [22]. Since Ang II is known to stimulate aldosterone production by the adrenal gland, we hypothesized that some of the morphologic effects exerted by Ang II are caused by Ang II-induced aldosterone generation. In the present study, we evaluated the occurrence of proteinuria and podocyte injury in high-dose aldosterone-infused native rats. To evaluate the effect of aldosterone independent of its hypertensive effect, comparable levels of blood pressure were maintained by using three types of antihypertensive agents, a MR-specific blocker (eplerenone, EPL), an angiotensin II receptor blocker (telmisartan, TEL) and a calcium channel blocker (amlodipine, AML).
Materials and methods
Animals
All experimental procedures were approved by the ethics committee for animal experiments of Wuhan University. Thirty male Sprague–Dawley rats (Animal Center, Wuhan University School of Medicine, China) weighing 140–170 g were used. Rats were bred under pathogen-free conditions in laminar flow rooms and had free access to a standard rodent diet(0.7% NaCl) and tap water. After a 1-week adaptation, osmotic mini-pumps (Alzet 2004, Alza, Palo Alto, CA, USA) filled with either vehicle (2.5% DMSO, Sigma, St Louis, MO, USA) or aldosterone (Sigma, St Louis, MO, USA) were inserted subcutaneously, as described previously [22]. The dose of aldosterone administered was 1.5 μg/h which was determined in a pilot experiment. After recovery from surgery, animals were randomized to receive one of the following 28-day treatments: vehicle (control rats, CR); aldosterone (ARR); aldosterone + eplerenone [23] (100 mg/kg/day, Pfizer, USA) (A/ERR); aldosterone + telmisartan [24] (3 mg/kg/day, Boehringer Ingelheim, Germany) (A/TRR); and aldosterone + amlodipine [25,26] (10 mg/kg/day, Pfizer) (A/ARR) (n = 6 rats/group). The daily dose of reagents was dissolved in 60% acetonitrile (Sigma, St Louis, MO, USA) and diluted in distilled water before gavage.
Blood pressure measurement and sample collection
Twenty-four-hour urine was collected in metabolic cages, and systolic blood pressure was recorded by the tail-cuff method at Day 0, 7, 14, 21 and 28 after implantation of the osmotic mini-pumps. At the end of study, animals were anaesthetized with pentobarbital (65 mg/kg, i.p., Sigma). Blood samples were collected from the left ventricle into heparin and non-heparin tubes, centrifuged at 2000 rpm for 10 min at room temperature and stored at − 80°C. The right kidney from each rat was removed and sectioned through a long axis into two slices. The tip of one slice in a diameter of 1 mm was fixed in 2.5% glutaraldehyde and processed for transmission electron microscopy. The remaining piece was fixed in 4% phosphate-buffered paraformaldehyde and embedded in paraffin. The cortex of the left kidney was separated and immediately snap-frozen in liquid nitrogen. Frozen samples were stored at − 80°C for RNA isolation.
Blood and urine parameters
Serum concentrations of creatinine, sodium and potassium were assayed using an automatic biochemical analyser (7170 A; Hitachi, Tokyo, Japan). Urinary albumin was determined with a competitive ELISA kit (Shibayagi, Shibukawa, Japan), and urinary albumin excretion rate (UAER) was calculated. Plasma aldosterone and angiotensin II concentrations were evaluated with radioimmunoassay kits (North Institute of Biological Technology products, Beijing, China) according to standard procedures.
Morphological analysis
Histopathological changes were evaluated using light microscopy and transmission electron microscopy. Kidney sections (2 μm thick) stained with periodic acid–Schiff (PAS) were used to evaluate glomerular lesions such as glomerular cell proliferation, expansion of the mesangial matrix and segmental glomerulosclerosis by two pathologists who were unaware of the experimental design. A semi-quantitative grading system was utilized according to a method described previously [27]. In brief, the severity of injury for each glomerulus was scored from 0 to 4: 0, no lesion; 1, < 25% involvement of the glomerulus; 2, 25–50% involvement; 3, 50–75% involvement; and 4, > 75% involvement. Fifty glomeruli were analysed per kidney section. A glomerular sclerosis score (GSS) per animal was calculated by multiplying each severity score (0–4 +) with the percentage of glomeruli displaying the same degree of injury and by summing these scores. Sections stained with uranyl acetate and lead citrate were photographed with a Hitachi H600 transmission electron microscopy (Hitachi, Tokyo, Japan). Podocyte lesions including effacement of foot processes, microvillus formation, apoptosis or depletion from glomerular basement membrane were analysed.
Podocyte apoptosis
Morphological evaluation of podocyte apoptosis in situ was carried out by the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end-labelling (TUNEL) assay. As described previously [22], de-waxed sections (4 μm) were blocked with 3% H2O2 followed by proteinase K digestion for 15 min at 37°C. Sections were washed and incubated with TUNEL reaction mixture (Roche Applied Science, Switzerland) for 1 h at room temperature. They were then exposed to streptavidin-peroxidase-conjugated anti-biotin-dUTP antibody; binding sites were visualized using diaminobenzidine (DAB, Dako). The slides were counterstained with haematoxylin stain. The omission of TdT was used as negative controls. Positive controls were processed by pre-treatment of sections with 0.1 U/μL deoxynuclease-1 before TdT staining. The ratio of apoptotic podocytes to the haematoxylin-stained cells on cross sections through the glomerulus was determined.
