

Abstract
Vitamin K carboxylase (VKC) is believed to convert vitamin K, in the vitamin K cycle, to an alkoxide-epoxide form which then reacts with CO2 and glutamate to generate γ-carboxyglutamic acid (Gla). Subsequently, vitamin K epoxide reductase (VKOR) is thought to convert the alkoxide-epoxide to a hydroquinone form. By recycling vitamin K, the two integral-membrane proteins, VKC and VKOR, maintain vitamin K levels and sustain the blood coagulation cascade. Unfortunately, NMR or X-ray crystal structures of the two proteins have not been characterized. Thus, our understanding of the vitamin K cycle is only partial at the molecular level. In this study, based on prior biochemical experiments on VKC and VKOR, we propose a hetero-dimeric form of VKC and VKOR that may explain the efficient oxidation and reduction of vitamin K during the vitamin K cycle.
Keywords: vitamin K cycle, vitamin K carboxylase, vitamin K epoxide reductase, hetero-dimer
1. Introduction
Vitamin K plays a key role in the blood coagulation cascade [1,2]. Through the vitamin K cycle, vitamin K helps to generate γ-carboxyglutamic acid (Gla) from glutamic acid (Glu). Gla enables several blood coagulation proteins, (factor II, factor VII, factor IX, factor X, protein C and protein S, …), to establish a fold necessary for anchoring to anionic lipid membranes in the presence of Ca2+ and thereby trigger and propagate the blood coagulation cascade [2–5].
At least, two trans-membrane (TM) proteins, vitamin K carboxylase (VKC, also referred to as GGCX) and vitamin K epoxide reductase (VKOR, also referred to as VKORC1), are involved in the vitamin K cycle at the endoplasmic reticulum (ER). The human sequences of VKC with 758 residues [6–8] and VKOR with 163 residues [9,10] have been identified. VKC has been suggested by Dowd to catalyze the formation of an activated form of vitamin K from the hydroquinone [11–13]. This alkoxide-epoxide form then abstracts a proton from glutamic acid (Glu) so that the resulting carbanion may react with CO2 to generate Gla. This is a 4 electron process. VKOR, on the other hand, converts the alkoxide-epoxide back to the quinone form (a 2 electron process), followed by reduction to the subsequent hydroquinone form (a fully reduced form) by VKOR (a 2 electron process) to complete the cycle. (See Supporting Materials) The last step remains controversial [14]. The mechanism for the reduction of the alkoxide-epoxide form to the quinone form has been studied both experimentally and theoretically [15,16]. The essence of the reduction mechanism involves a sequential participation of two cysteine residues. Thus, through a series of the oxidation and reduction processes, vitamin K is presumably recycled as shown in Fig.1.
Fig. 1.

The vitamin K cycle as regulated by the two TM enzymes, vitamin K carboxylase (VKC) and vitamin K epoxide reductase (VKOR).
Even though sequences and several functions of VKC and VKOR are known and the putative mechanisms of oxidation and reduction have been proposed, the structures and relative orientations of the two TM proteins have not been characterized. Then, several important questions arise: How does the largely hydrophobic vitamin K interact with the two TM proteins? How do these two TM proteins bind to the substrate, a vitamin K-dependent protein (VKD), both modifying vitamin K, to generate Gla from Glu on a specific domain? How can these two TM proteins control the vitamin K cycle efficiently within the bounds of a membrane?
Vitamin K is composed of a long hydrophobic chain and naphthoquinone unit. The human pool is very small (~100 μg) and the turnover rate is rapid (~1.5 days) [17], thus we can surmise that its use must be quite efficient. We can conjecture that once the lipophilic tail of a vitamin K is anchored in membrane, vitamin K would be restricted to lateral diffusion in the membrane. If VKC and VKOR were randomly separated, vitamin K and its intermediates would have to diffuse between geometrically separated active sites, (possibly involving Lys218 for VKC and Cys132, Cys135 for VKOR) to effect the oxidation/reduction process. Instead, if we envision that VKC and VKOR are co-localized as a hetero-dimer in the ER membrane, the inefficiency of diffusion of the vitamin K intermediates is minimized. We therefore propose the co-localization of VKC and VKOR to account for the observed efficient vitamin K cycle. A schematic co-localization model was previously proposed to account for the possible interaction of VKC/VKOR with calumenin in the ER lumen [18–20].
