

Summary
Objective
Resistin causes insulin resistance and diabetes in mice whereas in humans it is linked to inflammation and atherosclerosis. Few human genetic studies of resistin in inflammation and atherosclerosis have been performed. We hypothesized that the −420C>G putative gain-of-function resistin variant would be associated with inflammatory markers and atherosclerosis but not with metabolic syndrome or adipokines in humans.
Design and methods
We examined the association of three resistin polymorphisms, −852A>G, −420C>G and +157C>T, and related haplotypes with plasma resistin, cytokines, C-reactive protein (CRP), adipokines, plasma lipoproteins, metabolic syndrome and coronary artery calcification (CAC) in nondiabetic Caucasians (n = 851).
Results
Resistin levels were higher, dose-dependently, with the −420G allele (CC 5·9 ± 2·7 ng/ml, GC 6·5 ± 4·0 ng/ml and GG 7·2 ± 4·8 ng/ml, trend P = 0·04) after age and gender adjustment [fold higher for GC + GG vs. CC; 1·07 (1·00–1·15), P < 0·05)]. The −852A>G single nucleotide polymorphism (SNP) was associated with higher soluble tumour necrosis factor-receptor 2 (sol-TNFR2) levels in fully adjusted models [1·06 (95% CI 1·01–1·11), P = 0·01)]. The estimated resistin haplotype (GGT) was associated with sol-TNFR2 (P = 0·04) and the AGT haplotype was related to CRP (P = 0·04) in the fully adjusted models. Resistin SNPs and haplotypes were not associated with body mass index (BMI), fasting glucose, insulin resistance, metabolic syndrome, adipokines or CAC scores.
Conclusions
Despite modest associations with plasma resistin and inflammatory biomarkers, resistin 5′ variants were not associated with metabolic parameters or coronary calcification. This suggests that resistin is an inflammatory cytokine in humans but has little influence on adiposity, metabolic syndrome or atherosclerosis.
Introduction
Resistin was originally described as an adipose-derived protein in rodents that links obesity to insulin resistance.1,2 In mice, resistin expression in adipose tissue is increased in both diet-induced and genetic models of obesity and is related to measures of insulin resistance.3–5 In humans, however, the relationship between resistin and insulin resistance is disputed, with some studies suggesting a positive association6,7 and others finding no relationship.8–10 This may relate to species differences in gene homology and tissue expression. Unlike rodents, where resistin is adipocyte derived, resistin expression is myeloid restricted in humans11,12 and has an unclear relationship with adiposity, insulin resistance and metabolic syndrome, in lean nondiabetic subjects.13 Consistent with a myeloid origin in humans, recent studies suggest a positive relationship with chronic inflammatory states including rheumatoid arthritis,14 chronic kidney disease15 and atherosclerotic cardiovascular disease (CVD), although the strength and significance of this latter association remains uncertain.16
Up to two-thirds of plasma resistin variation may be attributable to heritable influences.17 Several studies, with conflicting results, have examined the relationship of resistin gene variation with insulin resistance, obesity and glucose homeostasis.18–21 Among these, a putative gain-of-function promoter single nucleotide polymorphism (SNP), −420C>G, has been associated with increased resistin levels.22,23 Little is known of the association of resistin gene variation with measures of inflammation and atherosclerotic CVD.21 We hypothesized that the −420C>G promoter SNP, and related haplotypes, would be related to inflammatory biomarkers and subclinical coronary atherosclerosis as measured by coronary artery calcification (CAC), but not with adiposity, markers of insulin resistance or plasma lipids in a nondiabetic Caucasian cohort.
Subjects and methods
Study subjects
The Study of Inherited Risk of Coronary Atherosclerosis (SIRCA) is a cross-sectional study of factors associated with CAC in a community-based sample of asymptomatic subjects and their families.24 Subjects were healthy adults, aged 30–75 years, with a family history of premature coronary artery disease (CAD) and were excluded if they had diabetes mellitus, serum creatinine > 3·0 mg/ml or if screening revealed evidence of CAD. The University of Pennsylvania Institutional Review Board approved the study protocol. Informed consent was given by all subjects. This work focused on nondiabetic, unrelated SIRCA Caucasian subjects (N = 851).
Evaluated parameters
Study subjects were evaluated at the General Clinical Research Center (GCRC) at the University of Pennsylvania Medical Center and procedures included a questionnaire, physical examination, electrocardiogram (ECG), electron beam tomography (EBT) and blood collection as described previously.24 Fasting plasma total and high density lipoprotein cholesterol (HDL-C), and triglyceride and glucose levels were measured enzymatically on a Cobas Fara II (Roche Diagnostic Systems Inc., NJ). Low density lipoprotein cholesterol (LDL-C) was calculated using the Friedewald formula. As described previously,24 plasma levels of resistin, adiponectin and leptin (Linco, St Charles, MO), as well as interleukin (IL)-6 and soluble tumour necrosis factor receptor 2 (sol-TNFR2) (R&D Systems, Minneapolis, MN) were assayed, in duplicate, by enzyme-linked immunosorbent assays (ELISAs). C-reactive protein (CRP) levels were assayed using an ultra high-sensitivity latex turbidimetric immunoassay (Wako Ltd, Osaka Japan).24 The intra- and interassay coefficients of variation (CVs) for pooled human plasma were 5·7% and 9·9% for adiponectin, 5·5% and 12·4% for leptin, 4·6% and 4·3% for resistin, 8·7% and 10·9% for IL-6, 5·3% and 12·1% for sol-TNFR2, and 8·0% and 8·3% for CRP, respectively. CAC scores, measured at EBT (Imatron, San Francisco, CA), were determined by the Agatston method as described previously.24 Framingham risk scores were calculated as described by Wilson et al.25 Subjects were classified as having the metabolic syndrome using the revised National Cholesterol Education Program (NCEP) definition,26 which encompasses the five elements of waist circumference, triglycerides, HDL-C, blood pressures, and glucose using the 100 mg/dl as the cut-point for abnormal fasting glucose. The homeostasis model assessment [HOMA-IR index = fasting glucose (mmol/l) × fasting insulin (μU/ml)/22·5]27 was used as a measure of insulin resistance.