Immunohistochemistry
Immunostaining for nephrin expression was performed on paraffin-embedded sections. Slides were deparaffinized and treated with 3% H2O2 for 30 min at room temperature. Antigen retrieval for nephrin was performed in microwaved citrate buffer (0.01 mol/L, pH 6.0) for 15 min. Endogenous peroxidase was blocked with 5% bovine serum albumin in 0.01 mol/L phosphate-buffered saline (PBS, pH 7.4) for 30 min, and sections were incubated with goat anti-nephrin antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) overnight at 4°C. Sections were washed in PBS, followed by biotinylated anti-goat secondary antibody and avidin–biotin peroxidase complex (Dako) for 30 min. After rinsing, the peroxidase activity was visualized by DAB (Dako), and sections were counter-stained with haematoxylin. Negative controls were performed by omitting the primary antibody and replacing it with PBS. Thirty randomly chosen glomeruli were analysed using Image-pro plus 5.10 software (Media Cybernetics) at a magnification × 400, and integrated optical density (IOD) was used as relative amount of positive staining.
Evaluation of nephrin mRNA expression
Total RNA from tissue specimens was extracted using TRIzol-Reagent (Invitrogen, Carlsbad, CA, USA). RNA was quantified and assessed for purity by UV spectrophotometry assay. All the reverse transcriptase reactions were performed using reverse transcriptase with oligo-dT priming. RT–PCR assay was performed using 2 μg of isolated RNA. First-strand cDNA synthesis was accomplished with an Oligo-(dT)18 primer (0.5 μg/μL) and RevertAidTM First Strand cDNA Synthesis Kit (MBI Fermentas, USA). Changes in nephrin mRNA levels were determined by real-time PCR with an iCycler IQ System (Bio-Rad Laboratories, Hercules, CA, USA). Real-time PCR analyses were performed in triplicate with 5 μM of both sense and anti-sense primers in a final volume of 25 μL using SYBR Green real-time PCR master mix plus (TOYOBO, Shanghai, China). The sequences of the specific primers (Invitrogen, Shanghai, China) were as follows: nephrin (AF161715.1) sense 5'-TGTCTGACGCTTTTGGCTTTC-3' and anti-sense 5'-AACAATCCCCGCCTATCTTCT-3'; and β-actin (EF156276.1) sense 5'-CGTTGACATCCGTAAAGACCTC-3' and anti-sense 5'-TAGGAGCCAGGGCAGTAATCT-3'. PCR conditions were 94°C for 2 min followed by 40 cycles of 15 s at 95°C, 15 s at 58°C and 45 s at 72°C. Systematic controls excluded amplification from contaminating genomic DNA. The formula used to quantify the relative changes in target genes over β-actin mRNAs is derived from the 2-ΔΔCt formula.
Statistical analysis
Data were presented as mean ± SEM or mean ± SD. Statistical analyses were performed using SPSS 11.5. Statistical comparisons were conducted using a one-way ANOVA followed by the least significant differences (LSD) test to compare differences among groups. Histological data were compared using non-parametric analysis with Kruskal–Wallis test, followed by Mann–Whitney U-test. Statistical significance was defined as a P-value < 0.05.
Results
Changes of systolic blood pressure and proteinuria
As shown in Figure 1, systolic blood pressure, albuminuria and proteinuria did not change in CR over 28 days. In contrast, ARR displayed higher blood pressure (172 ± 8 mmHg), albuminuria (10.31 ± 0.41 μg/min) and proteinuria (64.2 ± 3.64 mg/24 h) at Day 28. A/ERR, A/TRR and A/ARR showed amelioration of blood pressure during 28 days. However, TEL and AML treatment resulted in more reduction in BP when compared with EPL at Day 14. It is noteworthy that A/TRR exhibited a modestly higher BP at Day 28 when compared with Day 21, which was just opposite of A/ARR (Figure 1A). On the other hand, both EPL and TEL decreased albuminuria and proteinuria markedly when compared with AML (Figure 1B and C).
Fig. 1.
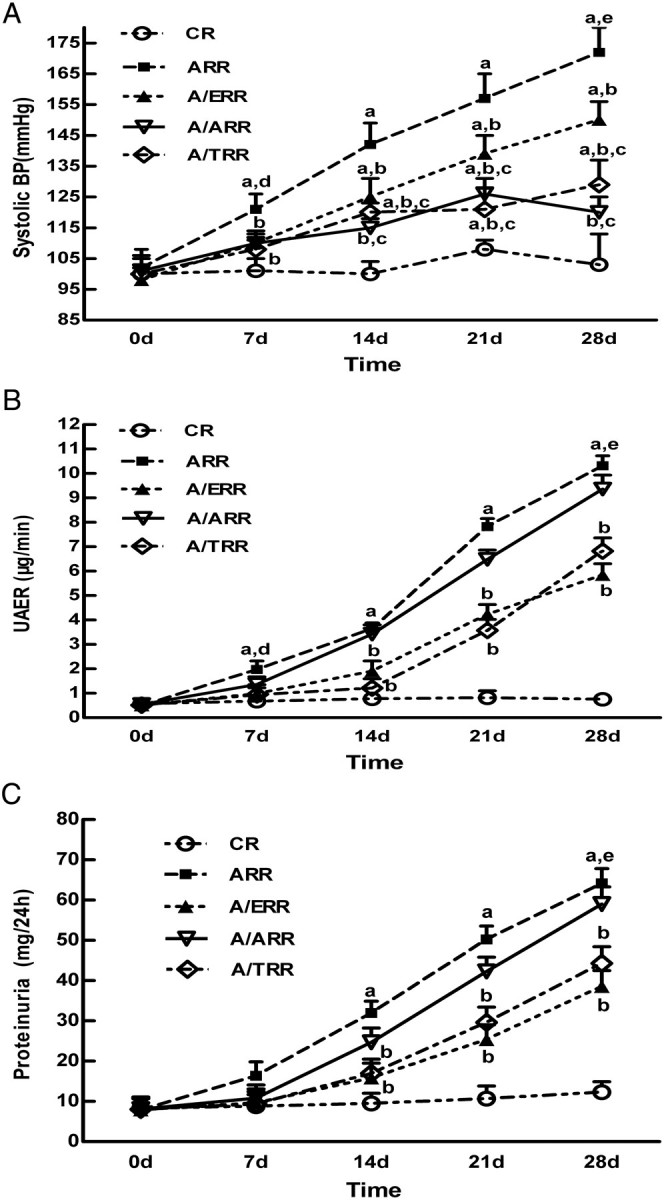
The temporal changes of systolic blood pressure, albuminuria and proteinuria in the different groups at 0, 7, 14, 21 and 28 days. A. Systolic blood pressure measured by tail-cuff plethysmography. B, C. Urinary albumin and proteinuria in the different groups, respectively. All graphs represent mean and SEM. CR, vehicle-treated; ARR, aldosterone-infused; A/ERR, A/ARR and A/TRR eplerenone-, amlodipine- and telmisartan-treated; UAER, urinary albumin excretion rate. a, P < 0.01 vs. CR; b, P < 0.01 vs. ARR; c, P < 0.05 vs. A/ERR at the same time point; d, P < 0.05 vs. 0 day; e, P < 0.05 vs. 14 days.