The formation of a hetero-dimer protein complex within membrane does exist in nature. For instance, a hetero-dimeric form of two TM proteins has been found in the maltose transport system. MalF and MalG, composed of eight TM helices and six TM helices respectively, form a hetero-dimer: two crescent shaped TM domains face each other in a concave manner and enclose a maltose molecule [21,22]. Similarly, a TM complex involving 4 subunits has been characterized for the ubiquinol oxidase system [23]. For a deeper understanding of the vitamin K cycle, clearly more accurate information about the 3D structures of VKC and VKOR will be ultimately required. Despite the absence of structural information of VKC and VKOR, however, there have been significant biochemical experiments on these two proteins. In this study, based on these prior biochemical experiments, computer-aided prediction of TM units, and quantum chemical study, we have built a rational model of the putative hetero-dimeric form of VKC and VKOR. We find that the hetero-dimer of VKC and VKOR is compatible with the intermediate vitamin K structures proposed in the Dowd mechanism. The model is also helpful in accounting for the observed non-competitive inhibition of VKOR by warfarin [24].
2. Methods
All the structures in Fig.1 were fully geometry optimized using Gaussian03 [25,26] with the method/basis of B3lyp/6-31+g(d,p) in gas-phase. In the preparation of the vitamin K and its derivatives, the methyl group (R1) and the long hydrophobic side chain (R2) after the first side-chain double bond were replaced by hydrogens [27]. Since the structure of VKC and VKOR have not been characterized, we used one of the TM helix units of sensory rhodopsin II (PDB : 1JGJ) [28] as a template in building our TM model. Since the sequences of VKC and VKOR that span the membrane range from 20 to 23 residues in our predicted model, the size of the rhodopsin template can be used for both VKC and VKOR. For the eight TM helix units (minimalist model) in our model, we used the rhodopsin TM helix unit with 25 residues (VGLTTLFWLGAIGMLVGTLAFAWAG). The lengths for TM helices in rhodopsin with more than 20 residues is measured to be 32.7 Å for Leu21-Thr43 (Cα-Cα; 22 residues) and 35.4 Å for Arg100-Ala124 (Cα-Cα; 24 residues) respectively. Thus, a reasonable length distribution for TM helices of VKC and VKOR in our predicted model is taken to be approximately 30 Å ~ 35 Å, consistent with the single rhodopsin helix we chose as a reference. In fact, recent molecular dynamics simulations by our group (unpublished) on VKOR models inserted in a model lipid membrane of this size (i.e. bilayer length : 38 Å ~ 40 Å) appear to be relatively stable for approximately 13ns. The radius (r) of TM helix backbone in our model was set to 3.5 Å. Our hetero-dimer model was built with VMD 1.8.7 [29].
3. Results and discussion
3.1 Location of putative active sites of VKC and VKOR
Of special significance, Lys218 has been implicated in playing a critical role in the deprotonation of the hydroquinone form of vitamin K and the subsequent initiation of the carboxylation reaction of VKC on the lumenal side of ER membrane [30]. Likewise, biochemical mutation experiments have revealed that Cys132 and Cys135 in the CXXC motif of VKOR are strong candidates for the redox center inside the membrane [31]. These putative active site residues of VKC (Lys218) and VKOR (Cys132, Cys135) therefore play a critical role in determining the relative position and orientation of vitamin K with respect to the two co-localized TM proteins. To locate the putative active sites of VKC and VKOR with respect to the ER membrane, we employed TM helix prediction programs for VKC and VKOR.
Among the various TM protein prediction programs available, we employed the Philius Transmembrane Prediction program [32], which is based on dynamic Bayesian network methodology and the consensus program TOPCONS [33] to predict topologies of TM helices of the two TM proteins. TOPCONS, in fact, uses a number of prediction programs, (SCAMPI-seq, SCAMPI-msa, PRODIV, PRO, and OCTOPUS), to produce a consensus result and thereby improve a reliability of prediction ability. To test the prediction reliability, we applied both methods to the case of the bacterial VKOR-like system, the structure of which was recently determined [34]. Both methods, in fact, predict a similar result, 5 TM helices, from the sequence information, a result consistent with the X-ray crystal structure of the bacterial VKOR-like protein. (see Supporting Material)
3.2 Topology of VKC based on prediction of TM helix units
