

Abstract
Regulatory T cells (Treg) induce robust neuroprotection in murine models of neuroAIDS, in part, through eliciting anti-inflammatory responses for HIV-1-infected brain mononuclear phagocytes (MP; macrophage and microglia). Herein, using both murine and human primary cell cultures in proteomic and cell biologic tests, we report that Treg promotes such neuroprotection by an even broader range of mechanisms than previously seen including inhibition of virus release, killing infected MP, and inducing phenotypic cell switches. Changes in individual Treg-induced macrophage proteins were quantified by iTRAQ labeling followed by mass spectrometry identifications. Reduction in virus release paralleled the upregulation of interferon-stimulated gene 15, an ubiquitin-like protein involved in interferon-mediated antiviral immunity. Treg killed virus-infected macrophages through caspase-3 and granzyme and perforin pathways. Independently, Treg transformed virus-infected macrophages from an M1 to an M2 phenotype by down- and up- regulation of inducible nitric oxide synthase and arginase 1, respectively. Taken together, Treg affects a range of virus-infected MP functions. The observations made serve to challenge the dogma of solitary Treg immune suppressor functions and provides novel insights into how Treg affects adaptive immunosurveillance for control of end organ diseases, notably neurocognitive disorders associated with advanced viral infection.
Keywords: regulatory T cells, bone marrow derived macrophages, cytotoxicity, human immunodeficiency virus type one (HIV-1), HIV-1 associated neurocognitive disorders, vesicular stomatitis virus, iTRAQ
Short abstract
Proteomic changes in HIV-1-infected murine macrophages following cocultivation with murine Treg, Tcon, or cells cultured alone were quantified by iTRAQ. Macrophage functions that were effected by Treg included upregulation of interferon stimulated proteins, apoptosis, and functional transformation in surviving cells from an M1 to an M2 phenotype. The latter ultimately led to reductions in progeny virus release and in neuroprotective activities.
Introduction
HIV-1 associated neurocognitve disorders (HAND) commonly follow progressive virus infection in the infected human host.(1) Clinical disease features range in severity from subtle deficits to incapacitating dementia and include asymptomatic neurocognitive impairment, mild neurocognitive disorder, and HIV-associated dementia (HAD).(2) Severe dementia is now rare since the use of antiretroviral therapy (ART).(3) Notably, cognitive impairment is associated, in measure, with immune suppression and a vicious cycle of viral replication, ingress of perivascular macrophages, and neuroinflammation.(4) Substantive neuropathological findings, in the era of ART, are diminished and described as neuronal dysfunction without loss.(5) The control and diagnosis of HAND remains poorly understood despite considerable research efforts.(6)
Recent research demonstrated a role for adaptive immunity and specifically CD4+ T cells in neurodegenerative diseases.7−11 Works performed by others and in our laboratories, both in murine models of HAND and Parkinson’s disease (PD), demonstrated that CD4+CD25+ Treg serve to attenuate brain mononuclear phagocyte (MP; macrophage and microglia) inflammation and sustain neuroprotection.7,9 Nonetheless, the mechanism underlying such Treg-associated neuroprotective functions remains incompletely understood.(12) Primary roles for Treg rests in the cells abilities to provide self-tolerance.(13) Acting as a suppressor, its role in HIV disease remains highly controversial.14−17 Indeed, it has been suggested that Treg play detrimental roles since they suppress the HIV-specific immune response and inhibit virus clearance.18,19 In contrast, others posit a beneficial role for Treg through suppression of immune hyperactivation and control of viral load.20−23 In support of the latter is Treg’s known role in simian immunodeficiency virus (SIV) infection in sooty mangabeys and its function as an elite suppressor to human immunodeficiency virus (HIV) infection.24,25 The similarity shared in these reports rests in maintenance of Treg frequency and function to limit generalized immune activation commonly seen during advanced HIV disease.(26)
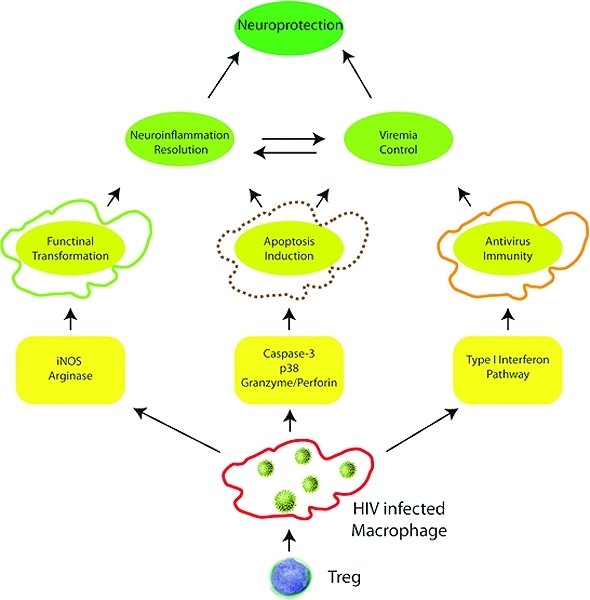
We now demonstrate a multifaceted role for Treg for HIV-1-infected MP. Such a role serves a range of regulatory functions acting to control disease such as suppression of viremia and modulation of HIV-1-infected macrophage neurotoxic activities. In support of such claims, we used isobaric tag for relative and absolute quantitation (iTRAQ) of proteins to identify differential protein expression between HIV-1-infected bone marrow-derived macrophages (BMM) with and without Treg. We found changes consistent with enhanced antiretroviral immunity through Treg-induced upregulation of interferon (IFN)-induced gene products. This was seen among a broad range of related protein expression changes linked to antiviral responses. Second, we showed that Treg utilizes caspase-3 and granzyme/perforin pathways to kill virus-infected cells. Third, Treg reduced neurotoxic secretions in vitro through its abilities to transform HIV-1-infected macrophages from an M1 to an M2 phenotype. These observations made in murine Treg-BMM cocultures were replicated separately in human Treg-monocyte-derived macrophages (MDM) cocultures. Taken together, these data demonstrate that Treg serve as effectors for virus-infected macrophages and suppressors for inflammation and as such, exert immune surveillance functions relevant to ongoing HIV infection and neuroAIDS.
Materials and Methods
Animals
C57BL/6J male mice (8−10 wk old) were purchased from The Jackson Laboratory and used for BMM and T cell isolations. All animal procedures were in accordance with the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center.
Isolation and Cultivation of BMM
Femurs of the mice were excised and flushed with Dulbecco’s phosphate buffered saline (DPBS) to obtain bone marrow-derived mononuclear cells. Cells were passed through a 40 μm cell strainer to remove the clumps and then centrifuged. Erythrocytes were removed using ACK lysis buffer (Gibco, Grand Island, NY). After washing twice with DBPS, cells were resuspended and plated in 6-well plates at 1 × 106 cells/mL in 3 mL complete medium [RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 10 mM HEPES, 1× nonessential amino acids, 50 μM 2-mercaptoethanol, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 μg/mL macrophage colony stimulating factor, MCSF (Pfizer, Cambridge, MA)]. After 7 days differentiation, cells were >98% CD11b+ as determined by flow cytometry (Supplemental Figure 1A).