Biochemical parameters
ARR presented with hypernatraemia and hypokalaemia, which were corrected only by EPL administration. EPL treatment increased mean serum potassium levels (P < 0.01) and decreased mean serum sodium levels (P < 0.01) in ARR. Since there was a comparable level of plasma aldosterone among all the aldosterone-receiving animals, these biochemical differences have to be attributed to EPL. In addition, a higher concentration of plasma Ang II was detected in A/TRR when compared with A/ERR and A/ARR. The concentrations of serum creatinine and blood urea nitrogen and the creatinine clearance were comparable among all groups (Table 1).
Table 1.
Comparison of biochemical parameters in different groups (n = 6)
| Scr (μmol/L) | BUN (mg/dL) | Ccr (mL/min/100 g BW) | K (mmol/L) | Na (mmol/L) | ALD (pmol/L) | Ang II (pmol/L) | |
|---|---|---|---|---|---|---|---|
| CR | 47 ± 6 | 34.2 ± 2.8 | 0.57 ± 0.17 | 4.51 ± 0.32 | 136 ± 5 | 36.4 ± 9.7 | 399.7 ± 100.9 |
| ARR | 69 ± 8 | 40.3 ± 1.2 | 0.45 ± 0.12 | 3.87 ± 0.33* | 146 ± 8 * | 234.6 ± 35.6* | 56.5 ± 10.8* |
| A/ERR | 56 ± 7 | 35.5 ± 3.1 | 0.56 ± 0.13 | 4.61 ± 0.51** | 135 ± 5** | 227.4 ± 27.0* | 51.4 ± 6.2* |
| A/ARR | 62 ± 11 | 38.8 ± 2.1 | 0.46 ± 0.16 | 3.91 ± 0.41* | 141 ± 6* | 233.2 ± 28.8* | 66.4 ± 13.0* |
| A/TRR | 61 ± 8 | 36.5 ± 1.9 | 0.50 ± 0.11 | 3.85 ± 0.38* | 143 ± 8* | 241.8 ± 21.6* | 516.2 ± 77.8*,** |
Values represent group mean ± SEM.
Scr, serum plasma creatinine; BUN, blood urea nitrogen; Ccr, creatinine clearance; K, serum potassium; Na, serum natrium; ALD, plasma aldosterone; Ang II, plasma angiotensin; CR, vehicle-treated; ARR, aldosterone-infused; A/ERR, A/ARR and A/TRR: eplerenone-, amlodipine- and telmisartan-treated.
P < 0.01 vs. CR.
P < 0.01 vs. ARR.
Histopathological characteristics
Control rats did not show any glomerular abnormalities (Figure 2A). On the other hand, ARR rats showed expansion of the glomerular tuft, mild cellular proliferation and accumulation of extracellular matrix (Figure 2B). Mean GSS in ARR was significantly higher when compared with control rats (P < 0.01); however, mean GSS significantly decreases in A/ERR, A/TRR and A/ARR. Notably, A/ERR showed an almost one-half decrease of mean GSS when compared with ARR (P < 0.01) (Figure 2F). Thereafter, the glomerular damage was investigated under a transmission electron microscopy. In the ARR, interdigitating foot processes were deranged and fused segmentally, and podocytes were occasionally enriched in nuclear euchromatin along with irregular nuclear membrane (Figure 3B and C). These findings are consistent with an early ultrastructural characteristic of apoptotic cells [28].
Fig. 2.
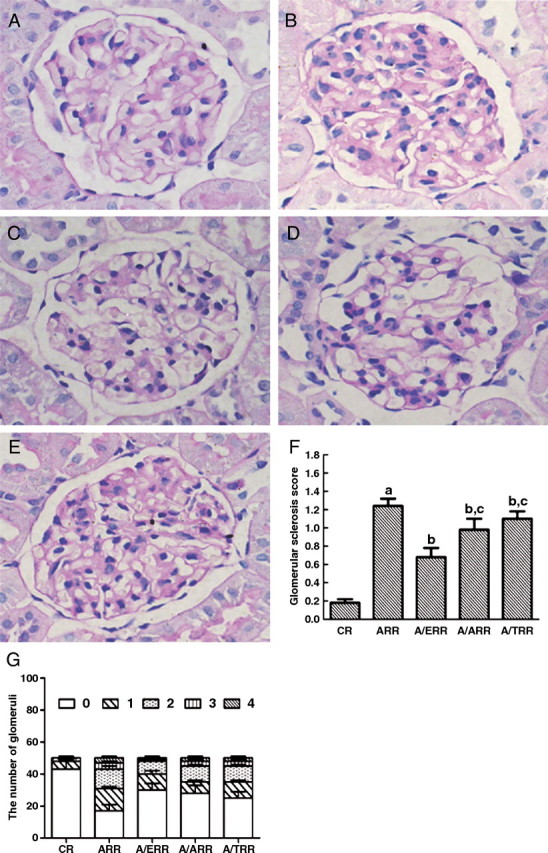
Histological findings of glomeruli from different group rats. A–E. Representative photomicrographs of glomerular sections from vehicle-treated controls (CR), aldosterone-infused rats (ARR), and eplerenone (A/ERR)-, amlodipine (A/ARR)- and telmisartan (A/TRR)-treated animals, respectively. F. Result of the semi-quantitative analysis. G. Distribution of glomeruli with different glomerular sclerosis score. Means and SD are given with arbitrary units. a, P < 0.01 vs. CR; b, P < 0.05 vs. ARR; c, P < 0.05 vs. A/ERR (PAS stain, original magnification × 400).
