

Abstract
Using assembled expressed sequence tags (ESTs) from 50 different cDNA libraries, we have identified contigs that represent the complete coding sequences of 850 known human genes, and have scanned these for high quality sequence substitutions. We report the identification and characteristics of 201 candidate single nucleotide polymorphisms found in the coding sequences (cSNPs) of 165 of these genes. Using a conservative calculation, coding region nucleotide diversity (the average number of differences between any pair of chromosomes) was found to be 3 per 10,000 bp based on this data. This analysis reveals that assembled ESTs from multiple libraries may provide a rich source of comparative sequences to search for cSNPs in the human genome.
There is great interest in developing a third generation genetic map of the human genome composed of single nucleotide polymorphism (SNP) markers (Collins et al. 1997). SNPs are the most common form of sequence variation (Nickerson et al. 1998; Wang et al. 1998), and it is likely that highly dense human genetic maps containing more than 100,000 markers can be developed from the human genome. Since SNPs are found in both coding and non-coding regions of the genome, randomly distributed markers as well as markers clustered in genes can be discovered (Collins et al. 1997; Harding et al. 1997; Nickerson et al. 1998). The majority of SNPs found in coding regions (cSNPs) are single base substitutions that may or may not lead to amino acid substitutions. Some cSNPs alter a functionally important amino acid residue, and these are of interest for their potential links with phenotype. Other cSNPs may prove useful for their potential links to functional cSNPs via linkage disequilibrium mapping (Collins et al. 1997).
There are many approaches available to find SNPs in the human genome, but all involve some form of comparative analysis of the same DNA segment from different individuals or from different haplotypes within the same individual. For example, DNA segments amplified by PCR can be compared based on conformation analysis, melting temperature analysis, the ability to be differentially cut with enzymes or chemicals (Cotton 1993), or by direct sequence analysis (Harding et al. 1997; Nickerson et al. 1998; Wang et al. 1998). It is also possible that large numbers of SNPs could be identified as high quality mismatches between overlapping clone sequences from the human genome project (Taillon-Miller et al. 1998). Most such variants are likely to be located in noncoding regions that form the bulk of genomic DNA sequence. Another available resource of sequences from different individuals is expressed sequence tags (ESTs), single-pass sequence reads obtained from cDNAs. Large-scale analysis of ESTs has resulted in a useful gene expression resource of both known and unknown genes (Adams et al. 1991; Hillier et al. 1996; Gerhold and Caskey 1996). Since cDNA libraries from multiple sources (representing different individuals) have been sequenced, there is substantial redundancy in the EST data that can be exploited to find SNPs (Buetow et al. 1999; Picoult-Newberg et al. 1999). We report the use of assembled ESTs, representing the complete coding sequences for 850 known human genes, to scan for coding region SNPs (cSNPs). Our analysis has identified 201candidate cSNPs, of which 87 are predicted to lead to amino acid changes.
RESULTS
Identification of cSNPs using ESTs
Human ESTs from 50 libraries (containing 574,401 sequence reads) were base-called with phred (Ewing et al. 1998) and assembled with phrap (Green 1994). SNP analysis focused on a subset of the contigs that matched the complete coding sequences of known human genes (see Methods). A total of 850 full-length coding sequences averaging 748 bp in size (range 204–2433 bp) and spanning a total of 637,497 bp were covered by EST contigs. An approach similar to that described by Picoult-Newberg et al. (1999), but systematically applying phred quality scores (Ewing and Green 1998) to help automate the analysis, was then used to identify single nucleotide mismatches in these genes. After filtering, 223 candidate SNPs were identified. Visual inspection of the traces for these candidate SNPs revealed two types of errors resulting from base-calling (n = 21) and misalignment (n = 1) problems. The remaining 201 candidate cSNPs were verified to have high quality mismatches, with a minimum of two reads for each of the alternative alleles. Of the 850 genes examined, 165 genes contained candidate SNPs. Twenty-nine of these genes contained more than one candidate cSNP, that is, 23 genes with two candidate cSNPs, 5 genes with 3 candidate cSNPs, and 1 gene with 4 candidate cSNPs.
Using a probabilistic approach, we estimated the nucleotide diversity across the scanned sequences to be 3 polymorphic differences per 10,000 bp between two randomly selected chromosomes. As candidate polymorphisms were required to have two sequences representing each candidate allele, this is likely to be an underestimate because it does not include the more rare alleles represented only once in the set of ESTs. Diversity estimates of 2 and 6 polymorphic differences per 10,000 bp were obtained for positions with the potential to give rise to nonsynonymous and synonymous substitutions, respectively.