Generation of HIV-1/VSV Pseudotype Virus and Assay of Viral Infectivity
VSV pseudotyped HIV-1, YU2 (HIV-1/VSV) was used to circumvent the required cellular receptors necessary for HIV-1 to infect mouse cells. The HIV-1YU2/VSV pseudotypes were generated by cotransfection of 3 μg of pYU2 and 1 μg of pHIT/G into 293T cells (per 1 × 106 cells) using the FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN). Pseudotyped virus stock was collected after 72 h post-transfection. Because the pseudotyped virus only contains HIV-1YU2 genes, but not the glycoprotein gene of VSV, they could only enter the mouse BMM once. The concentration of the virus stock was determined by HIV-1p24 ELISA kit (PerkinElmer, Boston, MA). We used a range of infective viral doses including 1, 2, and 3 pg of HIV-1p24/cell to infect BMM. This was done to ensure a data set independent of multiplicity of infection (MOI). After 24 h, the percentage of p24 positive cells was determined by immunohistochemistry. As shown in Supplemental Figures 1B and 1C at 1 pg of HIV-1p24/cell >95% HIV-1p24 immunoreactivity was observed. Further, the level of HIV-1p24 staining increased in parallel to the infective dose. In subsequent experiments, 1 pg of HIV-1p24/cell was used to infect BMM. Virus was placed into the media for 24 h then removed by vigorous washings with phosphate buffered saline (PBS).(27)
Isolation and Expansion of Mouse Tcon and Treg
CD4+ T cell subsets were isolated using previously described techniques.28,29 Briefly, immune cells were isolated from the spleens and lymph nodes (inguinal, brachial, axillary, cervical and mesenteric) by dissection, followed by mechanical dissociation by pushing the organs through a 70 μm mesh filter. Cells were washed with DPBS, erythrocytes were removed by the ACK lysis buffer (Gibco) and cells washed twice more with PBS. The processed cells were then passed through a mouse T cell enrichment column (R&D Systems, Minneapolis, MN) for T cell negative selection, followed by passage of the purified T cells through a mouse T cell CD4 subset column (R&D Systems) for negative selection of CD4+ T cells. CD4+ T cells were then labeled with monoclonal antimouse CD25-PE antibody, then reacted with anti-PE MicroBeads (Miltenyi Biotec, Auburn, CA) and passed through the MACS column in a magnetic field for positive selection of CD25+ cells. The flow through containing CD4+CD25- T cells was used in these experiments and propagated as CD3/CD28-activated conventional T cells (Tcon) (see below). The isolated CD4+CD25+ cells retained on the column and then eluted were used as Treg (Supplemental Figure 2A, Supporting Information). T cell isolates were >91% (CD4+CD25+FoxP3+) and >96% (CD4+CD25+) pure (Supplemental Figure 2B−D, Supporting Information), respectively. Treg and Tcon were expanded ex vivo using CD3/CD28 T cell expander Dynabeads (Invitrogen Dynal, Oslo, Norway). The Dynabeads were added to cells plated in 24 well plates at 1 × 106 cells/mL in complete RPMI along with 1000 U/mL mouse rIL-2 (R&D Systems) for Treg cultures; whereas, 100 U/mL mouse rIL-2 was used for Tcon cell cultures. Cells were expanded in culture for 10 days with half-media changes every 2−3 days. Treg remained >83% pure after expansion as determined by CD4+FoxP3+ staining (Supplemental Figure 2C, D, Supporting Information). The function of Treg was evaluated by lymphocyte proliferation suppression assay as described below (Supplemental Figure 2E, F, Supporting Information). Mouse and human T cells were cocultured only with same species mouse and human macrophages.
Isolation and Culture of Human MDM
Peripheral blood mononuclear cells (PBMC) from HIV-1, HIV-2, and hepatitis seronegative human donors were obtained by leukophoresis in full compliance and approval of the University of Nebraska Medical Center Institution Review Board. Monocytes were purified by countercurrent centrifugal elutriation.(30) Cells were cultured in complete RPMI medium and maintained in a 37 °C and 5% CO2 atmosphere. On days 2 and 5, half of the medium was exchanged. After seven days in culture, MDM were infected with HIV-1ADA (a macrophage tropic viral strain) at a MOI of 1 or left untreated (controls) in media devoid of MCSF. On day 1 after infection, a full medium exchange was performed followed by a half medium exchange every 2−3 days thereafter in medium devoid of MCSF. On day 7 after infection, cells were cocultured with activated Tcon or Treg (1:1) for an additional 24 h. The 1:1 ratio of T cells to macrophages was used based upon our prior data from effector cell optimization.7−9 Moreover, further additions of Tregs in virus-infected macrophage cocultures would eliminate all virus-infected cells and preclude any subsequent proteomic testing (data not shown).
Isolation and Expansion of Human T Cell Subsets
CD4/CD8 T cell ratios from donor blood were determined prior to starting all experiments with human cells. Tcon and Treg were isolated from the same donor as cultured MDM. Human Tregs were isolated from PBMC using the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec). Briefly, human Tregs were isolated by first depleting all non-CD4+ cells by indirect magnetic labeling of cells with a cocktail of biotin-conjugated monoclonal antihuman antibodies against CD8, CD14, CD16, CD19, CD36, CD56, CD123, TCRγ/δ and CD235a; adding antibiotin magnetic MicroBeads; and then passing cells through a magnetic column and collecting the CD4+ enriched fraction. Then CD4+CD25+ T cells were isolated by positive selection using magnetic MicroBeads conjugated to monoclonal anti-CD25 antibody and passing the cells over the magnetic column. The eluted CD4+CD25- cells were used as Tcon, and the retained CD4+CD25+ cells were used as Treg in these experiments. Purity was determined by CD25-PE staining and detection by flow cytometry (Supplemental Figure 3A, Supporting Information). Treg and Tcon cells were expanded and activated ex vivo using human Treg Expander Dynabeads (Invitrogen, Carlsbad, CA). The function of expanded Treg was evaluated by lymphocyte proliferation suppression assay (Supplemental Figure 3B, Supporting Information). Human T cells were only cocultured with human monocytes from the identical donors.