Fig. 3.
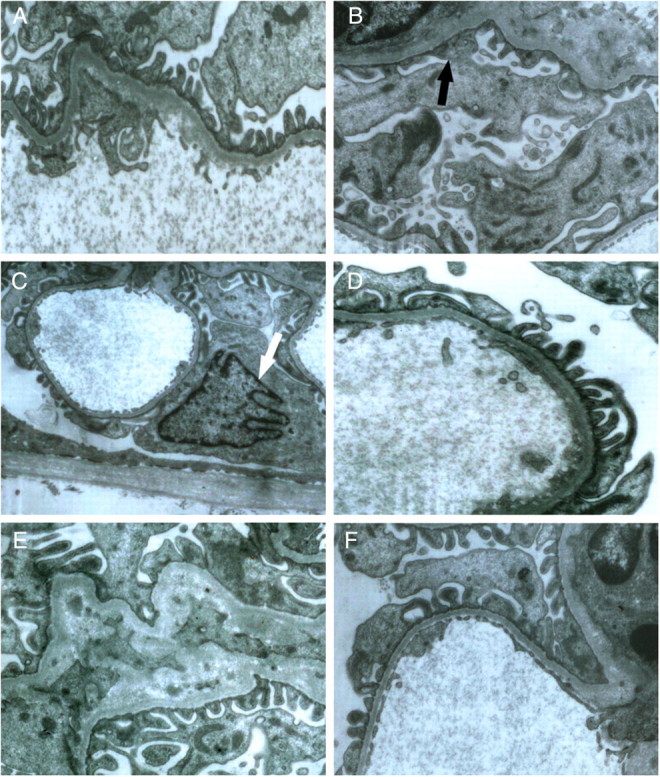
Representative micrographs of ultrastructural changes in glomeruli under transmission electron microscopy. The foot process in control rats appeared normal (A), while aldosterone infusion caused the lost of arrangement of foot processes and segmental fusion of foot process (black arrow, B), and enriched euchromatin along with shrinkaged nuclear membrane (white arrow) of podocyte was detected in aldosterone-infused rats (C). D–F display the changes of foot process after using eplerenone, amlodipine and telmisartan, respectively (A, B, D, E, and F × 10 000; C × 5000).
Apoptosis studies
TUNEL assay was performed to evaluate apoptotic cells in the glomeruli. In line with the ultrastructural morphology, TUNEL-positive podocytes and other resident glomerular cells were detected in glomerular cross sections in ARR (Figure 4B). Quantitation analysis revealed that occurrence of podocyte apoptosis (podocyte apoptosis ratio, ARP) in ARR was significantly attenuated by EPL (P < 0.01). However, TEL and AML showed a relatively limited ability to provide protection against ALD-induced podocyte apoptosis (Figure 4F).
Fig. 4.
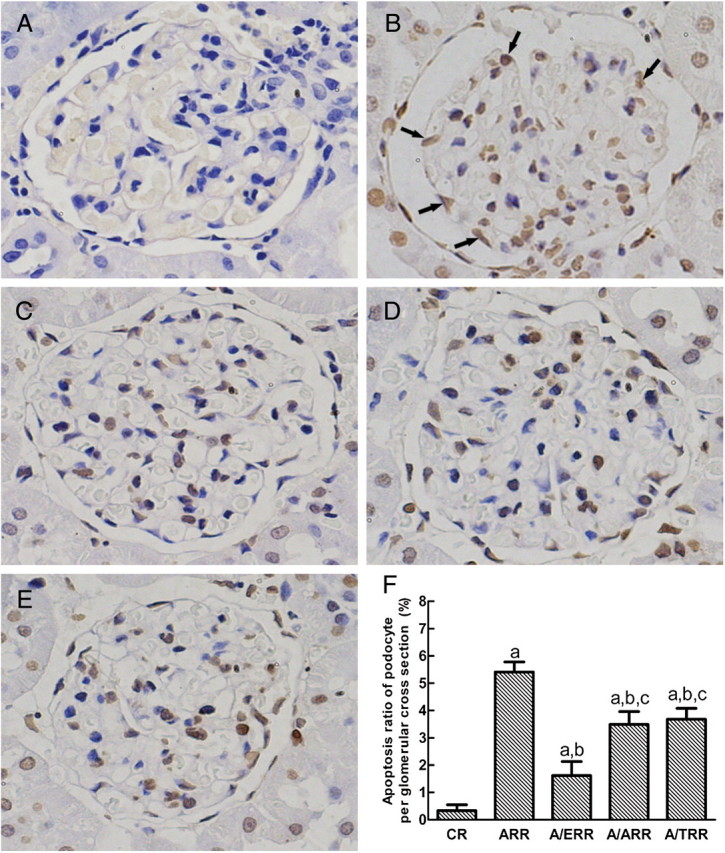
TUNEL assay of glomerular cell apoptosis. A–E display the representative micrographs of TUNEL-stained glomeruli from vehicle-treated controls (CR), aldosterone-infused rats (ARR), and eplerenone (A/ERR)-, amlodipine (A/ARR)- and telmisartan (A/TRR)-treated animals, respectively. Black arrows indicate apoptotic podocytes in aldosterone-infused rats. F. Comparison of apoptosis ratio of podocyte on a glomerular cross section. Values represent means ± SD. a, P < 0.01 vs. CR; b, P < 0.01 vs. ARR; c, P < 0.01 vs. A/ERR (original magnification × 400).