Characteristics of cSNPs
Candidate cSNPs were classified according to substitution type (A/C, A/G, A/T, C/G, C/T, and G/T) as shown in Figure 1a. As expected, transitions (69%) were more common than transversions (31%) among the sequence changes (Cooper and Krawczak 1990); 41% of the substitutions were found to occur at a CpG site, a dinucleotide known for its high mutability (Cooper and Youssoufian 1988).
Figure 1.
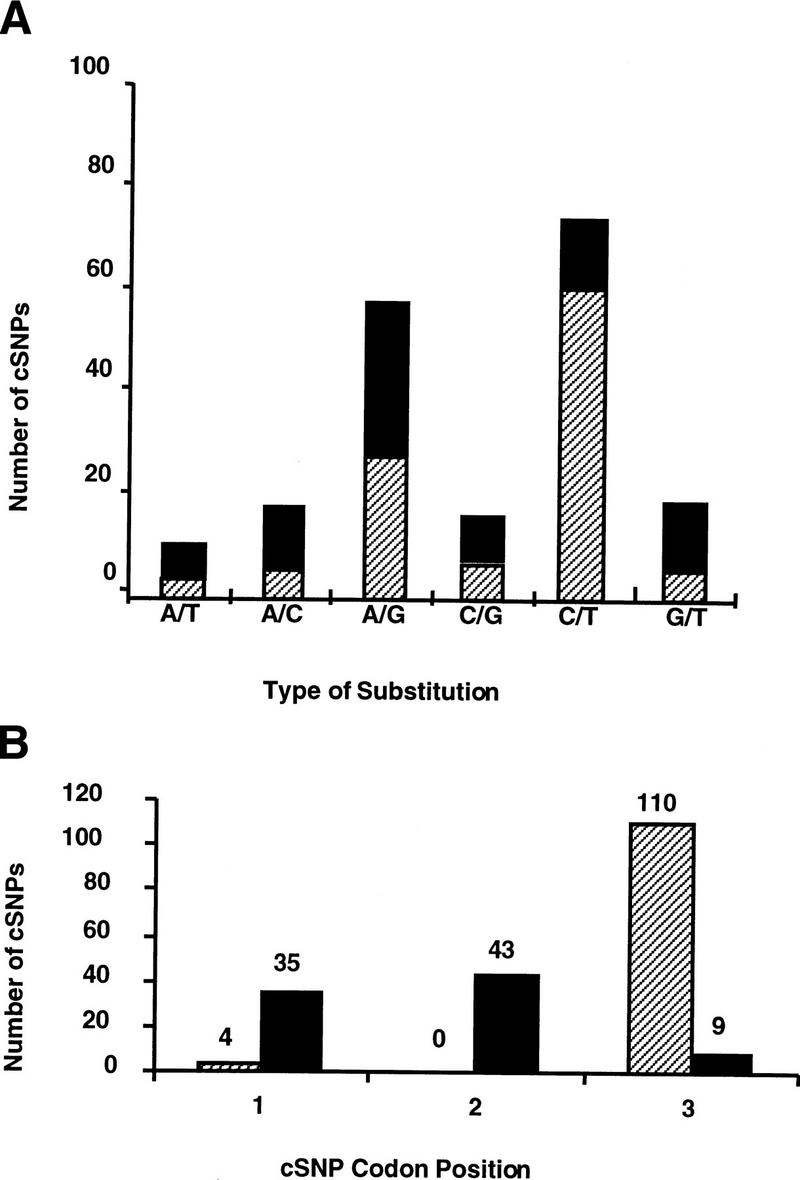
Distribution of candidate cSNPs by type of nucleotide substitution (A) and codon position (B), for synonymous (hatched bars) and nonsynonymous (solid bars) amino acid substitutions.
We also determined the position of each candidate cSNP in the codon, and whether the predicted change was synonymous (silent) or nonsynonymous (replacement). The majority of the candidate cSNPs (59%) were found to occur at the third codon position, and as expected most (92%) of these were synonymous (see Fig. 1b for distribution of candidate SNPs according to codon position). A number of changes were also identified in codon positions 1 and 2 (Fig. 1b) and these accounted for most of the nonsynonymous changes. In all, 87 of the candidate SNPs (43%) are predicted to result in nonsynonymous changes.