Flow Cytometry
Cells (1 × 106) were labeled with fluorescently labeled antibodies to CD3, CD4, CD8, CD11b (eBioscience, San Diego, CA), CD25 (BD Pharmingen, San Diego, CA), and CD39 (R&D Systems). For CD39 staining, Alexa Fluor 488 donkey antisheep secondary antibody (Invitrogen) was used. For intracellular staining, cells were fixed, permeabilized (FoxP3 staining kit, eBioscience, San Diego, CA) and stained for HIV-1p24 (Beckman Coulter, Brea, CA), mouse FoxP3 (eBioscience), granzyme A (Santa Cruz Biotechnology, Santa Cruz, CA), granzyme B (eBioscience) and perforin (eBioscience). All the data were acquired on FACSCalibur flow cytometer and analyzed with CellQuest Software (Becton Dickinson, Franklin Lakes, NJ). For lymphocyte proliferation suppression assay (Supplemental Figures 2E, F, and 3B, Supporting Information), Tcon were incubated with 1 μM carboxyfluorescein diacetate, succinimidyl ester (CFSE) per 10 × 106 cells for 15 min and washed twice with RPMI 1640 culture medium. Excluding the control group, Tcon were cultured alone or with Treg at 2-fold escalating ratios in flat-bottom 96-well plates with addition of Dynabeads Mouse T-Activator CD3/CD28. After 72 h, cells were collected, Dynabeads were removed by DynaMag−15 magnet (Invitrogen), and CFSE was measured by flow cytometry.
Culture Fluids and Cell Lysates
After 7 days differentiation, BMM were replated at 2 × 106 cells per well in 6-well plates. HIV-1-infected BMM were cocultured with Dynabead-activated Tcon or Treg at a ratio of 1:1. Twenty-four hours later, culture supernatants were collected as conditioned medium 1 (CM1), which contained soluble molecules from macrophages and/or T cells. T cells were removed by PBS washing and 2 mL fresh medium were added to each well. After another 24 h, culture supernatants were collected as conditioned medium 2 (CM2), which contained soluble molecules from macrophages only. The cytokines and virions in conditioned medium were detected by Cytometric Bead Array Mouse Inflammation kit (BD Biosciences, San Jose, CA) and Alliance HIV-1p24 ELISA kit (PerkinElmer, Boston, MA), respectively, according to manufactures’ protocols. For the cell lysates, macrophages were washed using PBS and then air-dried in a laminar flow hood. For each plate, cells were lysed by 500 μL ice-cold lysis buffer containing 0.05% sodium dodecyl sulfate (SDS, Bio-Rad, Hercules, CA) and 0.5 M triethylammonium bicarbonate (TEAB, Sigma-Aldrich, St. Louis, MO). Lysates were clarified by centrifugation at 14 000× g at 4 °C for 45 min.
Confocal Microscopy and Immunocytochemical Assays
For HIV-1p24 immunochemistry staining, mouse BMM or human MDM were fixed and permeabilized with acetone: methanol (1:1, precooled to −20 °C) for 15 min at room temperature (RT). Cells were washed 3 times with DPBS for 5 min each, then blocked (10% normal goat serum in DPBS) for 30 min at RT. Cells were incubated with mouse anti-HIV-1 p24 antibody (1:20, Dako, Carpinteria, CA) in blocking buffer at 4 °C overnight. Cells were washed with DPBS and incubated with secondary antibody according to EnVision Kits (Dako). Briefly, samples were incubated with horse radish peroxidase (HRP) conjugated polymer for 30 min at RT. After washing with DPBS, samples were incubated with HRP substrate 3,3′-diaminobenzidine (DAB) substrate-chromogen for 5−10 min at RT, which resulted in brown-colored precipitate in HIV-1/VSV infected groups. Pictures were taken with a light microscope (Nikon Eclipse TS100). For detection of intracellular proteins after treatment, BMM were fixed by 4% paraformaldehyde (PFA) at 37 °C for 15 min and permeabilized by 0.2% Triton X-100 in DPBS at 4 °C for 30 min. Nonspecific activity was blocked by incubation in 5% bovine serum albumin (BSA) in DPBS at room temperature for 1 h. Cells were incubated with fluorescence labeled antibodies (granzyme B and perforin) and unlabeled primary antibodies (tubulin and granzyme A) at RT for 3 h followed by staining with Alexa Fluor 488 goat antimouse IgG or Alexa Fluor 546 goat antimouse IgG (Invitrogen), respectively, at RT for 2 h. As for actin staining, we used Alexa Fluor 546 phalloidin. After washing, cells were mounted with ProLong Gold antifade reagent (Invitrogen) with DAPI and analyzed by Zeiss 510 Meta Confocal Laser Scanning Microscope. Images were acquired using the same parameters within groups stained with the same protein. Human MDM detection of Apo2.7 was also stained similarly using a phycoerythrin (PE) conjugated primary antibody (1:10, Beckman Coulter, Brea, CA). For T cell intracellular staining of granzyme A, B and perforin, 2 × 105 cells were sedimented on Shandon microscope slides (Thermo Fisher, Waltham, MA) by centrifugation at 1200 rpm for 5 min then covered with ProLong Gold antifade mounting solution (Invitrogen). For the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, TUNEL and FLICA caspase assays, cells were replated at 2 × 105 cells/well in 8-well polystyrene culture slides for 24 h. Cells were infected with HIV-1/VSV for 24 h, then cells were washed and cocultured with T cells for 24 h before adding probes. The cytotoxic effects of Tcon versus Treg secreted factors on HIV-1/VSV infected BMM after 24, 48, and 72 h in transwell cocultures were determined by MTT (Sigma) assay.(10) Mitochondrial membrane integrity was assessed by JC-1 (Calbiochem, San Diego, CA) cell stainings following the manufacturer’s suggested protocol. Caspase 1 and caspase 3/7 activities were measured using the carboxyfluoroescein FLICA assay (Immunochemistry Technologies, Bloomington, MN) according to the manufacturer’s protocol. TUNEL was performed using the in situ cell death detection kit, AP (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s protocol. Briefly, BMM were washed by DPBS, fixed by 4% PFA and permeabilized by 0.1% Triton X-100 in 0.1% sodium citrate. Subsequently, cells were incubated with TUNEL working solution, which contains Terminal Deoxynucleotidyl Transferase (TdT) and fluorescein-dUTP, at 37 °C for 1 h. Apoptotic cells were identified as green fluorescent TUNEL positive cells by fluorescence microscopy and were normalized to total number of cells as determined by bright field images. In addition, the cell death was measured by trypan blue staining.
iTRAQ Labeling
Protein concentrations of the cell lysates were quantified with 2-D Quant Kit (GE Healthcare, Piscataway, MA). Eighty micrograms of total protein was used from each group. All samples were diluted to the same volume for consistency. Proteins were further reduced, alkylated, digested and iTRAQ labeled using iTRAQ Reagents Multiplex Kit (Applied Biosystems, Carlsbad, CA) according to the manufacturer’s protocol. Briefly, proteins were reduced with 5 mM tris-(2-carboxyethyl)phosphine (TCEP) at 60 °C for 1 h and blocked with 10 mM methyl methanethiosulfonate (MMTS) at RT for 10 min. Then, the proteins were digested by 10 μg trypsin at 37 °C for 16−18 h. Peptides were then labeled with iTRAQ reagent (114 for control, 115 for HIV-1/VSV, 116 for HIV-1/VSV cocultured with Tcon, 117 for HIV-1/VSV cocultured with Treg) at RT for 1 h, followed by adding 100 μL Milli-Q water to each group to stop labeling reaction. The labeled peptides from the four groups were combined and dried in a Speed Vac (Thermo, Waltham, MA) to obtain a brown pellet, indicating efficient labeling.