Nephrin mRNA and protein expression
As nephrin is a podocyte-specific protein and essential for the integrity of the slit diaphragm and podocyte function, we next analysed glomerular expression of nephrin mRNA and protein. Interestingly, the protein level of nephrin assessed by immunohistochemical studies was upregulated in ARR. Conversely, administration of EPL corrected the increase of nephrin expression. However, the similar modification of nephrin expression was not detected in A/TRR or A/ARR (Figure 5). Additionally, real-time PCR was performed to confirm the results of protein expression, and a similar expression pattern was detected (Figure 6).
Fig. 5.
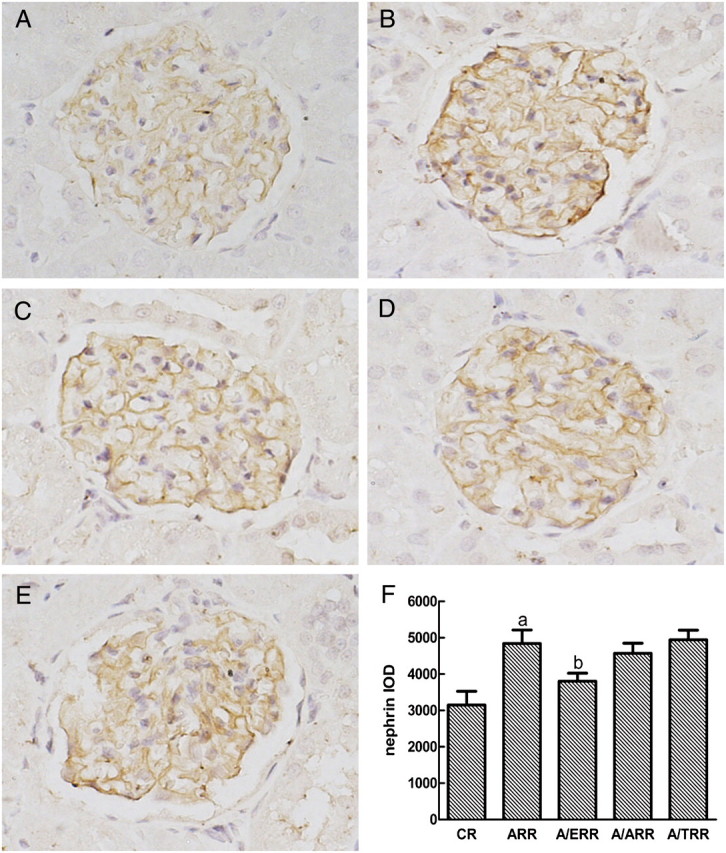
Immunostaining for nephrin expression in glomeruli from different groups. A–E indicate the representative photomicrographs of nephrin-stained glomeruli from vehicle-treated controls (CR), aldosterone-infused rats (ARR), and eplerenone (A/ERR)-, amlodipine (A/ARR)- and telmisartan (A/TRR)-treated animals, respectively. F. Analysis of the intensity of nephrin staining with mean integrated optical density (IOD) expressed as means ± SD (arbitrary units). a, P < 0.01 vs. CR; b, P < 0.01 vs. ARR (original magnification × 400).
Fig. 6.

The mRNA expression of nephrin analysed by real-time PCR. Amplification of β-actin served as a control, and the levels were compared with CR kidneys taken as 100% (n = 6). a, P < 0.01 vs. CR; b, P < 0.01 vs. ARR.
Discussion
In the present study, we have demonstrated that aldosterone-treated rats resemble features of human PA, including hypertension, elevated plasma aldosterone levels and decreased plasma Ang II levels. ARR also developed progressive albuminuria and glomerular injury in general and podocyte injury in particular. However, these biochemical and renal morphological alterations were attenuated by EPL, a mineralocorticoid receptor antagonist.
These results confirmed the hypothesis that the kidney is a target of aldosterone-induced damage, which is consistent with previous studies in PA patients and experimental animals [11,12]. However, the animal studies had confounding factors such as in the case of aldosterone/salt-hypertensive animals with uninephrectomy, which did not allow assessment of the direct effect of aldosterone in the pathogenesis of kidney injury [7,9]. In preliminary studies, we found that animals infused with the low dose (0.75 μg/h) of aldosterone, without uninephrectomy or high salt diet, did not develop hypertension, proteinuria and renal hypertrophy at Week 4. It is noteworthy that renal ablation and high salt intake are both independent risk factors for the development of hyperfiltration, a phenomenon known to induce proteinuria as well as progression of renal lesions [13,15,29,30]. Nevertheless, a large body of clinical data implicates that high dietary salt intake is a synergistic factor in the process of aldosterone-induced proteinuria [31]. Therefore, in order to mimic the aldosterone excess state of PA patients, a long-term exposure to a higher aldosterone level (1.5 μg/h infusion) was utilized in the present study. This experimental design has allowed us to explore the specific role of aldosterone in the development of renal injury.