For VKC (GenBankTM accession number ACF70731.1, 758 residues), both of the prediction methods show somewhat divergent features for the TM topology (Fig.2.A). The prior biochemical experimental observation that N-terminus is located at the cytoplasm of the ER (inside) [6] and the C-terminus at the lumen of the ER (outside) [6] requires an odd number of helices in the VKC topology. These experiments were based on insertion of fragments of the VKC sequence predicted to possibly contain TM domains. The Philius Transmembrane Prediction program predicts 5 TM helices, while TOPCONS predicts 7 TM helices. However, experiments have supported a 5 TM helix model for VKC topology [6,35]. Thus, we tentatively adopt 5 TM helices for VKC. In building the model of VKC, one important structural data point for VKC was considered. According to experiment [35], a disulfide bond between Cys99 and Cys450 exists in the lumen of the ER. This disulfide bond could help to define and stabilize the 3-dimensional arrangement of the TM helix units of VKC [36]. The Philius Transmembrane Prediction program was unable to accommodate the existence of the disulfide bond in the lumen [36] as it predicts the location of the two cysteines on the opposite sides of the ER membrane. For compatibility with the experiment on the disulfide bond [35,36], we choose the result of TOPCONS and set the residues of the first TM helix unit to be 62–82 in our model as shown Fig.2A. This, along with the N terminus, places Cys99 in the lumen. Subsequently, the second and third TM helix units in our model were chosen according to the prediction result by TOPCONS since they were in agreement with the experiment [6]. A most important factor in choosing the fourth and fifth TM units in our model is the location of the putative catalytic base, Lys218. According to a K218A mutation experiment [30], the results of which strongly suggest that Lys218 is involved in the deprotonation of vitamin K, Lys218 is located on the lumenal side of the ER membrane, consistent with earlier experiment [6]. Considering that the precursor VKD proteins to be post-translationally modified, and the Glu-interacting part of VKC must be located in the lumen [37,38], the surface of the membrane on the lumenal side is the proper location for Lys218 to be in contact with vitamin K. This is a key hypothesis. The prediction results of TOPCONS satisfy the requirement for the lumenal location of Lys218. For that reason, we have set the fourth and fifth TM helix units as residues 154–176 and 197–217, respectively. The location of Lys218, near the lumen surface of membrane, is consistent with both prediction programs. Thus, in our minimalist model, the N terminus of VKC is located in the cytoplasm to span the membrane with 5 TM helices, ending with the C terminus located in the lumen of the ER. It is not ruled out that there are two additional TM domains beyond residue 217, as suggested in Ref. [6]. TOPCONS predicts two more TM helices 252–272 and 293–313. Philius predicts TM domains at 253–272 and 283–305. The prediction for the sixth TM helix (252–272) in VKC by TOPCONS and a TM domain there by Philius gives a possible concurrence. If we include the TM helix (253–272) in building our VKC model, it is also required to add one more TM helix so that the required odd number (7 TM helices) is preserved. As a candidate for the seventh TM helix, we would chose 293–313. As shown in Fig.2, that TM helix supported by the experiment [6] is also predicted by TOPCONS. Thus, we could build an extended 7 TM helix model for VKC by adding two more TM helix (252–272, 293–313). Basically, this extended model would preserve the termini topology of 5TM VKC model. We will proceed, however, using the minimum number of TM domains that appear to define the active site since we have no mutational data as yet that substantiates a 7 TM choice. The disulfide bond (Cys99-Cys450) would be located on the lumenal side in either a 5 or 7 TM model.
Fig. 2.

The topology of VKC and VKOR surmised from experiments and computer prediction. Summarized are the prediction of TM helices units by experiments, TOPCONS and Philius Transmembrane Prediction. A: summary of the topology of transmembrane helices of VKC. B: summary of the topology of TM helices of VKOR.
3.3 Topology of VKOR based on prediction of TM helix units
For VKOR (163 residues), on the other hand, both programs give a consistent picture of the VKOR topology: 3 TM helices as shown in Fig.2.B. In addition, the two prediction results are consistent with experiment [39]. Unlike the complications of VKC, the active site participants of VKOR, Cys132 and Cys135, are predicted to be located inside the membrane. The N-terminus of VKOR starts from the lumen of the ER to span the membrane with 3 TM helices. Therefore, the C-terminus becomes located in the cytoplasm of the ER [39]. The topologies of VKC and VKOR by experiments and computer prediction are summarized in Fig.2. We assigned numbers (1–5) to each TM helix units of VKC. To avoid confusion, we assigned letters (A–C) to each TM unit of VKOR: the first helix unit A (9–29), the second helix unit B (104–124), and the third C (127–150). For the hetero-dimer with 8 TM helices (5⊕3: 5 TM helices from VKC and 3 TM helices from VKOR), and using 7Å as the estimated diameter for each helix backbone, we estimated the diameter of the “idealized pocket” is 12Å and the subtended area 110Å2. (Fig.3) The “pocket” in our hetero-dimer model must be able to accommodate the intermediate structures of the vitamin K and the parts of peptide chain of the VKD protein that is to be carboxylated.
Fig. 3.