Resistin SNPs were chosen based on prior evidence for association with resistin levels (−420C>G, rs1862513) and to provide additional linkage disequilibrium coverage of the 5′ region (−852A>G, rs3760678, and +157C>T, rs3219177). Genotyping was performed at the Broad Institute of the Massachusetts Institute of Technology by allele-specific multiplex primer extension of polymerase chain reaction (PCR)-amplified products with detection by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectroscopy using the Sequenom MassARRAY™ platform. Genotyping assays were designed using the Sequenom Assay Design program utilizing hME chemistry in pools of up to six SNPs. Genotype call rates were 94·8%, 99·3% and 97·8% for −852A>G, −420C>G and +157C>T, respectively, and each SNP was in Hardy–Weinberg equilibrium (P = 0·32, 0·96 and 0·16, respectively). Pairwise linkage disequilibrium (LD) between SNPs was estimated by Haploview Version 3·32 (Broad Institute http://www.broad.mit.edu/mpg/haploview).
Statistical analysis
Power estimates (Genetic Power Calculator, http://pngu.mgh.harvard.edu/~purcell/gpc/28) suggest that a SIRCA sample size of 851 provided > 90% power, at alpha 0·01, to detect a common SNP allele [minor allele frequency (MAF) = 0·3] effect accounting for 2% of the variance in a quantitative trait (e.g. plasma resistin levels or inflammatory and metabolic biomarkers).
Statistical analyses of SNPs were performed using Stata 9·0 software (Stata Corp., College Station, TX) and R 2·4·1 software (http://www.r-project.org/). In crude analysis of continuous traits, we performed a nonparametric test for trend across three-level genotypes ordered by the number of mutant alleles using Cuzick’s Wilcoxon-type test (nptrend command in Stata). Multivariable resistin SNP associations with inflammatory, metabolic and CAC phenotypes were tested using linear and logistic regression. Models were adjusted for age and gender and further for family history of premature CAD, body mass index (BMI), exercise (none vs. any), tobacco (current vs. never or ex-smokers) and alcohol use (drinks per week), use of medications (aspirin, niacin and statins) and finally, when not related to outcome, the metabolic syndrome and Framingham risk scores. There was no interaction by gender, assessed by the likelihood-ratio test in fully adjusted models, for the association of resistin genetic data with any phenotype. Results are presented for resistin alleles assuming a dominant model. For analysis of each SNP, all available genotype data were used; findings were similar when analyses were restricted to the subgroup with complete data for all three SNPs.
The package hapassoc implemented in the R (http://cran.r-project.org) was used to evaluate the association between traits and haplotypes adjusting for covariates. The procedure uses the Expectation Maximization (EM) algorithm to address haplotype ambiguity and perform maximum likelihood inference in generalized linear models. The EM algorithm provides an iterative approach to compute maximum likelihood estimates in problems involving missing data. In the context of haplotype analysis, the genotypes with linkage phase information constitute the missing data while SNP genotypes, phenotypes and covariates are observed. The E-step of the EM algorithm computes the conditional expectation of the complete data log-likelihood given the observed data and current estimates of the unknown parameters. The M-step maximizes the resulting conditional. Details of the method and its implementation can be found in Burkett and Graham.29 We used haplotypes of > 5% frequency, relative to the most common haplotype against which others were compared. A main effect model including indicator variables for haplotypes, age and gender (and additional covariates for fully adjusted model) was fit. Associations with CAC were evaluated using a linear regression model on log(CAC + 1). For all other continuous phenotypes, linear regression models were fit either on the original scale or the log scale to satisfy the assumption of normality.
Results
Characteristics of the SIRCA sample
As described previously,24 the SIRCA sample has low-average CVD risk factors (Table 1). Despite low Framingham risk, there were higher than expected age- and gender-based percentile CAC scores, consistent with recruitment based on an enriched family history of CVD. SNP allele frequencies and pairwise LD are shown in Table 2. Haplotype ACC was the most common (65·4%) whereas AGC, AGT and GGT haplotypes were estimated to each have frequencies of 9·1%, 10·5% and 10·2%, respectively. All other haplotypes had a frequency of < 5%.
Table 1.