OFFGEL Fractionation and Peptide Cleanup
For isoelectric point (pI)-based peptide separation, we used the 3100 OFFGEL Fractionator (Agilent Technologies, Santa Clara, CA, USA) with a 12-well setup. Prior to electrofocusing, pooled peptides were desalted using a mixed mode cation exchange cartridge (Waters, Milford, MA). Peptides were resuspended in a final volume of 1.8 mL 1× OFFGEL sample solution. The IPG gel strip (Agilent Technologies, Santa Clara, CA) with a 3−10 linear pH range was rehydrated with rehydration solution for 15 min according to the protocol of manufacturer. Then, 150 μL of prepared sample was loaded in each well. Electrofocusing of the peptides was performed using program 12-PE00. After fractionation, samples were cleaned up using C-18 Spin Column (Pierce, Rockford, IL). Finally, samples were dried in a Speed Vac and stored at −80 °C until ready for analysis by matrix-assisted laser desorption/ionization tandem mass spectroscopy (MALDI-MS/MS).
Matrix-Assisted Laser Desorption/Ionization (MALDI-MS/MS)
Peptide samples were spotted using the off-line Tempo C MALDI Spotting System (Applied Biosystems, Carlsbad, CA). Then, MS and MS/MS were performed using the 4800 MALDI-TOF/TOF (Time Of Flight) mass spectrometer (Applied Biosystems). Mass spectrometry data were analyzed using ProteinPilot 2.0 software (Applied Biosystems). The Tempo LC−MALDI robotic spotting system equipped with a C18 reversed phase capillary column (AB Sciex, Foster City, CA) was used to further fractionate peptides from OFFGEL fractions, followed by data acquisition using 4800 MALDI TOF/TOF (AB Sciex, Foster City, CA) as previously published.(31) Briefly, after using an in-house packed C18 column to separate fractions by HPLC gradient based on hydrophobicity, LC fractions were spotted onto MALDI 1232-spot format plates, with a spotting interval of 24 s and applying 2.8 kV plate voltage. Data was acquired from LC−MALDI spot fractions using 4800 MALDI TOF/TOF equipped with a 200 Hz repetition rate Nd:YAG laser. Spectrum from a total of 800 laser shots was accumulated for each TOF MS spectrum between 800−4000 m/z. Data dependent MS/MS mode was operated using CID gas and 2 kV collision energy; programmed laser stop conditions were employed for the accumulation of MS/MS spectra from 800−4000 laser shots.
Primary Mouse Neuronal Cytotoxicity Assays
Eighteen-day-old embryonic fetuses were obtained from terminally anesthetized pregnant C57BL/6J mice.(9) Cerebral cortices were dissociated and digested into a single-cell suspension, which was then cultured at a density of 2.0 × 105 cells/well in poly-d-lysine-coated 24-well plates with complete neurobasal medium supplemented with 2% B27, 1% penicillin/streptomycin, and 0.5 mM l-glutamine (Invitrogen, Carlsbad, CA). After 8−12 days, neuron-enriched cultures showed typical branching dendrites. Mature neurons were treated with 10% CM1 and CM2, respectively. Twenty-four hours later, neurons were fixed/permeabilized and then stained for tubulin. Dendrite morphology was observed by fluorescent microscopy (Zeiss, Thornwood, NY).
Western Blot Analyses
For Western blots, 10 μg of protein from each total cell lysate was electrophoretically separated on NuPAGENovex 4−12% Bis-Tris Gels (Invitrogen) and electrotransfered to polyvinylidene membranes (Bio-Rad, Hercules, CA). Membranes were incubated with respective primary antibodies at 4 °C overnight and HRP-conjugated secondary antibodies (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA) at RT for 1 h. HRP activity was visualized by using a SuperSignal West Pico Chemiluminescent substrate (Pierce, Rockford, IL). Band intensity was measured using ImageJ and normalized to β-actin (1:1000, Santa Cruz Biotechnology). Antibodies used for Western blot assays included murine anti-iNOS (1:500) and Arg-1 (1:1000, BD Transduction Laboratories, Franklin Lakes, NJ), Caspase-1 (1:500, Santa Cruz Biotechnology), Caspase-3 (1:1000, Cell Signaling, Danvers, MA), NDUFS3 (1:1000, Invitrogen, Carlsbad, CA, USA), Phospho-p38 (1:500, Santa Cruz Biotechnology), perforin (1:500, eBioscience, San Diego, CA, USA), STAT1 (1:2000, Santa Cruz Biotechnology), ISG15 (1:1000, Cell Signaling, Danvers, MA, USA), and HIV-1p24 (1:500, Dako, Carpinteria, CA).
Assays for HIV-1 p24, cAMP and NO Detection
Detection of HIV-1p24 by ELISA from culture supernatants was used to assay viral particle release according to the manufacturer’s protocol (PerkinElmer, Boston, MA). Intra- and extracellular cAMP was quantified using the Direct Cyclic AMP Enzyme Immunoassay (Assay Designs, Plymouth Meeting, PA). NO was measured in culture supernatants using the Griess reagent system (Promega, Madison, WI).
Statistical Analyses
Data presented are representative of three or more individual experiments. Statistical analyses were performed by one-way ANOVA with Bonferroni posttest and Student’s t test by using GraphPad Prism version 4.0c for Macintosh (GraphPad Software, San Diego, CA). Strict filtering of iTRAQ raw data was accomplished by log-transforming the raw data from 3 independent experiments then normalizing using a two-stage linear model macros written by Mayo Clinic.(32) Protein/peptide records were excluded from further analysis if the corresponding confidence values were less than 50, or less than 10 copies of peptide records across all three experiments per corresponding proteins were available. These filtering procedures resulted in a subset of 1394 proteins for later analysis. A one-way ANOVA with unequal variances was fitted to the corresponding normalized data for each protein. The associated p-values from ratios of labeled samples (from t test) were adjusted controlling for False Discovery Rate (FDR) via a method proposed by Benjamini and Hochberg(33) and those less than 0.05 were reported as differentially regulated proteins (strictly filtered data).