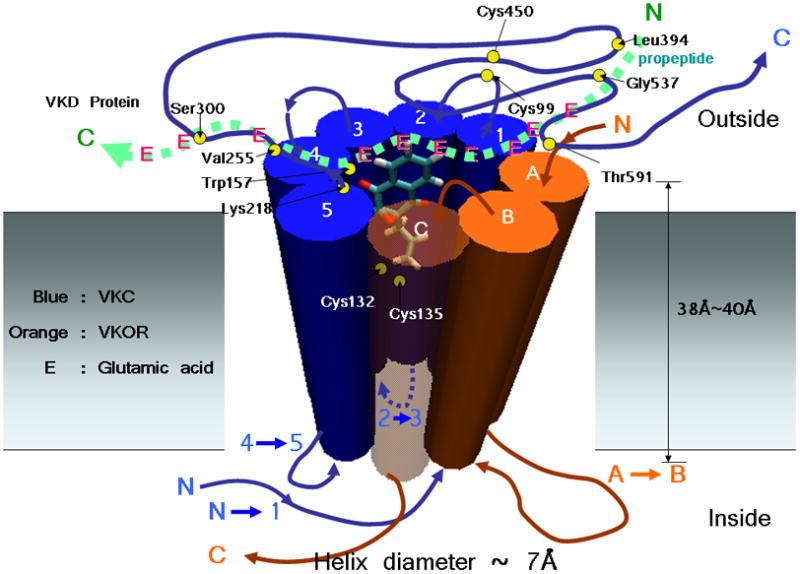
The model of hetero-dimeric form of VKC and VKOR. A possible relative position and orientation of the naphthoquinone unit of vitamin K epoxide is shown. Loop size and position are arbitrary. E represents Glu of a VKD, the substrate of VKC.
3.4 Quantum chemical study
According to the Dowd mechanism for the carboxylation of Glu, one of the intermediates of vitamin K is a glutamate carbanion, a key element in the “base amplification” process [11,27]. The quantum mechanically energy-optimized model structure for the transition state involving the carbanion is shown in Fig.4. This transition state is composed of three different moieties: vitamin K alkoxide-epoxide, a water molecule, and idealized glutamic acid (propionic acid). In Fig.4, we also indicated a putative backbone chain as a thick arrow. We surmise that the oxygen (oxy-3 denoted in Fig.1) of the vitamin K alkoxide-epoxide (geometry-optimized with the method/basis B3lyp/6-31+g(d,p)) will be located near Lys218 (VKC) to initiate the vitamin K cycle. Then, as a next step, we further surmise that another oxygen, oxy-2 denoted in Fig.1, will be located near the putative active site, Cys132 and Cys135, of VKOR for reduction of alkoxide-epoxide form to the quinone form during the vitamin K cycle. The near contact of the epoxide oxygen (oxy-2) with the two cysteines leaves the naphthoquinone unit of the vitamin K alkoxide-epoxide tilted in a clockwise manner as shown in Fig.3. The two constraints -(oxy-3:Lys218 and oxy-2:Cys132, Cys135)- require that the naphthoquinone unit of the vitamin K alkoxide-epoxide is almost immersed into membrane. Fig.5.A. shows a model of the hetero-dimeric form of VKC and VKOR with the vitamin K from top view. Fig.5.B gives a schematic view of a hetero-dimeric form VKC and VKOR with the intermediate structure of the Gla generation according to the Dowd mechanism, from a top view. According to a recent model quantum calculation [25], the chemical process for the generation of Gla in the vitamin K cycle favors aqueous environment rather than lipid environment. It is consistent with prior biochemical experiment that the deprotonation of the hydroquinone form of vitamin K by Lys218 and subsequent initiation of the carboxylation reaction of VKC takes place near the lumen side of the ER [30].
Fig. 4.

The fully energy-optimized structures of the epoxide-alkoxide intermediate in the proposed Dowd mechanism at the level of B3lyp/6–31+g(d,p). The intermediate is composed of three different moieties, vitamin K epoxide, a water molecule and propionic acid, a model for glutamic acid.
Fig.5.

A top view of a hetero-dimeric form of VKC and VKOR. A: Relative position and orientation of the naphthoquinone unit of vitamin K epoxide. For clarity, loops are not shown in this schematic picture. The diameter of the “pocket” is estimated to be 12Å and the area 110Å2. B: Relative position and orientation of the intermediate from Fig.4 with a respect to the hetero-dimer.
3.5 Prior biochemical mutation experiments
The hetero-dimer model concept can be tested by examining the growing body of mutational data. An interesting case is the VKC hydrophobic residue, Trp157. According to our hetero-dimer model, Trp157 is near Lys218 (VKC) at the surface of membrane in the lumen. With this placement, Trp157 could be involved in the vitamin K catalysis rather than binding of the substrate. This is consistent with W157R mutation experiment [40], in which the enzyme activity of VKC is greatly reduced to 8%–10% of the wild type. However, a patient with the mutation of W157R did not respond to vitamin K supplementation, which indicates, according to Ref.[40] that the mutation does not affect the binding of VKD substrates [40]. On the other hand, His160, a possible candidate for the catalytic base for the deprotonation of the hydroquinone form of vitamin K [30], may not be as near to oxy-3 as Lys218 since the H160A mutation shows the activity of wild-type VKOR [30]. Another residue, Thr591, putatively involved in both carboxylation and epoxidation activities [40], was predicted to be located in the lumen of the ER by both TM Prediction programs. Likewise, a L394R mutation experiment on VKC suggests that Leu394 is involved in propeptide binding [41], presumably at the lumen, and it thus consistent with our model. Recently, mutations in VKC have been reported to be linked to pseudoxanthoma elasticum (PXE), an autosomal recessive multi-system disorder with dystrophic mineralization of soft connective tissues [42]. Patients with G537Y and G558R mutations in VKC showed PXE-like clinical features [42]. Mutation experiments at these sites (predicted in the lumen in our model) in VKC decreased the affinity of VKC for its substrates [42]. Similarly, mutations S300F and V255M (also located in the lumen in the model) showed PXE-like clinical features. An analysis of in vitro VKC enzyme assay experiments on these mutations suggests that these mutations affect substrate binding [43]. The locations of the biochemically interesting sites are shown in our hetero-dimer model in Fig.3.