Characteristics of the study sample
| Characteristic | Men (n = 458) | Women (n = 393) |
|---|---|---|
| Age (years) | 46 (41–51) | 51 (44–57) |
| Blood pressure (mmHg) | ||
| Systolic | 126 (119–136) | 123 (111–135) |
| Diastolic | 79 (74–85) | 75 (68–82) |
| Smoking (%) | 11·8 | 10·8 |
| Body mass index (kg/m2) | 27·5 (25·2–30·3) | 25·5 (22·55–29·9) |
| Total cholesterol (mg/dl) | 199·5 (175–224) | 211 (183–235) |
| HDL cholesterol (mg/dl) | 42 (36–50) | 58 (46–68) |
| Triglycerides (mg/dl) | 125· ·5 (91–179) | 114 (80–148) |
| Waist circumference (cm) | 95·3 (88·9–104·1) | 81·3 (73·7–91·4) |
| Metabolic syndrome* (%) | 35 | 25 |
| Resistin (ng/ml) | 5·2 (3·87–6·87) | 5·8 (4·42–7·84) |
| C-reactive protein (mg/dl) | 1·1 (0·5–2) | 1·4 (0·6–3·7) |
| Soluble TNF receptor 2 (ng/ml) | 1·64 (1·41–1·95) | 1·68 (1·37–2·0) |
| Interleukin-6 (pg/ml) | 1·24 (0·79–1·87) | 1·29 (0·82–2·1) |
| Leptin (ng/ml) | 5·59 (3·28–8·75) | 16·20 (9·76–27·21) |
| Adiponectin (μg/ml) | 13·09 (8·92–17·78) | 21·16 (14·95–28·40) |
| Coronary artery calcification (CAC) | ||
| Mean score (± SD) | 128·14 (335·46) | 43·1 (136·23) |
| Median (IQR) | 7 (1–80) | 1 (0–14) |
| > 70th percentile (%) | 40·1 | 39·1 |
HDL, high density lipoprotein; TNF, tumour necrosis factor; IQR, interquartile range.
Values are given as median (IQR) unless stated otherwise.
Metabolic syndrome according to the revised National Cholesterol Education Program (NCEP) definition using glucose cut-point of 100 mg/dl.
Table 2.
Genotype frequencies and linkage disequilibrium (LD) of SNPs in the resistin gene
| R2 (D′) | SNP | Frequency (%) | |||||
|---|---|---|---|---|---|---|---|
| 0·25(0·67) |
|
0·19(0·75) |
|
−852A>G | AA (75·7) |
AG (22·0) |
GG (2·3) |
| 0·52(0·92) |
|
−420C>G | CC (46·1) |
CG (44·1) |
GG (9·8) |
||
| +157C>T | CC (61·3) |
CT (33·1) |
TT (5·6) |
||||
Each SNP was in Hardy–Weinberg equilibrium (P = 0·32, 0·96 and 0·16 for −852, −420 and +157, respectively). Pairwise LD between SNPs was estimated using Haploview Version 3·32 (Broad Institute http://www.broad.mit.edu/mpg/haploview).
Association of resistin SNPs with inflammatory and metabolic variables and CAC
Overall, levels of resistin and all inflammatory markers, but not metabolic parameters, tended to be higher in subjects with variant resistin alleles. In crude analysis, plasma resistin levels were dose-dependently higher with the G allele of the −420 polymorphism (CC 5·9 ± 2·7 ng/ml, GC 6·5 ± 4·0 ng/ml and GG 7·2 ± 4·8 ng/ml, trend P = 0·04) (Fig. 1). The T allele of SNP +157, which is in moderate LD with −420C>G (r2 = 0·52 in HapMap Caucasians; http://www.hapmap.org), also showed borderline statistical association with higher plasma resistin. In addition, the −852G allele was related to higher sol-TNFR2 (AA 1710 ± 518 pg/ml, AG 1837 ± 570 pg/ml and GG 1827 ± 563 pg/ml, trend P = 0·02). After adjusting for age and gender, and additionally for BMI, Framingham risk score and NCEP metabolic syndrome status, the −420G allele was associated with higher plasma resistin levels (P = 0·05) and the −852G allele with higher sol-TNFR2 levels (P = 0·015) (Table 3). The association of −852G with sol-TNFR2 was not attenuated even after further adjusting for plasma resistin [fold increase 1·06 (95% CI 1·01 to 1·11), P = 0·016]. There was no evidence of association between any resistin SNP and BMI, metabolic syndrome status, plasma lipids or circulating levels of leptin or adiponectin (Table 3). Resistin SNPs also showed no evidence of association with fasting glucose or HOMA-IR. For example, in age- and gender-adjusted models, resistin SNPs did not relate to fasting glucose [absolute change and 95% CI in mg/dl of −0·6 (−2·84 to 1·64), −1·69 (−3·54 to 0·15) and −1·30 (−3·19 to 0·60)] or HOMA-IR [fold change 0·99 (0·88 to 1·11), 0·93 (0·84 to 1·03) and 0·92 (0·82 to 1·02)] for −852A>G, −420C>G and +157C>T, respectively. Finally, CAC scores did not vary across resistin genotypes in unadjusted or adjusted models (Table 3). There was no evidence of interaction between resistin SNPs and resistin levels in the association with inflammatory markers, lipids and CAC (P interaction > 0·3 for all interactions). However, our study sample may be underpowered to detect modest interaction effects.
Fig. 1.