Based on sequence information alone it is difficult to predict the effect of an amino acid substitution on protein function. One potential way to obtain information about functional constraint is via evolutionary comparisons, as it is generally believed that conserved residues are more likely to be functionally significant than nonconserved residues. We examined sequence conservation at candidate nonsynonymous cSNP positions using orthologous protein sequences from mouse (Mus musculus), which were available for 38 of the genes containing 41 of the 87 nonsynonymous candidate cSNPs. It was found that candidate cSNPs were disproportionately over-represented among the nonconserved positions. Although only 17% of the amino acid residues were nonconserved (not identical) in the 38 mouse–human pairs, these included 42% (17/41) of the nonsynonymous candidate cSNPs. This is consistent with the idea that a high fraction of nonsynonymous polymorphic substitutions occurs at positions that are not functionally constrained on evolutionary timescales, particularly since a fraction of the “conserved” positions are presumably not under functional constraint but have by chance not mutated in the mouse and human lineages. However, some of the candidate cSNPs may have the potential to alter function. Although none were predicted to occur in an active site [as annotated in SwissProt (http://www.expasy.ch/sprot)] in proteins with enzymatic or receptor activity, eleven candidate cSNPs were found to lie in SH3 domains, transmembrane regions, binding sites, signal sequences, or other residues associated with specific functions (Table 1).
Table 1.
Candidate cSNPs Identified in Functional Regions as Indicated by Swiss Prot
| Accession | Gene | Change | Pos. | Domain/site | Domain/site pos. |
|---|---|---|---|---|---|
| M55067 | NCF1 | D → N | 166 | SH3 (potential) | 156–215 |
| X59445 | SOD2 | A → V | 16 | mitochondrion | 1–24 |
| K01500 | AACT | A → T | 9 | signal | 1–23 |
| M65128 | FKBP2 | R → Q | 7 | signal | 1–21 |
| L25085 | — | I → F | 88 | transmembrane (potential) | 70–90 |
| X97544 | — | V → I | 113 | transmembrane (potential) | 113–133 |
| X14829 | LGALS1 | W → X | 68 | β galactoside binding (potential) | 68–74 |
| Y00503 | KRT19 | G → A | 60 | filament head | 1–70 |
| M13755 | G1P2 | S → N | 83 | ubiquitin-like | 79–165 |
| L35233 | AMFR | I → V | 181 | cytoplasmic (potential) | 138–323 |
| M16961 | AHSG | T → S | 256 | O-glycosylated | 256 |
Two of the predicted changes among candidate cSNPs would lead to the substitution of an amino acid by a stop codon, and could affect function depending on where the protein is terminated. One predicted termination was located at a Trp residue (position 68 of 134) at the start of the binding domain in the beta-galactoside binding protein (LGALS1: SwissProt database: accession no. P09382). The other nonsense mutation was found at position 219 (Glu) of 245 residues of the 14-3-3 protein (SwissProt database: accession P27348). In each case, the reads containing the termination codon were contributed by only one cDNA library (although the libraries for the two cases were different) and that library also contained the “wild-type” allele.
Confirmation of Nonsynonymous cSNPs
By scanning the existing polymorphism literature, we were able to verify 17 of the 87 nonsynonymous candidate polymorphisms (Table 2). In some of these cases allele frequencies had been estimated. Although most of these cSNPs were common (average heterozygosity was calculated to be 0.33 for 12 sites; Table 2), three of them, K02215 and 16581 (alternative allele frequency 8%) and APOAI (alternative allele frequencies 0.05%), were relatively rare among the tested populations. In addition to the cSNPs identified as such in the literature, four nonsynonymous substitutions were indicated as “conflicts” of unknown origin in the SwissProt database (Table 2). Thus, nearly a quarter of the nonsynonymous candidate cSNPs (21 of 87) could be verified with data from other laboratories (Table 2). The phred quality of the bases for the previously known cSNPs we identified ranged from 21 to 51 with an average quality of 33 at the mismatch. This is similar to the average quality at the mismatch for the entire set of candidate cSNPs, which was 32.
Table 2.