3.6 Warfarin
Warfarin is a well-known anticoagulant, acting as a vitamin K antagonist [44,45]. It is generally believed that warfarin inactivates VKOR by blocking the formation of the quinone forms of vitamin K through non-competitive inhibition [24]. Although warfarin has been widely used as a medication for preventing thrombosis, the molecular basis of the inhibition mechanism of VKOR is still elusive. There are two enantiomeric forms, R- and S- warfarin (see Supplemental Materials). S-warfarin is the biologically effective enantiomer [14]. We geometry-optimized S-warfarin with the method/basis of B3lyp/6-31+g(d,p) similar to that performed for vitamin K transients [25]. The putative binding motif of warfarin to VKOR is thought, based on a body of data summarized in Ref.[46], to be at residues 138–140 (TYA), which are positioned inside the membrane, three residues removed from the putative vitamin K active site, cyteines and toward the center of the membrane in our model (See Fig. S3 in Supporting Material). The displacement of the warfarin binding site away from the vitamin K binding site (by at least one turn of the TM helix) is consistent with the observation that warfarin is a non-competitive inhibitor [24]. The fully extended phenyl group of S-warfarin may interact with the phenyl fragment of Tyr139 of VKOR in a stacking configuration [47]. It is interesting that there is structural evidence [47] that in the binding of S-warfarin to P4502C9 (CYP2C9), the phenyl group of S-warfarin binds to Phe476, one of binding pocket residues of CYP2C9, with a pi-pi stacking interaction. (see Supporting Material)
The mechanism for the reduction of the epoxide to the hydroquinone form of vitamin K in the vitamin K cycle is not well understood [14]. A possibility for the reduction to the hydroquinone could involve a flip-flop action of lipids in the ER membrane. The flip-flop of lipids is thought to take place through either a pore-mediated process [48] or of enzyme (flippase or floppase)-mediated process [49]. In this way, the naphthoquinone unit of the quinone form of vitamin K could traverse a section of the pore by being coupled to the flip-flop of lipids in the ER membrane and thereby the naphthoquinone unit could be reduced to the hydroquinone form by reducing agents in or near the cytoplasm of the ER. After reduction, the reduced form could cycle back via flip-flop to the lumen of the ER for the initiation phase of the vitamin K cycle. The binding of S-warfarin to VKOR in the presence of vitamin K could modify, i.e. reduce the rate of flip-flop of lipid membrane, ultimately leading to the decrease of the amount of the vitamin K hydroquinone form.
An interesting recent development has been the discovery of warfarin-resistant VKOR mutations in humans. Seven of these have been tabulated [50]: V29L, D36V, V45A, R58G, V66M, L128R. Our co-localization hetero-dimer model would place all of these residues, except V29 and L128, in the cytoplasm. A structural model for human VKOR, based on the X-ray crystal structure of a bacterial analog of VKOR [51], however, would place all of these residues in the ER lumen (See Supporting Material). This latter model has 4 VKOR TM domains with the N terminus in the cytoplasm; our model has 3 VKOR TM domains with the N terminus located in the lumen. Both models predict essentially the same final two TM domains (direction, sequence). Given that vitamin K supplementation is a standard treatment for warfarin poisoning [52,53], that warfarin-resistant mutations exist [50], that warfarin inhibition of the vitamin K cycle is non-competitive [24], and that lipid-protein charge interactions can control the topology of TM domain [54,55], we are led to suggest that the number of TM sequences in the ER membrane of residues 1–90 of VKOR may be changed from 1 to 2; or some additional partial helix as seen in the X-ray structure of the bacterial VKOR-like protein [34], so that additional TM-TM interactions are created which modulate the warfarin binding site.