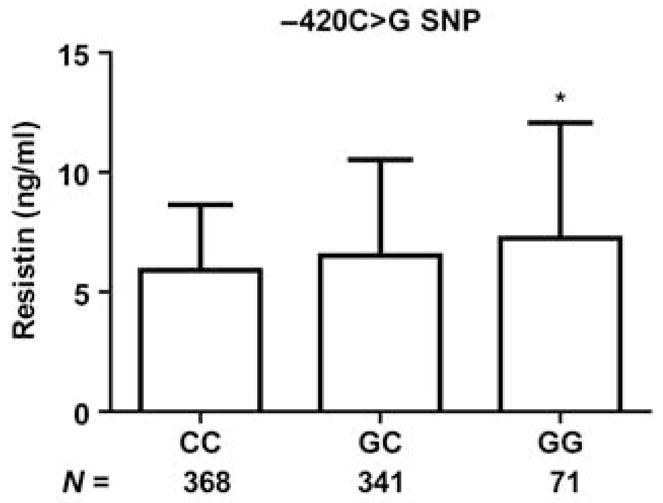
Variation in resistin levels by −420 genotypes. Mean ± SD values of resistin levels (ng/ml) are shown for −420C>G: *P for trend = 0·04; P = 0·009 and P = 0·02 for GG or GC, respectively, vs. CC.
Table 3.
Association of resistin SNPs with inflammatory and metabolic variables in age- and gender-adjusted and fully adjusted* models
| −852A>G | −420C>G | +157C>T | |
|---|---|---|---|
| Age-and gender-adjusted | |||
| Inflammatory markers | |||
| Resistin | 1·03 (0·95–1·12) | 1·07 (1·00–1·15) | 1·06 (0·99–1·14) |
| Soluble TNF receptor 2 | 1·07 (1·02–1·12) | 1·02 (0·98–1·06) | 1·02 (0·98–1·06) |
| Interleukin-6 | 1·04 (0·92–1·17) | 1·03 (0·93–1·14) | 0·98 (0·88–1·09) |
| C-reactive protein | 1·08 (0·89–1·31) | 1·12 (0·95–1·32) | 1·10 (0·93–1·29) |
| Metabolic variables | |||
| Body mass index | 0·98 (0·96–1·01) | 0·99 (0·97–1·02) | 0·99 (0·97–1·02) |
| Triglycerides | 1·02 (0·94–1·11) | 0·99 (0·92–1·06) | 1·00 (0·93–1·08) |
| HDL cholesterol | 0·97 (0·92–1·01) | 1·01 (0·97–1·05) | 0·98 (0·94–1·02) |
| Leptin | 0·96 (0·85–1·08) | 0·98 (0·88–1·08) | 0·92 (0·83–1·03) |
| Adiponectin | 0·94 (0·86–1·03) | 1·04 (0·96–1·13) | 1·00 (0·92–1·08) |
| Metabolic syndrome† | 1·06 (0·73–1·55) | 1·00 (0·73–1·37) | 1·16 (0·84–1·60) |
| Coronary artery calcium‡ | 1·04 (0·75–1·42) | 1·00 (0·77–1·31) | 1·04 (0·79–1·37) |
| Fully adjusted* | |||
| Inflammatory markers | |||
| Resistin | 1·03 (0·94–1·11) | 1·07 (1·00–1·14) | 1·06 (0·99–1·13) |
| Soluble TNF receptor 2 | 1·06 (1·01–1·11) | 1·02 (0·98–1·06) | 1·01 (0·97–1·06) |
| Interleukin-6 | 1·01 (0·90–1·14) | 1·03 (0·93–1·13) | 0·97 (0·88–1·07) |
| C-reactive protein | 1·05 (0·87–1·27) | 1·12 (0·96–1·31) | 1·08 (0·92–1·27) |
| Metabolic variables | |||
| Body mass index | 0·98 (0·95–1·05) | 1·00 (0·97–1·02) | 0·99 (0·97–1·02) |
| Triglycerides | 1·03 (0·96–1·10) | 0·99 (0·93–1·05) | 1·00 (0·94–1·06) |
| HDL cholesterol | 0·97 (0·94–1·01) | 1·00 (0·97–1·03) | 0·99 (0·95–1·02) |
| Leptin | 0·94 (0·84–1·05) | 0·97 (0·88–1·06) | 0·92 (0·83–1·01) |
| Adiponectin | 0·96 (0·88–1·05) | 1·04 (0·97–1·12) | 1·02 (0·94–1·10) |
| Metabolic syndrome† | 0·98 (0·64–1·51) | 1·04 (0·73–1·50) | 1·27 (0·88–1·83) |
| Coronary artery calcium‡ | 1·04 (0·76–1·41) | 1·00 (0·79–1·32) | 1·05 (0·80–1·37) |
HDL, high density lipoprotein; TNF, tumour necrosis factor; CI, confidence interval.
The results are presented as the fold increase and the 95% CI for each continuous outcome or odds ratio, for the metabolic syndrome, associated with variant resistin alleles assuming a dominant model (e.g. for −420, GG and GC vs. CC).
Fully adjusted models included age, gender, tobacco and alcohol use, exercise, aspirin/statin/niacin use, family history of CAD and, when not part of the model outcome, body mass index, the metabolic syndrome and Framingham risk score. Log-transformed continuous variables were used as outcomes in linear regression models.
The presence or absence of National Cholesterol Education Program (NCEP)-defined metabolic syndrome was used as outcome in logistic regression models.
Coronary artery calcium expressed as the natural log of (CAC + 1) was used in linear regression models.
Numbers in bold signify statistically significant values.