Confirmed Candidate cSNPs
| Gene | MRNA | Substitution | Amino acid | Codona | Libraryb | Freq.c | Referencesd | |
|---|---|---|---|---|---|---|---|---|
| 1 | AACT | K01500 | GCT → ACT | A → T | 9 | M | Poller et al. (1993) | |
| 2 | AGT | K02215 | ACG → ATG | T → M | 207 | S | 0.92 | Jeunemaitre et al. (1992) |
| 3 | AGT | K02215 | ATG → ACG | M → T | 268 | S | 0.64 | Jeunemaitre et al. (1992) |
| 4 | AHSG | M16961 | ACG → ATG | T → M | 248 | M | 0.67 | Osawa et al. (1997) |
| 5 | AHSG | M16961 | ACC → AGC | T → S | 256 | M | 0.67 | Osawa et al. /1997) |
| 6 | APOA1 | M11791 | AAG → ATG | K → M | 131 | S | >0.99 | von Eckardstein et al. (1990) |
| 7 | APOH | S80305 | GTA → TTA | V → L | 266 | S | 0.76 | Steinkasserer et al. (1993) |
| 8 | C1NH | M13690 | GTG → ATG | V → M | 480 | M | Bock et al. (1986) | |
| 9 | COMT | M65212 | GTG → ATG | V → M | 158 | M | 0.82 | Li et al. (1997) |
| 10 | GC | M12654 | ACG → AAG | T → K | 436 | S | 0.74 | Braun et al. (1992) |
| 11 | GLO1 | S83285 | GAG → GCG | E → A | 111 | M | Ridderstrom and Mannervik (1996) | |
| 12 | IGFBP1 | X15002 | ATA → ATG | I → M | 113 | M | 0.60 | Luthman et al. (1989) |
| 13 | MTH1 | D16581 | GTG → ATG | V → M | 83 | S | −0.92 | Wu et al. (1995) |
| 14 | PCMT1 | D25545 | ATA → GTA | I → V | 120 | S | 0.77 | Tsai and Clarke (1994) |
| 15 | RPL10 | M73791 | AGT → AAT | S → N | 202 | M | P27635 | |
| 16 | SFTPC | J03553 | AAT → ACT | N → T | 138 | S | P11686 | |
| 17 | SOD2 | X59445 | GCT → GTT | A → V | 16 | M | 0.50 | Rosenblum et al. (1996) |
| 18 | TCF6L1 | M62810 | AGT → ACT | S → T | 12 | M | Q00059e | |
| 19 | FDFT1 | S76822 | AGG → AAG | R → K | 45 | M | P37268e | |
| 20 | LGALS3 | M36682 | CAT → CCT | H → P | 64 | M | P17931e | |
| 21 | LGALS3 | M36682 | CCC → ACC | P → T | 98 | M | P17931e |
From the start codon.
Multiple (M) or single (S) library confirmation.
Frequency of the most frequent allele.
Swiss Prot accession number (http://www.expasy.ch/sprot) or reference reporting cSNP.
cSNPs listed as conflicts of unknown origin in the Swiss Prot database.
DISCUSSION
ESTs previously have been proven useful for finding SNPs associated with cDNA sequences (Buetow et al. 1999; Picoult-Newberg et al. 1999). We have shown that SNPs in coding regions (cSNPs) can also be identified using ESTs. Although our approach is similar to that described by Picoult-Newberg, we focused entirely on coding sequences. There was only a small overlap of five SNPs detected by each approach. Approximately 640,000 bp of coding sequence were examined across 850 genes and 201 sequence-confirmed candidate cSNPs identified. Our estimate of the number of cSNPs is highly conservative because we required two representatives for each allele. Despite this stringency, our probabilistic estimates of the nucleotide diversity (3 × 10−4) in coding regions is broadly consistent with estimates obtained by more direct approaches (Cargill et al. 1999; Halushka et al. 1999).
One problem in identifying SNPs using assembled ESTs, is discriminating mismatches due to base-calling error from those due to variation in the sequence. This can be problematic for ESTs because of the high estimated error rate (2%) associated with these single-pass sequences (Hillier et al. 1996). For preliminary screening, we applied a previously developed heuristic approach but systematically used sequence quality obtained from the phred base-caller to help sort base-calling errors from SNPs. To avoid missing true polymorphisms, we found it useful to set a relatively modest quality threshold (q > 20, corresponding to an error probability < 1%) but to require 2 reads for each allele. As the quality threshold is set higher, fewer bases in a read meet the cut-off and fewer candidate SNPs are identified.