Results
Treg Enhance Interferon Related Protein Expression and Proteins Involved in Apoptosis
Treg subverts brain MP functions and elicits neuroprotective activities in a HIV-1/VSV murine model of HIV encephalitis (HIVE) and in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s.8,9 To determine the protein composition up- or down-regulated in infected BMM cocultivated with Treg we used iTRAQ assays followed by OFFGel fractionation and MALDI TOF/TOF mass spectrometry. HIV-1/VSV infected BMM in solitary culture and with Tcon were also used for comparisons (Supplemental Table 2, Supporting Information). Analyses of proteomic changes in cocultures were done using comparisons to HIV-infected BMM alone in order to distinguish changes in the proteome that occur in the setting of HIV due to T cell interactions/responses from those that occur solely because of HIV infection. A combination treatment with Treg and Tcon was not used as this would represent total CD4+ T cell populations and preclude analyses of CD4+ T cell subsets. A total of 3553 unique proteins were identified from the three separate experiments performed. After strict data filtering (described in Materials and Methods) we identified 293 proteins changed upon HIV-1/VSV infection compared to uninfected BMM, 21 proteins changed in Treg cocultures compared to HIV-1/VSV infection alone, and 20 proteins were differentially regulated in Tcon cocultures compared to HIV-1/VSV infection alone. Proteome alterations due to Treg treatment were strongly associated with antiviral immune responses and apoptosis. In addition, proteins involved in cell shape/motility and metabolism were also uncovered (Table 1 and Supplementary Table 2). These changes pointed to an underappreciated role for Treg as effector cells and as such led us to explore aspects of antiviral immunity and induced apoptosis for the infected BMM as the proteome changes suggested.
Table 1. Treg-Induced HIV-1/VSV BMM Proteins Identified After Strict Filtering of iTRAQ Dataa.
| name of protein | NCBI accession number | M.W. [kDa] | 117/115b | P-valuec | function |
|---|---|---|---|---|---|
| Granzyme A | 6754102 | 28.6 | 2.1574 | 0.0040 | Induces apoptosis of pathogen infected cells |
| Granzyme B | 7305123 | 27.5 | 4.0877 | 0.0000 | Induces apoptosis of pathogen infected cells |
| Granzyme D | 3334466 | 27.6 | 3.9000 | 0.0000 | Induces apoptosis of pathogen infected cells |
| ISG15 ubiquitin-like modifier | 80476034 | 17.9 | 1.6406 | 0.0024 | Ubiquitin-like protein that is conjugated to intracellular target proteins after IFN-alpha or IFN-beta stimulation. May also be involved in autocrine, paracrine and endocrine mechanisms, as in cell-to-cell signaling, possibly partly by inducing IFN-gamma secretion by monocytes and macrophages. Seems to display antiviral activity during viral infections |
| Interferon-induced guanylate-binding protein 2 | 37999845 | 66.7 | 2.6956 | 0.0000 | IFN immune response. Binds GTP, GDP and GMP. |
| Guanylate-binding protein 5 | 37999681 | 66.9 | 2.2865 | 0.0039 | IFN immune response |
| Interferon-inducible GTPase 1 | 88909159 | 47.6 | 1.9876 | 0.0139 | May play a role in resistance to intracellular pathogens. Has low GTPase activity. Has higher affinity for GDP than for GTP. |
| Immune-responsive gene 1 protein | 119364598 | 53.8 | 1.9306 | 0.0262 | Immune response - IFN-inducible gene |
| Tryptophanyl-tRNA synthetase, cytoplasmic | 51704226 | 54.4 | 1.1543 | 0.0117 | IFN-gamma induced protein. Protein synthesis |
| Prolow-density lipoprotein receptor-related protein 1 | 81867523 | 504.7 | 0.7061 | 0.0077 | Endocytic receptor involved in endocytosis and in phagocytosis of apoptotic cells. Involved in cellular lipid homeostasis. |
| Twinfilin-1 | 92090648 | 40.1 | 0.9064 | 0.0040 | Actin-binding protein involved in motile and morphological processes |
| Sialic acid binding Ig-like lectin 1, sialoadhesin | 187954777 | 183.1 | 0.8250 | 0.0186 | Macrophage-restricted adhesion molecule that mediates sialic-acid dependent binding to lymphocytes. |
| Pyruvate dehydrogenase (lipoamide) beta | 63101525 | 38.9 | 0.8850 | 0.0276 | Metabolism |
Protein quantitation was made using iTRAQ as performed on cell lysates of BMM from three separate experiments.
These experiments compared HIV-1/VSV infected murine BMM that were cocultured with murine Treg (117 label) to HIV-1/VSV infected murine BMM cultured alone (115 label).
P-value after Benjamini and Hochberg FDR adjustment.
Treg Suppression of Virus Release is Linked to the Upregulation of Interferon Stimulated Gene 15 (ISG15)
To elucidate the mechanisms by which Treg could alter functional outcomes for HIV-1-infected macrophages, we first focused on validating the enhanced antiviral immune response supported by protein changes observed after strict filtering of the iTRAQ data. Treg coculture resulted in upregulation of BMM expression of signal transducer and activator of transcription 1 (STAT1) and ISG15 (Figure 1A, B) found in type I IFN antiviral signaling.(34) Since ISG15 was reported to inhibit release of HIV-1 virions, we assayed whether the increased ISG15 expression observed resulted in reduced virion release.(34) HIV-1p24 levels in BMM cell lysates and culture supernatants were measured by Western blotting and ELISA. No changes in HIV-1p24 were observed between HIV-1/VSV infected BMMs and HIV-1/VSV infected BMMs cocultured with CD3/CD28-activated Treg or Tcon in cell lysates (Figure 1C). This reflected that the pseudotyped virus is not capable of spreading viral infection, so the level of HIV-1p24 is dependent only on the initial infective dose (Supplemental Figures 1B, C, Supporting Information). However, ELISA tests of culture supernatant fluids showed both Tcon and Treg inhibited virus release by reductions in HIV-1p24 concentrations (Figure 1D). Moreover, Treg more than Tcon (the inhibition rate is 79 versus 45%, respectively) inhibited HIV-1p24 release in culture fluids. We posit that such findings could result from stimulation of autocrine/paracrine pathways linked to innate IFN antiviral immunity regulated by Treg (Figure 1E).
Figure 1.