Haplotype analysis
In fully adjusted models (Table 4), relative to the most common estimated haplotype (ACC), the AGT haplotype was associated with higher CRP levels (P = 0·04), and the GGT haplotype was associated with sol-TNFR2 levels (P = 0·04). When resistin was included in the model, there was no significant attenuation of the AGT haplotype association with CRP [fold change 1·21 (95% CI 1·00 to 1·45), P = 0·04) and a modest attenuation of the GGT association with TNF [1·04 (0·99 to 1·08), P = 0·1]. Haplotypes with G alleles at positions −852 and −420 tended to have higher plasma resistin levels although this did not reach nominal statistical significance (P = 0·09 for GGT). By contrast, there was no association of any variant haplotypes with BMI, metabolic syndrome status, plasma lipids or circulating levels of leptin and adiponectin. Similar to the individual SNP analysis, in age- and gender-adjusted models, resistin haplotypes were not associated with fasting glucose [absolute change and 95% CI in mg/dl of −0·6 (−3·1 to 1·8), −1·3 (−3·5 to 1·0) and −1·2 (−3·4 to 1·0)] or HOMA-IR [fold change and 95% CI of 0·96 (0·84 to 1·10), 0·89 (0·59 to 1·01) and 0·95 (0·85 to 1·08)] for AGC, AGT and GGT, respectively. Similarly, there was no association with CAC scores (Table 4).
Table 4.
Association of estimated haplotypes in resistin with inflammatory and metabolic variable and coronary artery calcification
| AGC | AGT | GGT | |
|---|---|---|---|
| Inflammatory markers | |||
| Resistin | 1·06 (0·97–1·16) | 1·02 (0·94–1·11) | 1·06 (0·98–1·15) |
| Soluble TNF receptor 2 | 1·01 (0·96–1·06) | 0·98 (0·93–1·03) | 1·05 (1·00–1·10) |
| Interleukin-6 | 1·02 (0·89–1·16) | 1·03 (0·91–1·16) | 0·95 (0·84–1·06) |
| C-reactive protein | 1·02 (0·83–1·26) | 1·23 (1·01–1·49) | 0·99 (0·82–1·19) |
| Metabolic variables | |||
| Body mass index | 1·00 (0·97–1·04) | 1·00 (0·98–1·03) | 0·99 (0·96–1·02) |
| Triglycerides | 0·97 (0·90–1·05) | 0·97 (0·91–1·05) | 1·03 (0·96–1·11) |
| HDL cholesterol | 1·00 (0·96–1·04) | 0·99 (0·96–1·03) | 0·98 (0·95–1·02) |
| Leptin | 1·01 (0·89–1·14) | 0·95 (0·85–1·07) | 0·94 (0·84–1·05) |
| Adiponectin | 1·09 (0·99–1·20) | 0·99 (0·91–1·09) | 0·97 (0·89–1·06) |
| Metabolic syndrome† | 0·96 (0·59–1·57) | 1·14 (0·72–1·79) | 1·01 (0·64–1·60) |
| Coronary artery calcium‡ | 0·78 (0·55–1·11) | 0·94 (0·68–1·29) | 1·05 (0·77–1·44) |
HDL, high density lipoprotein; TNF, tumour necrosis factor; CI, confidence interval.
The results are presented as the fold change (or the odds ratio for a logistic regression model) and the 95% CI in each outcome for variant resistin haplotype relative to the referent most common haplotype (ACC) in multivariate models, which include age and gender, tobacco and alcohol use, aspirin/statin/niacin use, family history and, when not part of the model outcome, body mass index, metabolic syndrome and Framingham risk score.
Log-transformed continuous variables were used as outcomes in linear regression models.
The presence or absence of National Cholesterol Education Program (NCEP)-defined metabolic syndrome was used as outcome in logistic regression models.
Coronary artery calcium expressed as the natural log of (CAC + 1) was used in linear regression models.
Numbers in bold signify statistically significant values.
Discussion
Little is known of the association of resistin gene variation with measures of inflammation in humans.21 We found that carriers of the G allele at position −420C>G had higher plasma resistin levels but, unlike large published studies in Asian subjects, this variant only accounted for a minor variation in circulating resistin. We report for the first time that the G allele of the −852A>G SNP and haplotypes with G alleles at −852 and/or −420 were associated with higher levels of the cytokine marker sol-TNFR2. By contrast, we found no relationships between resistin gene variation and any metabolic measure including BMI, metabolic syndrome, plasma lipids or the adipokines, leptin and adiponectin. There was no association of resistin SNPs or haplotypes with CAC, an established surrogate of coronary atherosclerotic burden and CVD risk in humans.