Approximately 52% (n = 105) of our 201 candidate cSNPs have sequence-based confirmation from multiple cDNA libraries, in the sense that each allele is represented in at least two libraries. It is possible that a portion of the remaining 48% could represent somatic rather than germline mutations; distinguishing between these sources of variation would require testing DNA from the contributing individual (currently not available) or systematic genotyping of a large sample. Only 8 of 52, or 15%, of the single-library nonsynonymous candidate cSNPs had been previously identified and confirmed by other approaches, a much smaller fraction than for the multiple-library candidate nonsynonymous cSNPs (13 of 35, or 37%). This could indicate that either the single-library candidates are rarer or that some of them are somatic mutations. However, only one of the single-library cSNPs originated from a tumor cell cDNA library.
SNPs identified in coding regions are useful for association studies because of their potential linkage disequilibrium with functional variants in those genes (Collins et al. 1997). In this respect, several of the candidate cSNPs we uncovered have been associated with disease susceptibility (Jeunemaitre et al. 1992; Arngrimsson et al. 1993; Lachman et al. 1996; Li et al. 1997; Morgan et al. 1999). All of the candidate cSNPs reported here have been deposited in dbSNP (http://www.ncbi.nlm.nih.gov/SNP) and at our web site (http://droog.mbt.washington.edu) and are available for further analysis with regard to their distribution within and among human populations and their potential functional relevance.
METHODS
EST Assembly
Chromatogram files for human ESTs (574,401 reads from 50 cDNA libraries) were downloaded from the Washington University EST web site (ftp://genome.wustl.edu). Based on the limited descriptive information available for the source of these libraries, it appears that sequences from at least 53 different individuals (106 chromosomes) are represented. Traces were base-called with phred (Ewing et al. 1998, v. 0.961028) and assembled with phrap (Green 1994, v. 0.971106). Assembly was carried out in two stages. In the first stage, the collection of ESTs was divided into subsets of 100,000 to 200,000, and each subset was independently assembled. In the second stage, the sets of contigs and unincorporated singletons from the first stage were assembled together, producing the final set of contigs, and all the ESTs were aligned to the contig sequences (C. Wilson and P. Green, unpubl.).
Identifying EST Contigs Matching Known Coding Sequences
A set of nonredundant full length coding sequences of human genes was prepared as follows (M. Robinson, unpubl.). Entries with an annotated full-length coding sequence (CDS), starting with NTG and ending with a stop codon, were extracted from GenBank (release 103.0, files gbpri1.seq and gbpri2.seq). Entries that contained any of the words “mutant,” “variant,” “antisense,” “rearranged,” or “T-cell receptor,” or that did not contain the word “RNA” in the locus field were discarded. Redundant sequences were then deleted and the 5′ and 3′ untranslated regions were removed, leaving a total of 4883 intact coding sequences.
Cross_match (Green 1993) was then used to compare EST contigs to the CDSs, and each CDS that aligned >96% of its length to matching EST contig was identified. There were 954 such CDSs. Phrap permits mismatches if similarity between the reads is high, and may assemble together ESTs from paralogous genes and genes from multigene families. Some of these show such a high degree of similarity that it is difficult to distinguish between alleles and paralogs. To minimize the risk of paralogs contributing spuriously to the candidate cSNPs, we eliminated those CDSs for which the number of high quality discrepancies (quality ≥ 30) between the contig and the CDSs exceeded 1% of the length of the alignment; this left 896 CDS. We then further eliminated 20 CDSs whose associated contigs had <3 or >500 reads, 23 CDSs belonging to ribosomal protein families, and 3 CDSs belonging to lipocalin, DEFA, and CGB gene families. This left a total of 850 CDSs that were then scanned for SNPs.