Renal hyperfiltration induced by high blood pressure and volume expansion has long been considered as a major determinant of urinary albumin loss in PA. In patients with PA, renal hyperfiltration was ameliorated after suppression of aldosterone excess with adrenalectomy and MR blockade [32]. However, the present results demonstrated that high dose of aldosterone induced systemic high blood pressure and relatively low creatinine clearance which indicated that the globally renal hyperfiltration was not present. In the present study, ARR developed glomerular lesions characterized by cell proliferation, accumulation of extracellular matrix and even glomerular sclerosis under light microscopy. If a considerable number of glomeruli were sclerotic, whether the other glomeruli might be exposed to glomerular hypertension was not ascertained by direct methods in the present study. Whether the renal dysfunction with albuminuria is a marker of a dynamic or structural renal defect remains controversial [33,34]. Accumulating animal studies have suggested that aldosterone contribute to renal inflammation and nephrosclerosis, and the pathologic alterations were ameliorated by aldosterone antagonists. Furthermore, Zhou et al. introduced renal micropuncture to evaluate the effect of aldosterone antagonist eplerenone on glomerular dynamics, and the study demonstrated that eplerenone attenuated proteinuria and nephrosclerosis independent of systemic and glomerular dynamics [35]. These studies suggest that non-haemodynamic and direct intrarenal actions of aldosterone contribute to the development of progressive proteinuria in animals, which is at variance with the long-held tenet that hypertension in patients with PA is relatively benign. Although there was an isolated case report of PA showing some degree of renal damage characterized by arteriolosclerosis, clear evidence of renal injury in the literature is limited [36]. Indeed, the discrepancy requires further investigation.
Mineralocorticoid receptor is mainly localized in the renal tubular distal epithelial cells. Under physiological conditions, aldosterone primarily binds to the MR to increase sodium reabsorption and enhance potassium excretion, and thus promote volume expansion and increase of blood pressure. Moreover, experimental evidence suggests that excessive MR-inducing signalling also mediates profibrotic effects on renal and cardiovascular cells [5,37]. In the rat kidney, MR has been localized to pre-glomerular vasculature, mesangial cells, fibroblasts and distal tubular cells [1]. Aldosterone stimulates the synthesis of extracellular matrix proteins in cultured mesangial and proximal tubular cells. However, these matrix synthetic effects were inhibited by the MR blockers [5]. Similarly, the present study showed that accumulation of mesangial matrix was a typical pathological feature in aldosterone-infused animals, which was reversed by eplerenone (MR blocker). In addition, aldosterone-mediated production of ROS and upregulation of inflammatory molecules have been demonstrated to contribute to the progression of renal fibrosis [5,7,9,38]. Aldosterone-induced generation of ROS has also been incriminated for the induction of renal cells apoptosis, both mesangial and proximal tubular cells [1,39,40]. Recently, Shibata and his colleagues have demonstrated that podocytes consistently express MR, and their interaction with aldosterone led to the production of ROS in cultured podocytes as well as in aldosterone/salt-administrated rats. In the present and our recently published study, we have confirmed that aldosterone is capable of inducing podocyte apoptosis in vivo and in vitro [41]. We presume that this pro-apoptotic effect of aldosterone might be mediated through podocyte ROS generation.
Of note, protein and mRNA expression of nephrin were upregulated in our rat model, which was contrary to that observed in aldosterone/salt-infused rats [9] and in the early nephropathy of SHR/NDmcr-cp, a rat model of metabolic syndrome [42]. The aldosterone signalling has been implicated in podocyte injury in above two animal models. Accumulating data demonstrate that nephrin regulates a number of podocyte intracellular signalling [20], including activation of the phosphoinositide 3-OH kinase–PKB pathway to prevent podocyte apoptosis [43]. Moreover, proper localization of nephrin within specialized lipid rafts at the plasma membrane is known to be essential for correct slit diaphragm signalling [21,44]. Our previous data demonstrated that Ang II exerted a bimodal effect on nephrin expression in Ang II-infused rats, and enhanced protein and mRNA level of nephrin with altered expression pattern were detected in the early phase [22]. Compared with the presented biological and pathological characteristics in the studies of Shibata and Nagase, the degree of renal damage in our rats at Week 4 was more modest. Upregulated expression of nephrin might be a compensatory response to prevent aldosterone-induced apoptosis. However, mislocalized nephrin was not capable of exerting the anti-apoptosis effect. Thus, the different stages of the development of aldosterone-induced kidney damage may contribute to the discrepancy of nephrin expression between the present study and the works of Shibata and Nagase.
We also evaluated the effects of a MR antagonist (eplerenone), an angiotensin II receptor blocker (telmisartan), and a calcium channel blocker (amlodipine) on the renal injury in our model and found that eplerenone was less capable of lowering BP than telmisartan and amlodipine. However, UAER and proteinuria were decreased only by eplerenone and telmisartan, and not by amlodipine. Indeed, eplerenone not only prevented podocyte apoptosis but also inhibited aldosterone-induced nephrin upregulation, whereas no similar effects were detected in telmisartan- and amlodipine-treated rats. Thus, the protective effects of eplerenone may be mainly attributable to blockade of MR signalling to prevent the specific cytotoxic effects of aldosterone on renal injury, not to the decrease of systemic blood pressure. In the case of A/TRR, although intraglomerular haemodynamics was not evaluated in the present study, it was reported that in vitro addition of aldosterone may exert a direct and rapid vasoconstrictive effect on the efferent renal arteriole [45]. It is possible that modulation of renal hyperfiltration may contribute to the reduction of UAER and no improvement of pathologic injury in telmisartan-treated rats, but this hypothesis requires further investigation.
In summary, the present investigation demonstrates that aldosterone-infused rats developed proteinuria and renal injury characterized by podocyte apoptosis. Eplerenone had only a modest effect on blood pressure; nevertheless, it attenuated proteinuria, podocyte apoptosis and nephrin expression. It appears that eplerenone may provide protection against aldosterone-induced renal injury through its anti-apoptotic effect and the modulation of podocyte nephrin expression.
Acknowledgments
This work was supported by the National Nature Science Foundation Grant of China (30370656, 30871167 to G.D. and 30900688 to C.C.). A portion of this work was presented at the 41st Annual Renal Week Meeting of the American Society of Nephrology, November 2008, Philadelphia, PA, USA.