Prior studies, mostly of Asian populations, have reported a relationship between the −420G allele and plasma resistin levels.22,30 For example, in a Japanese sample of over 2000 individuals, Osawa et al. found a robust relationship with the −420G allele and resistin levels30 and estimated that this genotype accounted for approximately 26% of the variation in circulating resistin. Menzaghi et al. (N = 737)17 and Dolinkova et al. (N = 85),31 however, did not observe this relationship in Caucasians, although these studies lacked power. By contrast, our study had adequate power to detect small to moderate effects of resistin SNPs on quantitative traits including inflammatory and metabolic biomarkers. Although we replicated the −420G allele association with resistin levels in our Caucasian cohort, this SNP accounted for minor variation (about 1–2%) of circulating resistin, despite similar genotype frequencies in Caucasian and Asian populations. Consistent with this, Norata et al. recently reported a weak, nonsignificant, trend towards higher resistin levels with the −420G allele in an Italian sample.21 The molecular mechanism underlying this apparent ethnic difference is uncertain but might relate to reported variation in regulation of mRNA transcription across ethnicity.32
The −420C>G SNP appears to confer a gain of function for Sp1/3 transcription factor binding, thus enhancing resistin transcription and plasma protein levels.23 Insulin signalling promotes gene transcription in part through Sp transcription factors,33 suggesting that myeloid expression of resistin may be modulated specifically in insulin-resistant states.34 Conversely, resistin activates nuclear factor-kappa B (NF-κB) inflammatory signalling and attenuates insulin signalling in adipocytes.34,35 Thus, leucocyte and adipose tissue macrophage-derived resistin induced in hyperinsulinaemic states may act in an endocrine or paracrine manner to promote adipocyte inflammation and disruption of insulin signalling. Several studies, however, including our work, suggest that resistin is not significantly associated with adiposity or metabolic traits, at least in the absence of obesity or type 2 diabetes.8,9
Kunnari et al. reported an association of −420G with higher glucose levels in 258 Finnish subjects,36 and separate studies in China (N = 624),20 Europe (N = 585) and Quebec (N = 590) related this SNP to glucose intolerance and measures of adiposity.19,37 However, these results were not replicated in Scandinavian case–control and family-based populations,37 and others have not found associations with type 2 diabetes18 or insulin resistance.38 Inconsistent findings may reflect differences in the metabolic phenotypes and populations across these different studies. Norata et al. found weak associations of the −420G allele with obesity, plasma triglycerides and the metabolic syndrome, although they did not report multivariable analyses adjusted for confounders.21 In SIRCA, we did not replicate association of the −420G allele or other 5′ SNPs and haplotypes with any metabolic variables including BMI, waist circumference, NCEP-defined metabolic syndrome, plasma levels of leptin or adiponectin, HOMA or fasting glucose. Our negative findings suggest that the −420G allele and related promoter variation have limited impact on adiposity, metabolic syndrome and insulin resistance, at least in this relatively lean, nondiabetic European ancestry sample.
We previously reported that in humans, unlike in rodents, resistin is induced by lipopolysaccharides (LPS) and TNF-α in macrophages in vitro and is markedly increased in leucocytes and plasma in vivo during endotoxaemia.39 Furthermore, in asymptomatic SIRCA participants plasma levels of sol-TNFR2, a marker of TNF system activation, were correlated with resistin.13 We now report a modest association of the −852A>G variant, and haplotypes containing the G allele at −852 and −420, with higher sol-TNFR2 in SIRCA. Adjustment for plasma resistin did not attenuate the relationship of the −852G with sol-TNFR2, suggesting that it is unlikely that this association reflects induction of cytokines by circulating resistin.34,35 Systemic inflammation, however, may direct the collective transcription of multiple inflammatory proteins, including resistin and TNF.
Resistin has direct pro-inflammatory actions on macrophages and vascular cells, which may promote atherosclerosis, and limited studies in rodents and humans have suggested that resistin may be atherogenic.13,40 In SIRCA, despite genotype associations with plasma resistin and inflammatory biomarkers, we did not find any evidence of a relationship between resistin gene promoter variation and CAC in crude or adjusted analyses. Consistent with our results, a recent study of the −420 variant in Europeans found no correlation with carotid atherosclerosis in a population-based cohort and no association with myocardial infarction (MI) in a case–control study.21 We cannot exclude false-negative findings as our study has limited power to detect small genetic effects and we did not study all variations in the resistin gene. Indeed, heterogeneous effects may operate across race/ethnicity as a recent Chinese case–control study (N = 225 CAD patients) reported an association of the −420G allele with the presence of CAD at angiography.41
This study has several limitations. SIRCA is a cross-sectional study and causal inferences cannot be drawn and generalizability to nonobese, nondiabetic Caucasian populations is limited. We did not perform replication studies because we focused on resistin gene variation with prior evidence of functional significance and relationship to plasma resistin levels.23 Our study can therefore be viewed as replicating, for the first time in Caucasians, resistin gene associations with plasma resistin levels originally reported in Asian populations. Large-scale examination of additional variants that tag all common haplotype blocks is warranted to further examine the relationship with inflammatory phenotypes and to exclude modest associations with metabolic traits and atherosclerosis, and in particular to assess apparent ethnic heterogeneity.
In conclusion, we report that resistin gene variation is associated with plasma resistin levels and circulating inflammatory biomarkers but not measures of adiposity or metabolic syndrome. Furthermore, we found no relationship between resistin SNPs and haplotypes and coronary calcification, a validated measure of coronary atherosclerosis. Overall, our results support the concept that resistin is an inflammatory adipokine but argue against a significant role for resistin 5′ gene variation in adiposity, metabolic syndrome or atherosclerosis in Caucasians.
Acknowledgments
This work was supported by a Clinical and Translational Science Award (RFA-RM-06-002) from the National Center for Research Resources (NCRR) to the University of Pennsylvania, by U54 RR020278-01 from the NCRR to the Broad Institute Center for Genotyping and Analysis and by HL RO1073278, P50 HL-083799 (SCCOR) and W.W. Smith Charitable Trust (#H0204) to M.P.R.