SNP Identification
To identify candidate SNPs in the EST contigs, we reassembled each contig, including the sequence of the mRNA, as a reference sequence. The latter was assigned zero quality. Since several chromosomes are generally represented in the contigs, candidate SNPs can be identified as base mismatches between reads. Mismatches could also be due to copying errors introduced during the production or cloning of the cDNAs. To help minimize this problem, we required that mismatches be confirmed (≥ two reads for each alternative base; the reference sequence was permitted to be one of these). Other sources of mismatches are base calling errors and misalignments of the sequences. To decrease this problem, we required all identified “mismatched sites” to pass a series of filters: (1) phred quality >20; (2) average phred quality ≥20 over a window of 5 bases on either side of the site; (3) an exact sequence match at all the bases in a window of 5 on either side of suspected site; and (4) manual trace inspection using consed (Gordon et al. 1998). The last step was required to eliminate mismatches due to systematic errors in the base-calling by phred or alignment by phrap. With current versions of phred (0.980904) and phrap (0.990319), 21 base-calling errors (all due to the presence of multiple peaks at the site in question) and 1 misalignment case (due to inclusion of reads not belonging to the contig) were found. We only considered substitution type polymorphisms in our analysis because of their higher confirmation rate (Picoult-Newberg et al. 1999).
There were ∼7000 ESTs that matched these 850 genes but did not assemble together with the contigs selected for these genes. These ESTs were directly aligned to these genes and were scanned for the candidate SNPs using all the criteria described above, except for manual trace inspection. This resulted in an additional set of 264 possible candidates, a random subset of which was selected for closer inspection. Most of these turned out to be in ESTs showing a large percentage of high quality mismatches with the CDS, indicating that the EST was likely to be from a paralogous rather than allelic gene. There were also a number of cases in which there were large gaps in the alignment between the CDS and ESTs. Some of the candidate SNPs in such ESTs may be real, if the gaps are due to alternative or incomplete splicing. However, the gaps could also indicate that the EST belongs to a different gene or is chimeric. Due to the high risk of spurious ESTs, the candidate cSNPs from these unincorporated ESTs were not pursued further and are not reported in this paper.
Based on the reference CDS, each substitution mismatch was classified as to whether it leads to a synonymous or nonsynonymous substitution. Information on candidate cSNPs was stored in a relational database and was deposited in dbSNP (ss 5277 to ss 5477). A list of the candidate cSNPs is available at http://droog.mbt.washington.edu. Each candidate also has the following information available: mRNA accession number, contig with assembly information, SNP position in mRNA, codon change, amino acid change, gene name (if known), whether the SNP has single or multilibrary confirmation, the ESTs with their source (library), and 10 bp sequence on either side of the SNP.
Calculation of Sequence Diversity
For the diversity calculation, only reads from those libraries derived from single individuals (excluding the two pooled libraries Gessler Wilms tumor and Morton Fetal Cochlea) were considered. All the libraries except for four (Soares adult brain N2b4HB55Y, Soares adult brain N2b5HB55Y, Soares retina N2b4HR, and Soares retina N2b5HR) were considered to be from different individuals. At each coding sequence position in the 850 genes, all reads meeting the three quality and sequence match filters (see above, SNP identification) were determined. Positions at which there were fewer than four such reads, or where exactly one of the reads was discrepant with the others, were excluded from further consideration, leaving 443,440 positions that could be used for the diversity calculations.
At each position, the selected reads were grouped according to the individual of origin and the number of distinct chromosomes represented in each group was estimated as follows. Since each group derives from a single individual, the number of distinct chromosomes cannot exceed two. If two different bases (alleles) occur within the group, then there are exactly two chromosomes. Otherwise (only one allele present), the probability that there is one chromosome represented (all reads derive from the same chromosome) is 2 * 0.5n and the probability that there are two chromosomes represented is 1–2* 0.5n, where n is the number of reads in the group.
The expected number of pairs of distinct chromosomes for which the two alleles are the same (ns), and the expected number of pairs for which the two alleles are different (nd), can now be estimated within and between groups at each position. For example, if only a single allele is present for a group of n reads, then there is a fractional contribution of 1– 2* 0.5n (the probability that two chromosomes are present) to the value of ns and a contribution of 0 to nd for that group. The within and between group estimates are added at each position, and then added over positions to get overall estimates for ns and nd. The estimated diversity is then given by nd/(ns + nd).
Acknowledgments
We thank C. Wilson for providing the EST assembly, M. Robinson for the coding sequence database, and S. Taylor for helpful discussions. This work was supported by the National Science Foundation (NSFBIR9214821) to K.G. and D.A.N, the National Institutes of Health (HG00774) to P.G., and the Department of Energy (DE-FG03-97ER62385) to D.A.N.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL: kavitag@u.washington.edu; FAX (206) 685-7301.