Conflict of interest statement. None declared.
References
- 1.Del VL, Procaccio M, Vigano S, et al. Mechanisms of disease: the role of aldosterone in kidney damage and clinical benefits of its blockade. Nat Clin Pract Nephrol. 2007;3:42–49. doi: 10.1038/ncpneph0362. [DOI] [PubMed] [Google Scholar]
- 2.Funder JW. Mineralocorticoid receptor activation and oxidative stress. Hypertension. 2007;50:840–841. doi: 10.1161/HYPERTENSIONAHA.107.098012. [DOI] [PubMed] [Google Scholar]
- 3.Hollenberg NK. Aldosterone in the development and progression of renal injury. Kidney Int. 2004;66:1–9. doi: 10.1111/j.1523-1755.2004.00701.x. [DOI] [PubMed] [Google Scholar]
- 4.Ponda MP, Hostetter TH. Aldosterone antagonism in chronic kidney disease. Clin J Am Soc Nephrol. 2006;1:668–677. doi: 10.2215/CJN.00120106. [DOI] [PubMed] [Google Scholar]
- 5.Remuzzi G, Cattaneo D, Perico N. The aggravating mechanisms of aldosterone on kidney fibrosis. J Am Soc Nephrol. 2008;19:1459–1462. doi: 10.1681/ASN.2007101079. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006;70:2116–2123. doi: 10.1038/sj.ki.5001854. [DOI] [PubMed] [Google Scholar]
- 7.Blasi ER, Rocha R, Rudolph AE, et al. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003;63:1791–1800. doi: 10.1046/j.1523-1755.2003.00929.x. [DOI] [PubMed] [Google Scholar]
- 8.Chrysostomou A, Pedagogos E, Macgregor L, et al. Double-blind, placebo-controlled study on the effect of the aldosterone receptor antagonist spironolactone in patients who have persistent proteinuria and are on long-term angiotensin-converting enzyme inhibitor therapy, with or without an angiotensin II receptor blocker. Clin J Am Soc Nephrol. 2006;1:256–262. doi: 10.2215/CJN.01040905. [DOI] [PubMed] [Google Scholar]
- 9.Shibata S, Nagase M, Yoshida S, et al. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension. 2007;49:355–364. doi: 10.1161/01.HYP.0000255636.11931.a2. [DOI] [PubMed] [Google Scholar]
- 10.Tylicki L, Rutkowski P, Renke M, et al. Triple pharmacological blockade of the renin–angiotensin–aldosterone system in nondiabetic CKD: an open-label crossover randomized controlled trial. Am J Kidney Dis. 2008;52:486–493. doi: 10.1053/j.ajkd.2008.02.297. [DOI] [PubMed] [Google Scholar]
- 11.Rossi GP, Bernini G, Desideri G, et al. Renal damage in primary aldosteronism: results of the PAPY Study. Hypertension. 2006;48:232–238. doi: 10.1161/01.HYP.0000230444.01215.6a. [DOI] [PubMed] [Google Scholar]
- 12.Sechi LA, Novello M, Lapenna R, et al. Long-term renal outcomes in patients with primary aldosteronism. JAMA. 2006;295:2638–2645. doi: 10.1001/jama.295.22.2638. [DOI] [PubMed] [Google Scholar]
- 13.Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest. 1996;98:1063–1068. doi: 10.1172/JCI118867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piecha G, Koleganova N, Gross ML, et al. Regression of glomerulosclerosis in subtotally nephrectomized rats: effects of monotherapy with losartan, spironolactone, and their combination. Am J Physiol Renal Physiol. 2008;295:F137–F144. doi: 10.1152/ajprenal.00065.2008. [DOI] [PubMed] [Google Scholar]
- 15.Fox CS, Larson MG, Hwang SJ, et al. Cross-sectional relations of serum aldosterone and urine sodium excretion to urinary albumin excretion in a community-based sample. Kidney Int. 2006;69:2064–2069. doi: 10.1038/sj.ki.5000378. [DOI] [PubMed] [Google Scholar]
- 16.Nagase M, Shibata S, Yoshida S, et al. Podocyte injury underlies the glomerulopathy of Dahl salt-hypertensive rats and is reversed by aldosterone blocker. Hypertension. 2006;47:1084–1093. doi: 10.1161/01.HYP.0000222003.28517.99. [DOI] [PubMed] [Google Scholar]
- 17.Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- 18.Asanuma K, Mundel P. The role of podocytes in glomerular pathobiology. Clin Exp Nephrol. 2003;7:255–259. doi: 10.1007/s10157-003-0259-6. [DOI] [PubMed] [Google Scholar]
- 19.Durvasula RV, Shankland SJ. Podocyte injury and targeting therapy: an update. Curr Opin Nephrol Hypertens. 2006;15:1–7. doi: 10.1097/01.mnh.0000199012.79670.0b. [DOI] [PubMed] [Google Scholar]
- 20.Benzing T. Signaling at the slit diaphragm. J Am Soc Nephrol. 2004;15:1382–1391. doi: 10.1097/01.asn.0000130167.30769.55. [DOI] [PubMed] [Google Scholar]
- 21.Patrakka J, Tryggvason K. Nephrin—a unique structural and signaling protein of the kidney filter. Trends Mol Med. 2007;13:396–403. doi: 10.1016/j.molmed.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Jia J, Ding G, Zhu J, et al. Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis. Am J Nephrol. 2008;28:500–507. doi: 10.1159/000113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endemann DH, Touyz RM, Iglarz M, et al. Eplerenone prevents salt-induced vascular remodeling and cardiac fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension. 2004;43:1252–1257. doi: 10.1161/01.HYP.0000128031.31572.a3. [DOI] [PubMed] [Google Scholar]