References
- 1.Steppan CM, Bailey ST, Bhat S, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 2.Kim KH, Lee K, Moon YS, et al. A cysteine-rich adipose tissue-specific secretory factor inhibits adipocyte differentiation. Journal of Biological Chemistry. 2001;276:11252–11256. doi: 10.1074/jbc.C100028200. [DOI] [PubMed] [Google Scholar]
- 3.Rangwala SM, Rich AS, Rhoades B, et al. Abnormal glucose homeostasis due to chronic hyperresistinemia. Diabetes. 2004;53:1937–1941. doi: 10.2337/diabetes.53.8.1937. [DOI] [PubMed] [Google Scholar]
- 4.Satoh H, Nguyen MT, Miles PD, et al. Adenovirus-mediated chronic ‘hyper-resistinemia’ leads to in vivo insulin resistance in normal rats. Journal of Clinical Investigation. 2004;114:224–231. doi: 10.1172/JCI20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steppan CM, Lazar MA. The current biology of resistin. Journal of Internal Medicine. 2004;255:439–447. doi: 10.1111/j.1365-2796.2004.01306.x. [DOI] [PubMed] [Google Scholar]
- 6.Azuma K, Katsukawa F, Oguchi S, et al. Correlation between serum resistin level and adiposity in obese individuals. Obesity Research. 2003;11:997–1001. doi: 10.1038/oby.2003.137. [DOI] [PubMed] [Google Scholar]
- 7.McTernan PG, Fisher FM, Valsamakis G, et al. Resistin and type 2 diabetes: regulation of resistin expression by insulin and rosiglitazone and the effects of recombinant resistin on lipid and glucose metabolism in human differentiated adipocytes. Journal of Clinical Endocrinology and Metabolism. 2003;88:6098–6106. doi: 10.1210/jc.2003-030898. [DOI] [PubMed] [Google Scholar]
- 8.Lee JH, Chan JL, Yiannakouris N, et al. Circulating resistin levels are not associated with obesity or insulin resistance in humans and are not regulated by fasting or leptin administration: cross-sectional and interventional studies in normal, insulin-resistant, and diabetic subjects. Journal of Clinical Endocrinology and Metabolism. 2003;88:4848–4856. doi: 10.1210/jc.2003-030519. [DOI] [PubMed] [Google Scholar]
- 9.Utzschneider KM, Carr DB, Tong J, et al. Resistin is not associated with insulin sensitivity or the metabolic syndrome in humans. Diabetologia. 2005;48:2330–2333. doi: 10.1007/s00125-005-1932-y. [DOI] [PubMed] [Google Scholar]
- 10.Gerber M, Boettner A, Seidel B, et al. Serum resistin levels of obese and lean children and adolescents: biochemical analysis and clinical relevance. Journal of Clinical Endocrinology and Metabolism. 2005;90:4503–4509. doi: 10.1210/jc.2005-0437. [DOI] [PubMed] [Google Scholar]
- 11.Yang RZ, Huang Q, Xu A, et al. Comparative studies of resistin expression and phylogenomics in human and mouse. Biochemical and Biophysical Research Communications. 2003;310:927–935. doi: 10.1016/j.bbrc.2003.09.093. [DOI] [PubMed] [Google Scholar]
- 12.Kaser S, Kaser A, Sandhofer A, et al. Resistin messenger-RNA expression is increased by proinflammatory cytokines in vitro. Biochemical and Biophysical Research Communications. 2003;309:286–290. doi: 10.1016/j.bbrc.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Reilly MP, Lehrke M, Wolfe ML, et al. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111:932–939. doi: 10.1161/01.CIR.0000155620.10387.43. [DOI] [PubMed] [Google Scholar]
- 14.Migita K, Maeda Y, Miyashita T, et al. The serum levels of resistin in rheumatoid arthritis patients. Clinical and Experimental Rheumatology. 2006;24:698–701. [PubMed] [Google Scholar]
- 15.Axelsson J, Bergsten A, Qureshi AR, et al. Elevated resistin levels in chronic kidney disease are associated with decreased glomerular filtration rate and inflammation, but not with insulin resistance. Kidney International. 2006;69:596–604. doi: 10.1038/sj.ki.5000089. [DOI] [PubMed] [Google Scholar]
- 16.Burnett MS, Lee CW, Kinnaird TD, et al. The potential role of resistin in atherogenesis. Atherosclerosis. 2005;182:241–248. doi: 10.1016/j.atherosclerosis.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Menzaghi C, Coco A, Salvemini L, et al. Heritability of serum resistin and its genetic correlation with insulin resistance-related features in nondiabetic Caucasians. Journal of Clinical Endocrinology and Metabolism. 2006;91:2792–2795. doi: 10.1210/jc.2005-2715. [DOI] [PubMed] [Google Scholar]
- 18.Conneely KN, Silander K, Scott LJ, et al. Variation in the resistin gene is associated with obesity and insulin-related phenotypes in Finnish subjects. Diabetologia. 2004;47:1782–1788. doi: 10.1007/s00125-004-1537-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mattevi VS, Zembrzuski VM, Hutz MH. A resistin gene polymorphism is associated with body mass index in women. Human Genetics. 2004;115:208–212. doi: 10.1007/s00439-004-1128-4. [DOI] [PubMed] [Google Scholar]
- 20.Xu JY, Sham PC, Xu A, et al. Resistin gene polymorphisms and progression of glycaemia in southern Chinese: a 5-year prospective study. Clinical Endocrinology. 2007;66:211–217. doi: 10.1111/j.1365-2265.2006.02710.x. [DOI] [PubMed] [Google Scholar]