REFERENCES
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science. 1991;252:1651–1656. doi: 10.1126/science.2047873. [DOI] [PubMed] [Google Scholar]
- Arngrimsson R, Purandare S, Connor M, Walker JJ, Bjornsson S, Soubrier F, Kotelevtsev YV, Geirsson RT, Bjornsson H. Angiotensinogen: A candidate gene involved in preeclampsia? Nat Genet. 1993;4:114–115. doi: 10.1038/ng0693-114. [DOI] [PubMed] [Google Scholar]
- Bock SC, Skriver K, Nielsen E, Thogersen HC, Wiman B, Donaldson VH, Eddy RL, Marrinan J, Radziejewska E, Huber R, et al. Human C1 inhibitor: Primary structure, cDNA cloning, and chromosomal localization. Biochemistry. 1986;25:4292–4301. doi: 10.1021/bi00363a018. [DOI] [PubMed] [Google Scholar]
- Braun A, Bichlmaier R, Cleve H. Molecular analysis of the gene for the human vitamin D-binding protein (group-specific component): Allelic differences of the common genetic GC types. Hum Genet. 1992;89:401–406. doi: 10.1007/BF00194311. [DOI] [PubMed] [Google Scholar]
- Buetow KH, Edmonson MN, Cassidy AB. Reliable identification of large numbers of candidate SNPs from public EST data. Nat Genet. 1999;21:323–325. doi: 10.1038/6851. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Lane CR, Lim EP, Kalayanaraman N, Nemesh J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Collins FS, Guyer MS, Chakravarti A. Variations on a theme: Cataloging human DNA sequence variations. Science. 1997;278:1580–1581. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M. The mutational spectrum of single base-pair substitutions causing human genetic disease: Patterns and predictions. Hum Genet. 1990;85:55–74. doi: 10.1007/BF00276326. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- Cotton RG. Current methods of mutation detection. Mutat Res. 1993;285:125–144. doi: 10.1016/0027-5107(93)90060-s. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Gerhold D, Caskey CT. It's the genes EST access to human genome content. BioEssays. 1996;18:973–981. doi: 10.1002/bies.950181207. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Green P. cross_match. 1993. http://www.genome.washington.edu/UWGC/analysistools/phrap.htm ( http://www.genome.washington.edu/UWGC/analysistools/phrap.htm). ). [Google Scholar]
- Green P. phrap. 1994. http://www.genome.washington.edu/UWGC/analysistools/phrap.htm ( http://www.genome.washington.edu/UWGC/analysistools/phrap.htm). ). [Google Scholar]
- Halushka MK, Fan JB, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A. Patterns of single-nucleotide polymorphisms in candidate genes for blood pressure homeostasis. Nat Genet. 1999;22:239–247. doi: 10.1038/10297. [DOI] [PubMed] [Google Scholar]
- Harding RM, Fullerton SM, Griffiths RC, Bond J, Cox MJ, Schneider JA, Moulin DS, Clegg JB. Archaic African and Asian lineages in the genetic ancestry of modern humans. Am J Hum Genet. 1997;60:772–789. [PMC free article] [PubMed] [Google Scholar]
- Hillier L, Lennon G, Becker M, Bonaldo MF, Chiapelli B, Chissoe S, Dietrich N, DuBuque T, Favello A, Gish W, et al. Generation and analysis of 280,000 human expressed sequence tags. Genome Res. 1996;6:807–828. doi: 10.1101/gr.6.9.807. [DOI] [PubMed] [Google Scholar]
- Jeunemaitre X, Soubrier F, Kotelevtsev YV, Lifton RP, Williams CS, Charru A, Hunt SC, Hopkins PN, Williams RR, Lalouel JM, Corvolt P. Molecular basis of human hypertension: Role of angiotensinogen. Cell. 1992;71:169–180. doi: 10.1016/0092-8674(92)90275-h. [DOI] [PubMed] [Google Scholar]
- Lachman HM, Papolos DF, Saito T, Yu YM, Szumlanski CL, Weinshilboum RM. Human catechol-O-methyltransferase pharmacogenetics: Description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics. 1996;6:243–250. doi: 10.1097/00008571-199606000-00007. [DOI] [PubMed] [Google Scholar]