Treg inhibition of virus release from infected BMM is interferon-stimulated gene 15 pathway associated. (A) Western blotting showed increased STAT1 phosphorylation in HIV-1/VSV infected BMM cocultured with Treg group. Data shown are representative of three independent experiments. (B) Western blotting showed dramatically increased free ISG15 and ISGylated proteins in HIV-1/VSV infected BMM cocultured with Treg group. Data shown are representative of three independent experiments. (C) Western blotting showed no change of HIV-1 p24 expression between HIV-1/VSV infected BMM group, HIV-1/VSV infected BMM group cocultured with Tcon group and HIV-1/VSV infected BMM group cocultured with Treg group. In this study the pseudotyped virus we used could enter the mouse BMM once, so the level of p24 staining is dependent only on the infective dose. Data shown are representative of three independent experiments. (D) Both Tcon and Treg could inhibit HIV-1 release determined by HIV-1 p24 ELISA. Data shown are representative of three independent experiments. ***p < 0.001. (E) Proposed pathways involved in initiating antiviral immune response in HIV-1-infected macrophages, which was enhanced by Treg and Tcon. In macrophages, engagement of TLR3 by HIV derived dsRNA in endosomes mediates IFN-α/β/λ production. TLR3 induces a Trif-dependent pathway, which recruits kinases (TBK1, IKKϵ and IKKαβγ) that mediate activation of the transcription factors IRF3 and NF-κB. Together with IRF7, IRF3 and NF-κB translocate into the nucleus and bind to positive regulatory domains on the Ifn genes promoter, leading to Ifn transcription. Secreted IFNs signal through binding to their cognate receptors (IFNAR1 and IFNAR2) and then recruit kinases (TYK2 and JAK1) leading to translocation of phosphorylated transcription factors (STAT1 and STAT2) and IRF9 to the nucleus where binding to the enhancer region of the ifn promoter and isg promoter occurs, which leads to the activation and nuclear transport of ISGF3 and induction of ISRE to produce IFNs and ISGs. Both IFNs and ISGs could inhibit ubiquitination of HIV Gag and Tsg101, which results in less virus release. With unknown mechanisms, Treg exhibit much greater capacity to enhance the antiviral immune response than Tcon.
Treg Kills Infected BMM
In the past decade following Treg discovery, substantive research has focused on the mechanisms for immune tolerance by Treg through its inhibitory molecule expression and secretion, such as CTLA-4, GITR, IL-10 and TGF-β (reviewed by Zhu and Paul(35)). However, Tregs also exhibit perforin-dependent cytotoxicity against autologous target cells(36) indicating that Treg can act as natural cytotoxic cells, like natural killer cells and cytotoxic T lymphocytes. Based on such prior findings and our observations of granzymes in the infected BMM lysates treated with Treg by mass spectrometry, we hypothesized that Treg may induce HIV-1-infected macrophage death to deplete the cell source of neurotoxic factors. To test this hypothesis, we used terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay to assess the cytotoxicity of Treg against HIV-1/VSV infected BMM. Greater than 50% of BMMs were TUNEL positive in Treg treated cocultures (Figure 2A, B). The findings mirrored trypan blue exclusion tests (Figure 2A, B). Cytotoxicity of Treg was <10% in uninfected BMM (data not shown) supporting the notion that Treg are killers of activated or pathogen-altered BMM. Results were replicated in HIV-1ADA infected human MDM cocultured with human Treg (Supplemental Figure 4A−C, Supporting Information). In these assays human MDM were shrunken and detached following Treg cocultivation (Supplemental Figure 4C, Supporting Information). Taken together, the reductions seen in HIV-1p24 in macrophage culture supernatant fluids by Treg likely reflect both antiviral innate immunity and the killing of infected cells.
Figure 2.

Treg Induces Virus-Infected BMM Death. (A) Treg induces HIV-1/VSV-infected BMM death shown by TUNEL assay plus confocal immunofluorescence microscopy and trypan blue staining plus light microscopy. For TUNEL assay, bright field is shown in the top panel and the corresponding fluorescent field is shown in the middle panel. Trypan blue staining is shown in the bottom panel (trypan blue positive cells are indicated by yellow arrows). White scale bar represent 100 μm. (B) Graphs represent the mean percentage of TUNEL positive BMM and trypan blue positive BMM counted and calculated from five different fields (data were pooled from three independent experiments). Each bar represents a mean ± SD *p < 0.05, **p < 0.01, ***p < 0.001.
Treg Induces Infected BMM Apoptosis but not Pyroptosis
Based on these observations of Treg-induced control of viral infection, we next investigated molecular mechanisms underlying the Treg induced cytotoxic responses. Immunoblots for cleavage of procaspases-1 and 3 demonstrated that Treg induced HIV-1/VSV infected BMM apoptosis is caspase-3 specific. In contrast, Tcon induces infected BMM pyroptosis through caspase-1 (Figure 3A, B). HIV-1/VSV infection itself triggered caspase-1 activation in BMM and was enhanced by Tcon but was reduced by Treg (Figure 3A). Densitometric analysis of blots for caspase-1 subunits failed to reveal differences between caspase-1 p20 in HIV-1/VSV infected BMM alone versus infected BMM cocultured with Tcon (16 to 18% when compared to actin). However, there were differences in caspase-1 p10 observed between HIV-1/VSV infected BMM alone and HIV-1/VSV infected BMM cocultured with Tcon (11 to 17% when compared to actin). Futhermore, procaspase-1 was present in cell lysates from HIV-1/VSV infected BMM at 30% compared to actin, while 42% compared to actin from HIV-1/VSV infected BMM cocultured with Tcon. (Figure 3A). In addition, faint bands were observed in these blots for caspase-1 in the Treg treatment group and for caspase-3 in the Tcon treatement group, suggesting much limited activation. Apoptosis induction by Treg and pyroptosis by Tcon was detected independently using a Fluorochrome Inhibitor of Caspases Assay (FLICA) utilizing binding of fluorescent inhibitors to specific active caspases (Figure 3C). Replicate observations were also seen in human cells (Supplemental Figure 4A, Supporting Information).
Figure 3.

Treg and Tcon Induced Virus-Infected BMM Apoptosis and Pyroptosis. (A) Western blotting showed HIV-1/VSV infection triggers caspase-1 activation in BMM, which was enhanced by Tcon coculture while attenuated by Treg coculture. Data shown are representative of five independent experiments. (B) Western blotting showed Treg coculture activated caspase-3 pathway in HIV-1/VSV infection BMM. Data shown are representative of five independent experiments. (C) Caspase-1 activation was measured by FLICA assay plus immunofluorescence microscopy. White scale bars represent 50 μm. Caspase-3 and 7 activation was measured by FLICA assay plus immunofluorescence microscopy. White scale bars represent 50 μm. (D) Proposed mechanisms Treg and Tcon use to modulate HIV-1-infected macrophage functions. Upon infection, HIV-1 activates NF-κB and the inflammasome, which results in IL-1β release. Tcon worsened this response by secretion of proinflammatory cytokines. In addition, those proinflammatory cytokines increase mitochondrial activity, which could lead to energy exhaustion and pyroptosis. However, Treg use their anti-inflammatory cytokines to extinguish the inflammation resulting from virus infection. Treg induce infected BMM apoptosis. Compared with apoptosis, pyroptosis causes more ATP release. Excessive ATP is an important inflammatory molecule and could be hydrolyzed by ecto-nucleoside triphosphate diphosphohydrolase CD39 and CD73 expressed on Treg. Moreover, Treg could transport cAMP to infected BMM and contribute to inflammation resolution. The activation status of infected macrophage affects Th0 cell differentiation. For example, M1 induce Th1 and Th17, while M2 induce Th2 and iTreg. (E) Comparison of CD39 expression on Tcon and Treg was determined by flow cytometric analysis.