- 21.Norata GD, Ongari M, Garlaschelli K, et al. Effect of the −420C/G variant of the resistin gene promoter on metabolic syndrome, obesity, myocardial infarction and kidney dysfunction. Journal of Internal Medicine. 2007;262:104–112. doi: 10.1111/j.1365-2796.2007.01787.x. [DOI] [PubMed] [Google Scholar]
- 22.Cho YM, Youn BS, Chung SS, et al. Common genetic polymorphisms in the promoter of resistin gene are major determinants of plasma resistin concentrations in humans. Diabetologia. 2004;47:559–565. doi: 10.1007/s00125-003-1319-x. [DOI] [PubMed] [Google Scholar]
- 23.Osawa H, Yamada K, Onuma H, et al. The G/G genotype of a resistin single-nucleotide polymorphism at −420 increases type 2 diabetes mellitus susceptibility by inducing promoter activity through specific binding of Sp1/3. American Journal of Human Genetics. 2004;75:678–686. doi: 10.1086/424761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reilly MP, Wolfe ML, Localio AR, et al. C-reactive protein and coronary artery calcification. The Study of Inherited Risk of Coronary Atherosclerosis (SIRCA) Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23:1851–1856. doi: 10.1161/01.ATV.0000092327.60858.4A. [DOI] [PubMed] [Google Scholar]
- 25.Wilson PW, D’Agostino RB, Levy D, et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 26.Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome. An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Executive summary. Cardiology in Review. 2005;13:322–327. [PubMed] [Google Scholar]
- 27.Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 28.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- 29.Burkett K, Graham J. Likelihood inference of trait associations with SNP haplotypes and other attributes using the EM algorithm. [accessed 03 September 2008];The hapassoc package. 2007 Available at: http://stat-db.stat.sfu.ca:8080/statgen/research/hapassoc.
- 30.Osawa H, Tabara Y, Kawamoto R, et al. Plasma resistin, associated with single nucleotide polymorphism −420, is correlated with insulin resistance, lower HDL cholesterol, and high-sensitivity C-reactive protein in the Japanese general population. Diabetes Care. 2007;30:1501–1506. doi: 10.2337/dc06-1936. [DOI] [PubMed] [Google Scholar]
- 31.Dolinkova M, Krizova J, Lacinova Z, et al. Polymorphisms of adiponectin and resistin genes in patients with obesity and anorexia nervosa [in Czech] Casopis Lekaru Ceskych. 2006;145:562–566. [PubMed] [Google Scholar]
- 32.Spielman RS, Bastone LA, Burdick JT, et al. Common genetic variants account for differences in gene expression among ethnic groups. Nature Genetics. 2007;39:226–231. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaytor EN, Zhu JL, Pao CI, et al. Insulin-responsive nuclear proteins facilitate Sp1 interactions with the insulin-like growth factor-I gene. Journal of Biological Chemistry. 2001;276:36896–36901. doi: 10.1074/jbc.M104035200. [DOI] [PubMed] [Google Scholar]
- 34.Nagaev I, Bokarewa M, Tarkowski A, et al. Human resistin is a systemic immune-derived proinflammatory cytokine targeting both leukocytes and adipocytes. PLoS ONE. 2006;1:e31. doi: 10.1371/journal.pone.0000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steppan CM, Wang J, Whiteman EL, et al. Activation of SOCS-3 by resistin. Molecular and Cellular Biology. 2005;25:1569–1575. doi: 10.1128/MCB.25.4.1569-1575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunnari A, Ukkola O, Kesaniemi YA. Resistin polymorphisms are associated with cerebrovascular disease in Finnish type 2 diabetic patients. Diabetic Medicine. 2005;22:583–589. doi: 10.1111/j.1464-5491.2005.01480.x. [DOI] [PubMed] [Google Scholar]
- 37.Engert JC, Vohl MC, Williams SM, et al. 5′ flanking variants of resistin are associated with obesity. Diabetes. 2002;51:1629–1634. doi: 10.2337/diabetes.51.5.1629. [DOI] [PubMed] [Google Scholar]
- 38.Pizzuti A, Argiolas A, Di Paola R, et al. An ATG repeat in the 3′-untranslated region of the human resistin gene is associated with a decreased risk of insulin resistance. Journal of Clinical Endocrinology and Metabolism. 2002;87:4403–4406. doi: 10.1210/jc.2002-020096. [DOI] [PubMed] [Google Scholar]
- 39.Anderson PD, Mehta NN, Wolfe ML, et al. Innate immunity modulates adipokines in humans. Journal of Clinical Endocrinology and Metabolism. 2007;92:2272–2279. doi: 10.1210/jc.2006-2545. [DOI] [PubMed] [Google Scholar]
- 40.Lubos E, Messow CM, Schnabel R, et al. Resistin, acute coronary syndrome and prognosis results from the AtheroGene study. Atherosclerosis. 2007;193:121–128. doi: 10.1016/j.atherosclerosis.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 41.Tang NP, Wang LS, Yang L, et al. A polymorphism in the resistin gene promoter and the risk of coronary artery disease in a Chinese population. Clinical Endocrinology. 2008;68:82–87. doi: 10.1111/j.1365-2265.2007.03003.x. [DOI] [PubMed] [Google Scholar]