- Li T, Vallada H, Curtis D, Arranz M, Xu K, Cai G, Deng H, Liu J, Murray R, Liu X, Collier DA. Catechol-O-methyltransferase Val158Met polymorphism: Frequency analysis in Han Chinese subjects and allelic association of the low activity allele with bipolar affective disorder. Pharmacogenetics. 1997;7:349–353. doi: 10.1097/00008571-199710000-00002. [DOI] [PubMed] [Google Scholar]
- Luthman H, Soderling-Barros J, Persson B, Engberg C, Stern I, Lake M, Franzen SA, Israelsson M, Raden B, Lindgren B, et al. Human insulin-like growth-factor-binding protein. Low-molecular-mass form: Protein sequence and cDNA cloning. Eur J Biochem. 1989;180:259–265. doi: 10.1111/j.1432-1033.1989.tb14641.x. [DOI] [PubMed] [Google Scholar]
- Morgan T, Craven C, Lalouel JM, Ward K. Angiotensinogen thr235 variant is associated with abnormal physiologic change of the uterine spiral arteries in first trimester decidua. 1999. Am J Obstet Gynecol. 1999;180:95–102. doi: 10.1016/s0002-9378(99)70156-0. [DOI] [PubMed] [Google Scholar]
- Nickerson DA, Taylor SL, Weiss KM, Clark AG, Hutchinson RG, Stengard J, Salomaa V, Vartiainen E, Boerwinkle E, Sing CF. DNA sequence diversity in a 9.7 kb region of the human lipoprotein lipase gene. Nat Genet. 1998;19:233–240. doi: 10.1038/907. [DOI] [PubMed] [Google Scholar]
- Osawa M, Umetsu K, Okhi T, Nagasawa T, Suzuki T, Takeichi S. Molecular evidence for human alpha2-HS glycoprotein (AHSG) polymorphism. Hum Genet. 1997;99:18–21. doi: 10.1007/s004390050302. [DOI] [PubMed] [Google Scholar]
- Picoult-Newberg L, Ideker TE, Pohl MG, Taylor SL, Donaldson MA, Nickerson DA, Boyce-Jacino M. Mining SNPs from EST databases. Genome Res. 1999;9:167–174. [PMC free article] [PubMed] [Google Scholar]
- Poller W, Faber JP, Weidinger S, Tief K, Scholz S, Fischer M, Olek K, Kirchgesser M, Heidtmann HH. A leucine-to-proline substitution causes a defective alpha 1-antichymotrypsin allele associated with familial obstructive lung disease. Genomics. 1993;17:740–743. doi: 10.1006/geno.1993.1396. [DOI] [PubMed] [Google Scholar]
- Ridderstrom M, Mannervik B. Optimized heterologous expression of the human zinc enzyme glyoxalase I. Biochem J. 1996;314:463–467. doi: 10.1042/bj3140463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum JS, Gilula NB, Lerner RA. On signal sequence polymorphisms and diseases of distribution. Proc Natl Acad Sci. 1996;93:4471–4473. doi: 10.1073/pnas.93.9.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkasserer A, Dorner C, Wurzner R, Sim RB. Human beta 2-glycoprotein I : Molecular analysis of DNA and amino acid polymorphisms. Hum Genet. 1993;91:401–402. doi: 10.1007/BF00217367. [DOI] [PubMed] [Google Scholar]
- Taillon-Miller P, Gu Z, Li Q, Hillier L, Kwok PY. Overlapping genomic sequences: A treasure trove of single-nucleotide polymorphisms. Genome Res. 1998;8:748–754. doi: 10.1101/gr.8.7.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai W, Clarke S. Amino acid polymorphisms of the human L-isoaspartyl/D-aspartyl methyltransferase involved in protein repair. Biochem Biophys Res Commun. 1994;203:491–497. doi: 10.1006/bbrc.1994.2209. [DOI] [PubMed] [Google Scholar]
- von Eckardstein A, Funke H, Walter M, Altland K, Benninghoven A, Assmann G. Structural analysis of human apolipoprotein A-I variants: Amino acid substitutions are nonrandomly distributed throughout the apolipoprotein A-I primary structure. J Biol Chem. 1990;265:8610–8617. [PubMed] [Google Scholar]
- Wang DG, Fan JB, Siao C, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]
- Wu C, Nagasaki H, Maruyama K, Nakabeppu Y, Sekiguchi M, Yuasa Y. Polymorphisms and probable lack of mutation in a human mutT homolog, hMTH1, in hereditary nonpoliposis colorectal cancer. Biochem Biophys Res Commun. 1995;214:1239–1245. doi: 10.1006/bbrc.1995.2419. [DOI] [PubMed] [Google Scholar]