Comparisons between apoptosis and pyroptosis are illustrated in Figure 3D. Briefly, upon infection, HIV-1 activates nuclear factor-kappa beta (NF-κB) and the inflammasome in macrophages resulting in inflammatory cytokine release. Tcon enhanced this response (Figure 3C). In addition, as demonstrated, excessive activation commonly leads to energy exhaustion and pyroptosis. When compared to apoptosis, pyroptosis leads to increased adenosine triphosphate (ATP) release. Low concentration of ATP is a critical apoptotic cell find-me signal for scavenger recruitment. On the contrary, excessive ATP incites inflammation. Thus, enzymatic removal of excessive ATP is one strategy to resolve such inflammation, which could be achieved by Treg through its surface ectonucleoside triphosphate diphosphohydrolase, CD39 and CD73.(37) Indeed, Treg was found to have significantly greater CD39 expression than Tcon (Figure 3E).
Treg Uses Granzymes and Perforin to Kill Infected BMM
Previously, Grossman, et al. reported that activated human naturally occurring Treg utilized the perforin-granzyme A pathway to kill autologous target cells.(36) Meanwhile, they demonstrated that activated mouse naturally occurring Treg cells use perforin-granzyme B pathway to kill target cells. To compare the expression profile of granzymes in Tcon and Treg before adding them to the infected BMM, we used confocal microscopy and flow cytometry to evaluate granzyme A and B and perforin intensity. As shown in Supplemental Figure 5 (Supporting Information), both Tcon and Treg exhibited cytoplasmic granular staining of granzyme A, B and perforin. Treg showed larger size and more abundant granules than Tcon (Supplemental Figure 5A, Supporting Information). Flow cytometry results confirmed those observations (Supplemental Figures 5B, C, Supporting Information).
To verify that Treg cells inserted perforin into the HIV-1-infected cell membrane, an important step in transferring granzymes to target cells, we used immunoblotting assays to detect inserted perforin. A very clear band was observed in Treg treated cell groups, while a diminished one was in Tcon treated macrophages (Figure 4A). Next, we used intracellular staining and confocal microscopy to visualize granzymes and perforin in BMM. Both control and HIV-1/VSV infected BMM showed no staining, indicating BMM do not produce granzyme and perforin de novo. However, cytoplasmic granular staining of granzymes and perforin were seen in the Treg treated cells and less so in Tcon BMM cocultivations (Figure 4B).
Figure 4.

Treg Uses Granzyme A, B and Perforin to Kill Virus-Infected BMM. (A) Western blotting demonstrating increased p38 phosphorylation (upper panel) and perforin insertion (middle panel), which were normalized to β-actin (bottom panel) in HIV-1/VSV infected BMM cocultured with Treg compared with HIV-1/VSV infected BMM cocultured with Tcon groups. Data shown are representative of three independent experiments. (B) Confocal immunofluorescences of BMM with specific monoclonal antibodies against granzyme A (upper panel), granzyme B (middle panel) and perforin (bottom panel) show cytoplasmic granular staining of granzyme A (red), granzyme B (red) and perforin (red) in Tcon and Treg treated groups. Both control and HIV-1/VSV infected groups showed no staining of those antibodies, which means BMM did not produce granzyme and perforin. Higher intensity of granzyme and perforin were seen in the Treg treated group compared with the Tcon treated group. Data shown are representative of three independent experiments. White scale bars represent 5 μm. (C) Cleaved NDUFS3, the 30 kDa subunit of mitochondrial complex I and the important substrate of granzyme A, was shown in HIV-1/VSV infected BMM cocultured with Treg group by Western blotting. Data shown are representative of three independent experiments. (D) Membrane potential-dependent staining of mitochondria in HIV-1/VSV infected BMM by JC-1 visualized by fluorescence microscopy showed the loss of red JC-1 aggregates fluorescence and cytoplasmic diffusion of green monomer fluorescence following coculture with Treg.
It is reported that granzyme A accesses the mitochondrial matrix to cleave the novel identified substrate, NADH dehydrogenase [ubiquinone] iron−sulfur protein 3 (NDUFS3), through perforin to initiate caspase-independent cell death.38,39 Together with other subunits, NDUFS3 forms complex I of the electron transport chain in mitochondria. Its cleavage results in respiratory chain dysfunction and superoxide anion generation. Using Western blotting, we found a cleaved NDUFS3 band in HIV-1/VSV infected BMM cocultured with Treg. In addition, faint bands were observed on immunoblots of HIV-1/VSV infected BMM lysates with/out Tcon, indicating that NUDFS3 might be cleaved by other enzymes (Figure 4C). We also detected increased p38 phosphorylation in Treg treated BMM (Figure 4A). Since altered NUDFS3 and phosphorylated p38 suggest that mitochondrial function was affected, we used 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine iodide (JC-1) staining and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to evaluate its membrane potential and activity. Fluorescence microscopy showed the loss of red JC-1 aggregates fluorescence and cytoplasmic diffusion of green monomer fluorescence in BMM following Treg coculture (Figure 4D). Light microscopy showed reduced color intensities/cell and overall levels of MTT in Treg treated BMM. In contrast, for infected BMM cocultured with Tcon, MTT staining intensities were increased in infected cells (Supplemental Figure 6A, Supporting Information). Such Treg induced cytotoxicity on infected BMM was cell−cell contact dependent as demonstrated by transwell cocultures (Supplemental Figure 6B, Supporting Information).
Treg Induces Virus Infected BMM Phenotypic Switches by Intercellular Transportation of cAMP
We noted that among all cell death assays we used in this study, only around 50% of cells are killed by Treg. In addition, greater than 98% of BMM were infected as determined by p24 immunoflurescence staining (Supplemental Figure 1B, Supporting Information). So why did Treg only kill 50% of the cells? We observed that the privileged BMM underwent remarkable cytoskeletal rearrangement (Figure 5A), which may indicate its polarization switching.(40) As shown in Figure 5A, Treg induced changes in infected BMM morphology from a polarized to a rounded appearance with contracted processes showing redistribution of actin from focal adhesions to a cortical distribution. Such alterations paralleled functional outcomes in the BMM proteome (Table 1 and data not shown). We hypothesized that a polarization switch in this model could be due to either direct effects of Treg or in response to cytotoxicity induced by Treg. To test this, we used Western blotting to detect expression of inducible nitric oxide synthase (iNOS) and arginase-1 (Arg-1), which are putative markers for classical activation macrophage (M1) and alternative activation macrophage (M2), respectively. We found that Treg downregulated iNOS 35-fold, with a concomitant 18-fold upregulation of Arg-1 (Figure 5B). The decreased NO production by HIV-1/VSV-infected BMM cocultured with Treg supports this transformation. This is in stark contrast to the significantly increased NO production by infected BMM in the presence of Tcon (Figure 5C).
Figure 5.