4. Conclusions
We have built a hetero-dimer model of VKC and VKOR in the presence of vitamin K based on prior biochemical experiments, TM helix unit prediction methods, and quantum chemical study. In our hetero-dimer of VKC and VKOR model, Lys218 and Trp157 are located near the oxy-3 of vitamin K at the lumen side of the ER membrane. Two cysteines (Cys132 and Cys135) of VKOR are located near oxy-2 inside the membrane. We propose a “5⊕3” TM helix hetero-dimer model with a relatively open core for the mutual reactions of VKC and VKOR. However, an extended 7⊕3 model for VKC/VKOR could also be considered while still retaining the essential idea (limited diffusion of vitamin K and its intermediates) of the hetero-dimer model. Obviously, the TM helices do not have to be as tightly packed as in our “idealized” model, the helices could be at various angles to one another. The interaction of VKC or VKOR with the lipid molecules in the membrane environment will affect the dynamic arrangement of the TM helices, which will modulate the effective “pocket” volume. Thus, it appears that the dynamical “pocket” of the helices for the minimalistic 5⊕3 hetero-dimer model, which co-localizes VKC and VKOR, can provide sufficient space for the reactive glutamic acid side chains as well as water, vitamin K transients and/or warfarin.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (HL-06350) and the National Science Foundation (FRG DMR 084549). We thank a reviewer for very insightful suggestion.
Footnotes
Highlights. In this study, based on the biochemical experiments of VKC and VKOR, we proposed a hetero-dimeric form of VKC and VKOR to explain the efficient oxidation and reduction of vitamin K during the vitamin K cycle, which was compatible with various biochemical findings as well as our quantum chemical study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wright DJ, Morris DP, Stafford DW. Vitamin K-dependent γ-glutamyl carboxylase. In: High KA, Roberts HR, editors. Molecular basis of thrombosis and hemostasis. Marcel Dekker; New York: 1995. pp. 309–329. [Google Scholar]
- 2.Furie B, Furie BC. Molecular basis of vitamin K-dependent γ-carboxylation. Blood. 1990;75:1753–1762. [PubMed] [Google Scholar]
- 3.Suttie JW. Vitamin K-dependent carboxylase. Annu Rev Biochem. 1985;54:459–477. doi: 10.1146/annurev.bi.54.070185.002331. [DOI] [PubMed] [Google Scholar]
- 4.Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 5.Roberts HR, Tabares AH. Overview of the coagulation reaction. In: High KA, Roberts HR, editors. Molecular basis of thrombosis and hemostasis. Marcel Dekker; New York: 1995. pp. 35–50. [Google Scholar]
- 6.Tie J, Wu SM, Jin D, Nicchitta CV, Stafford DW. A topological study of the human gamma-glutamyl carboxylase. Blood. 2000;96:973–978. [PubMed] [Google Scholar]
- 7.Wu SM, Morris DP, Stafford DW. Identification and purification to near homogeneity of the vitamin K-dependent carboxylase. Proc Natl Acad Sci. 1991;88:2236–2240. doi: 10.1073/pnas.88.6.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu SM, Cheung WF, Frazier D, Stafford DW. Cloning and expression of the cDNA for human gamma-glutamyl carboxylase. Science. 1991;254:1634–1636. doi: 10.1126/science.1749935. [DOI] [PubMed] [Google Scholar]
- 9.Li T, Chang CY, Jin DY, Lin PJ, Khvorova A, Stafford DW. Identification of the gene for vitamin K epoxide reductase. Nature. 2004;427:541–544. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 10.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hörtnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EGD, Müller CR, Strom TM, Oldenburg J. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–541. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 11.Dowd P, Hershline R, Ham SW, Naganathan S. Vitamin K and energy transduction: a base strength amplification mechanism. Science. 1995;269:1684–1691. doi: 10.1126/science.7569894. [DOI] [PubMed] [Google Scholar]
- 12.Ham SW, Dowd P. On the mechanism of action of vitamin K. A new nonenzyme model. J Am Chem Soc. 1990;112:1160–1161. [Google Scholar]
- 13.Dowd P, Ham SW, Geib SJ. Mechanism of action of vitamin K. J Am Chem Soc. 1991;113:7734–7743. [Google Scholar]
- 14.Stafford DW. The vitamin K cycle. J Thromb Haemst. 2005;3:1873–1878. doi: 10.1111/j.1538-7836.2005.01419.x. [DOI] [PubMed] [Google Scholar]
- 15.Silverman RB. Chemical model studies for the mechanism of vitamin K epoxide reductase. Am Chem. 1981;103:5939–5941. [Google Scholar]