- 24.Younis F, Kariv N, Nachman R, et al. Telmisartan in the treatment of Cohen-Rosenthal Diabetic Hypertensive rats: the benefit of PPAR-gamma agonism. Clin Exp Hypertens. 2007;29:419–426. doi: 10.1080/10641960701578444. [DOI] [PubMed] [Google Scholar]
- 25.Oshima T, Ono N, Ozono R, et al. Effect of amlodipine and cilazapril treatment on platelet Ca2 + handling in spontaneously hypertensive rats. Hypertens Res. 2003;26:901–906. doi: 10.1291/hypres.26.901. [DOI] [PubMed] [Google Scholar]
- 26.Zhou MS, Jaimes EA, Raij L. Benazepril combined with either amlodipine or hydrochlorothiazide is more effective than monotherapy for blood pressure control and prevention of end-organ injury in hypertensive Dahl rats. J Cardiovasc Pharmacol. 2006;48:857–861. doi: 10.1097/01.fjc.0000238598.09152.30. [DOI] [PubMed] [Google Scholar]
- 27.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int. 1984;26:137–143. doi: 10.1038/ki.1984.147. [DOI] [PubMed] [Google Scholar]
- 28.Davis B, Dei CA, Long DA, et al. Podocyte-specific expression of angiopoietin-2 causes proteinuria and apoptosis of glomerular endothelia. J Am Soc Nephrol. 2007;18:2320–2329. doi: 10.1681/ASN.2006101093. [DOI] [PubMed] [Google Scholar]
- 29.Matavelli LC, Zhou X, Varagic J, et al. Salt loading produces severe renal hemodynamic dysfunction independent of arterial pressure in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;292:H814–H819. doi: 10.1152/ajpheart.00671.2006. [DOI] [PubMed] [Google Scholar]
- 30.Ritz E, Koleganova N, Piecha G. Role of sodium intake in the progression of chronic kidney disease. J Ren Nutr. 2009;19:61–62. doi: 10.1053/j.jrn.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Pimenta E, Gaddam KK, Pratt-Ubunama MN, et al. Relation of dietary salt and aldosterone to urinary protein excretion in subjects with resistant hypertension. Hypertension. 2008;51:339–344. doi: 10.1161/HYPERTENSIONAHA.107.100701. [DOI] [PubMed] [Google Scholar]
- 32.Ribstein J, Du CG, Fesler P, et al. Relative glomerular hyperfiltration in primary aldosteronism. J Am Soc Nephrol. 2005;16:1320–1325. doi: 10.1681/ASN.2004100878. [DOI] [PubMed] [Google Scholar]
- 33.Catena C, Colussi G, Sechi LA. Renal function in primary aldosteronism. Hypertension. 2006;48:e110–e111. doi: 10.1161/01.HYP.0000245660.75946.d0. [DOI] [PubMed] [Google Scholar]
- 34.Rossi GP, Mantero F, Pessina AC. Response to renal function in primary aldosteronism: is glomerular hyperfiltration a hallmark of primary aldosteronism? Further results from the Primary Aldosteronism Prevalence in Hypertension (PAPY) Study. Hypertension. 2006;48:e111–e112. [Google Scholar]
- 35.Zhou X, Ono H, Ono Y, et al. Aldosterone antagonism ameliorates proteinuria and nephrosclerosis independent of glomerular dynamics in L-NAME/SHR model. Am J Nephrol. 2004;24:242–249. doi: 10.1159/000077396. [DOI] [PubMed] [Google Scholar]
- 36.Oelkers W, Diederich S, Bahr V. Primary hyperaldosteronism without suppressed renin due to secondary hypertensive kidney damage. J Clin Endocrinol Metab. 2000;85:3266–3270. doi: 10.1210/jcem.85.9.6819. [DOI] [PubMed] [Google Scholar]
- 37.Lastra-Gonzalez G, Manrique-Acevedo C, Sowers JR. The role of aldosterone in cardiovascular disease in people with diabetes and hypertension: an update. Curr Diab Rep. 2008;8:203–207. doi: 10.1007/s11892-008-0035-9. [DOI] [PubMed] [Google Scholar]
- 38.Nishiyama A, Yao L, Nagai Y, et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension. 2004;43:841–848. doi: 10.1161/01.HYP.0000118519.66430.22. [DOI] [PubMed] [Google Scholar]
- 39.Patni H, Mathew JT, Luan L, et al. Aldosterone promotes proximal tubular cell apoptosis: role of oxidative stress. Am J Physiol Renal Physiol. 2007;293:F1065–F1071. doi: 10.1152/ajprenal.00147.2007. [DOI] [PubMed] [Google Scholar]
- 40.Mathew JT, Patni H, Chaudhary AN, et al. Aldosterone induces mesangial cell apoptosis both in vivo and in vitro. Am J Physiol Renal Physiol. 2008;295:F73–F81. doi: 10.1152/ajprenal.00435.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen C, Liang W, Jia J, et al. Aldosterone induces apoptosis in rat podocytes: role of PI3-K/Akt and p38MAPK signaling pathways. Nephron Exp Nephrol. 2009;113:e26–e34. doi: 10.1159/000228080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol. 2006;17:3438–3446. doi: 10.1681/ASN.2006080944. [DOI] [PubMed] [Google Scholar]
- 43.Huber TB, Hartleben B, Kim J, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol. 2003;23:4917–4928. doi: 10.1128/MCB.23.14.4917-4928.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Welsh GI, Saleem MA. Nephrin-signature molecule of the glomerular podocyte? J Pathol. 2010;220:328–337. doi: 10.1002/path.2661. [DOI] [PubMed] [Google Scholar]
- 45.Arima S, Kohagura K, Xu HL, et al. Nongenomic vascular action of aldosterone in the glomerular microcirculation. J Am Soc Nephrol. 2003;14:2255–2263. doi: 10.1097/01.asn.0000083982.74108.54. [DOI] [PubMed] [Google Scholar]