- 16.Davis CH, Deerfield D, II, Wymore T, Stafford DW, Pedersen LG. A quantum chemical study of the mechanism of action of Vitamin K epoxide reductase (VKOR) II: transition states. J Mol Graphics Modell. 2007;26:401–408. doi: 10.1016/j.jmgm.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Suttie JW. Vitamin K in Health and Disease. 1. CRC Press; Boca Raton: 2009. [Google Scholar]
- 18.Wallin R, Hutson SM, Cain D, Sweatt A, Sane DC. A molecular mechanism for genetic warfarin resistance in the rat. FASEB. 2001;15:2542–2544. doi: 10.1096/fj.01-0337fje. [DOI] [PubMed] [Google Scholar]
- 19.Wajih N, Sane DC, Hutson SM, Wallin R. The inhibitory effect of calumenin on the vitamin K-dependent γ-carboxylation system. J Biol Chem. 2004;279:25276–25283. doi: 10.1074/jbc.M401645200. [DOI] [PubMed] [Google Scholar]
- 20.Wallin R, Hutson SM. Warfarin and the vitamin K-dependent γ-carboxylation system. Trends Mol Med. 2004;10:299–302. doi: 10.1016/j.molmed.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Oldham ML, Khare D, Quinocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transport. Nature. 2007;450:515–522. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- 22.Khare D, Oldham ML, Orelle C, Davidson AL, Chen J. Alternating access in maltose transporter mediated by rigid-body rotations. Mol Cell. 2009;33:528–536. doi: 10.1016/j.molcel.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abramson J, Ristama S, Larsson G, Jasaitis A, Svensson-Ek M, Laakkonen L, Puustinen A, Iwata S, Wikström M. The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nature Struct Biol. 2000;7:910–917. doi: 10.1038/82824. [DOI] [PubMed] [Google Scholar]
- 24.Lasseur R, Longin-Sauvageon C, Videmann B, Billeret M, Berny P, Benoit E. Warfarin resistance in a french strain of rats. J Biochem Mol Toxicol. 2005;19:379–385. doi: 10.1002/jbt.20104. [DOI] [PubMed] [Google Scholar]
- 25.Wu S, Liu S, Davis CH, Stafford DW, Pedersen LG. Quantum chemical study of the mechanism of action of vitamin K carboxylase in solvent. Int J Quant Chem. 2010;110:2744–2751. doi: 10.1002/qua.22740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian03, revision C. 02. Gaussian, Inc; Pittsburgh, PA: 2004. [Google Scholar]
- 27.Davis CH, Deerfield D, II, Stafford DW, Pedersen LG. Quantum chemical study of the mechanism of action of vitamin K carboxylase (VKC). IV. Intermediates and transition states. J Phys Chem A. 2007;111:7257–7261. doi: 10.1021/jp068564y. [DOI] [PubMed] [Google Scholar]
- 28.Luecke H, Schobert B, Lanyi JK, Spudich EN, Spudich JL. Crystal structure of sensory rhodopsin II at 2.4 angstroms: insights into color tuning and transducer interaction. Science. 2001;293:1499–1503. doi: 10.1126/science.1062977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 30.Rishavy MA, Hallgren KW, Yakubemko AV, Shtofman RL, Runge KW, Berkner KL. Brønsted analysis reveals Lys218 as the carboxylase active site base that deprotonates vitamin K hydroquinone to initiate vitamin K-dependent protein carboxylation. Biochemistry. 2006;45:13239–13248. doi: 10.1021/bi0609523. [DOI] [PubMed] [Google Scholar]
- 31.Wajih N, Sane DC, Hutson SM, Wallin R. Engineering of a recombinant vitamin K-dependent γ-carboxylation system with enhanced γ-carboxyglutamic acid forming capacity: evidence for a functional CXXC redox center in the system. J Biol Chem. 2005;280:10540–10547. doi: 10.1074/jbc.M413982200. [DOI] [PubMed] [Google Scholar]
- 32.Reynolds SM, Käll L, Riffle ME, Bilmes JA, Noble WS. Transmembrane topology and signal peptide prediction using dynamic Bayesian networks. PLoS Comput Biol. 2008;4:1–14. doi: 10.1371/journal.pcbi.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernsel A, Viklund H, Hennerdal A, Elofsson A. TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 2009;37:1–4. doi: 10.1093/nar/gkp363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W, Schulman S, Dutton RJ, Boyd D, Beckwith J, Rapoport TA. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature. 2010;463:507–513. doi: 10.1038/nature08720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tie J, Mutucumarana VP, Heijne GV, Straight DL, Carrick KL, Pope RM, Stafford DW. Determination of disulfide bond assignment of human vitamin K-dependent γ-glutamyl carboxylase by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Biol Chem. 2003;278:45468–45475. doi: 10.1074/jbc.M309164200. [DOI] [PubMed] [Google Scholar]
- 36.Tie J, Zheng MY, Hsiao KLN, Perera L, Stafford DW, Straight DL. Transmembrane domain interactions and residue proline 378 are essential for proper structure, especially disulfide bond formation, in the human vitamin K-dependent γ-glutamyl carboxylase. Biochemistry. 2008;47:6301–6310. doi: 10.1021/bi800235r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlisle TL, Suttie JW. Vitamin K dependent carboxylase: subcellular location of the carboxylase and enzymes involved in vitamin K metabolism in rat liver. Biochemistry. 1980;19:1161–1167. doi: 10.1021/bi00547a019. [DOI] [PubMed] [Google Scholar]
- 38.Mutucumarana VP, Acher F, Straight DL, Jin DY, Stafford DW. A conserved region of human vitamin K-dependent carboxylase between residues 393 and 404 is important for its interaction with the glutamate substrate. J Biol Chem. 2003;278:46488–46493. doi: 10.1074/jbc.M307707200. [DOI] [PubMed] [Google Scholar]
- 39.Tie J, Nicchitta C, Heijne GV, Stafford DW. Membrane topology mapping of vitamin K epoxide reductase by in vitro translation/cotranslocation. J Biol Chem. 2005;280:16410–16416. doi: 10.1074/jbc.M500765200. [DOI] [PubMed] [Google Scholar]
- 40.Darghouth D, Hallgren KW, Shtofman RL, Mrad A, Gharbi Y, Maherzi A, Kastally R, LeRicousse S, Berkner KL, Rosa JP. Compound heterozygosity of novel missense mutations in the gamma-glutamyl-carboxylase gene causes hereditary combined vitamin K-dependent coagulation factor deficiency. Blood. 2006;108:1925–1931. doi: 10.1182/blood-2005-12-010660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mutucumarana VP, Stafford DW, Stanley TB, Jin DY, Solera J, Brenner B, Azerd R, Wu SM. Expression and characterization of the naturally occurring mutation L394R in human γ-glutamyl carboxylase. J Biol Chem. 2000;275:32572–32577. doi: 10.1074/jbc.M006808200. [DOI] [PubMed] [Google Scholar]
- 42.Vanakker OM, Martin L, Gheduzzi D, Leroy BP, Loeys BL, Guerci VI, Matthis D, Terry SF, Coucke PJ, Pasquali-Ronchetti I, De Paepe A. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J Invest Dermatol. 2007;27:581–587. doi: 10.1038/sj.jid.5700610. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Grange DK, Armstrong NL, Whelan AJ, Hurley MY, Rishavy MA, Hallgren KW, Berkner KL, Schurgers LJ, Jiang Q, Uitto J. Mutations in the GGCX and ABCC6 genes in a family with pseudoxanthoma elasticum-like phenotypes. J Invest Dermatol. 2009;129:553–563. doi: 10.1038/jid.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stahmann MA, Huebner CF, Link KP. Studies on the hemorrhagic sweet clover disease: identification and synthesis of the hemorrhagic agent. J Biol Chem. 1941;138:513–527. [Google Scholar]
- 45.Sadler JE. Medicine: K is for koagulation. Nature. 2004;427:493–494. doi: 10.1038/427493a. [DOI] [PubMed] [Google Scholar]
- 46.Schwarz R, Seibel PN, Rahman S, Schoen C, Huenerberg M, Müller-Reibel C, Dandekar C, Karchin R, Shultz J, Thobias M. Detecting species-site dependencies in large multiple sequence alignments. Nucleic Acids Res. 2009:1–10. doi: 10.1093/nar/gkp634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams PA, Cosme J, Ward A, Angove HC, Vinković DM, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 48.Matsuzaki K, Murase O, Fujii N, Miyajima K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. 1996;35:11361–11368. doi: 10.1021/bi960016v. [DOI] [PubMed] [Google Scholar]
- 49.Sanyal S, Menon AK. Flipping Lipids: Why an “What's reason for?”. Chem Biol. 2009;4:895–909. doi: 10.1021/cb900163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilms EB, Touw DJ, Conemans JMH, Veldkamp R, Hermans M. A new VKORC1 allelic variant (p.Trp59Arg) in a patient with partial resistance to acenocoumarol and phenprocoumon. J Thromb Haemst. 2008;6:1224–1226. doi: 10.1111/j.1538-7836.2008.02975.x. [DOI] [PubMed] [Google Scholar]
- 51.Schulman S, Wang B, Li W, Rapoport TA. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc Nat Acad Sci. 2010;107:15027–15032. doi: 10.1073/pnas.1009972107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Meer J, Hemker HC, Loeliger EA. Pharmacological aspects of vitamin K1: a clinical and experimental study in man. Thromb Diath Haemorrh Suppl. 29(1968):1–96. [PubMed] [Google Scholar]
- 53.Park BK, Scott AK, Wilson AC, Haynes BP, Breckenridge AM. Plasma disposition of vitamin K1 in relation to anticoagulant poisoning. 1984;18:652–662. doi: 10.1111/j.1365-2125.1984.tb02526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bogdanov M, Xie J, Dowhan W. Lipid-protein interactions drive membrane protein topogenesis in accordance with the positive inside rule. J Biol Chem. 2009;284:9637–9641. doi: 10.1074/jbc.R800081200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dowhan W, Bogdanov M. Lipid-dependent membrane protein topogenesis. Annu Rev Biochem. 2009;78:515–540. doi: 10.1146/annurev.biochem.77.060806.091251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.