Treg Transforms the Infected BMM. (A) Treg induces HIV-1/VSV infected BMM morphology change from elongation to roundness shown by confocal immunofluorescence microscopy (zoomed part is shown at the bottom panel). White scale bars represent 20 μm. (B) Western blotting showed Treg downregulated iNOS, while upregulated arginase-1. (C) The concentration of NO in supernatant was measured with Griess test. Treg inhibited NO production from HIV-1/VSV infected BMM, ***p < 0.001. (D) Concentrations of cAMP in the lysate (L) and supernatant (S) of Tcon or Treg were detected by ELISA, *p < 0.05 (E) Concentrations of cAMP in the lysate and supernatant of BMM with different treatments. The cAMP in the supernatant was undetectable, while its concentration in lysate increased in Treg treated group. ***p < 0.001.
To decipher the mechanisms underlying this phenotypic switch in a strictly cell contact-dependent manner, we measured the levels of cyclic adenosine monophosphate (cAMP) in the culture supernatants and cell lysates, based on two important findings: cAMP is reported to modulate iNOS and Arg-1 expression in macrophages;(41) and Treg is reported to transport cAMP to target cells via gap junctions.(42) Indeed, we found no detectable cAMP in T cell or BMM culture supernatants (Figure 5D, E). However, compared with Tcon, Treg harbors higher levels of cAMP. And most importantly, coculture of infected BMM with Treg resulted in increased intracellular cAMP (5-fold) but not with Tcon (Figure 5E). Since cAMP in supernatants is undetectable, we believe it may have traversed membranes via gap junctions.
An alternative hypothesis is that production of adenosine as a result of coculture of Treg expressing CD39 likely contributed to the increased intracellular cAMP production through adenosine receptor signaling. Both mechanisms could be involved and would be up to future studies to determine.
Treg Elicits Infected BMM-Mediated Neuroprotective Responses
Given the multiple mechanisms of Treg presented herein used to modulate HIV-1/VSV infected macrophages (Supplemental Figure 7, Supporting Information), we expected that Treg must have neuroprotective roles due to their abilities to inhibit viremia and resolve inflammation. Indeed, our lab recently showed the neuroprotective properties of Treg in an HIVE model.(9) To confirm this anticipation, we used conditioned medium to treat primary neurons in vitro. Neuroprotection is readily observed in Treg-treated BMM groups (Figure 6). Comparisons between neurons treated with conditioned media from HIV-1/VSV infected BMM in the absence or presence of Treg and Tcon are shown (Figure 6 and Supplemental Table 1, Supporting Information). Furthermore, addition of media from uninfected macrophages was not observed to result in any neurotoxicity (Figure 6 and data not shown).
Figure 6.

Treg-induced infected BMM elicits neuroprotection. Tubulin staining shows persevered neuronal dendrites in groups cultured with both CM1 (upper panel) and CM2 (bottom panel) from Treg treated HIV-1/VSV-infected BMM, which was not seen in Tcon derived CM. Moreover, CM2 from the Tcon treated group exhibits more toxicity than the infected group, which suggests that the proinflammatory cytokines are more destructive than viral proteins. White scale bar represents 50 μm.
Discussion
Broad functions of Treg were identified in the current report and beyond what is known for immune suppression. For the HIV-1-infected macrophage this includes enhancement of innate antiviral immunity, polarization in control of inflammatory activities and toxic secretory responses, and immunosurveillance in the killing of virus-infected macrophages. For the latter Treg utilize several mechanisms including granzyme A, B and perforin. In addition, Treg also altered redox biology to lower levels of oxidative stress.(9) These results, taken together, demonstrate a much broader range of functions of these regulatory cells than previously appreciated and help explain their control of disease. This of course includes HAND which is rarely seen during subclinical disease and immunocompetence.(43)
The abilities of ART in lowering HAND severity have long been appreciated.(44) The mechanisms described revolve around the number and function of CD8+ CTLs resulting in the elimination of infected cells. However, ART also preserves CD4+ T cell populations. CD4+ T cells orchestrate innate and adaptive immune responses to HIV. Importantly, Th1, Th2, Th17 and Treg are heterogeneous in function based, in large measure, on their cytokine secretion and specific transcription factor expression. Indeed, cytokine profiles, tissue environment, microbial infection or injuries affect the cells differential fates.(35) It has long been appreciated that Treg control immune responses to infections, but whether the ultimate outcome from Treg actions in disease is protective or destructive remains unresolved.14,45,46 For the better part of its existence Treg serve to limit damage caused by excessive virus-induced inflammatory responses.(47) However, for HIV-1 infection, its role can be seen as a double-edged sword. Treg-mediated suppression can lead to cessation of antiretroviral immune responses and in so doing allow productive viral replication.15,20,48 It is possible that effector T cell responses could prevent Treg-mediated suppression of antiviral responses; especially those mediated through soluble factors, but could still allow Treg killing of infected macrophages. Moreover, excessive immune activation can incite secondary disease manifestations.49,50 On the other hand, secondary end organ effects of virus that include kidney, liver, lung, and brain pathologies could be attenuated by Treg through control of inflammatory responses. It is of interest to note that changes in numbers and function of Treg in lymphoid tissues and blood during HIV-1 infection are evolutionary.26,51 Commonly, an increased frequency of Treg is observed in HIV-1-infected individuals and notably in those with readily detectable plasma viremia.(52) Thus, whether Treg are ultimately beneficial for HIV disease still remains an active source of discussion and debate.9,52−54
For HAND, the roles of Treg are likely better defined.(9) Indeed, our previous works in murine models of HIVE using infected bone marrow-derived macrophages showed that following adoptive transfer of Treg astrogliosis and microgliosis were attenuated with substantive neuroprotection.(9) Such observations parallel our laboratory’s results in an experimental 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of PD.7,28 The current study clearly illustrates control of inflammatory effector functions by CD4+CD25+ Treg for HIV-infected macrophages. Furthermore, Treg are effective in inhibiting HIV-1 replication and release.(9) Indeed, we recently demonstrated Treg abiltity to migrate to sites of inflammation in the brains of HIVE mice resulting in decreased HIV-1p24 and gliosis, with a concomitant increase in neurotrophins.(55) These functions support a multifaceted and beneficial role of Treg in HIV infection and help further explain the neuroprotection provided, notably during ART where adaptive immunity is largely preserved.(56)
The observations that Treg induce interferon-mediated antiviral immunity and kill HIV-1-infected MPs by caspase-3 and granzyme/perforin pathways, depleting cellular sources of neurotoxic factors is of importance. Indeed, Treg are capable of killing a variety of autologous target effector cells, including CD4+ and CD8+ T cells, CD14+ monocytes, and dendritic cells.(36) Furthermore, Treg preferentially kill activated T cells.(36) Our results demonstrate that HIV-activated M1 macrophages are also killed more so than uninfected cells. The phenotypic switch from M1 to M2 for the macrophages is attributed to Treg in the setting of HIV infection but the mechanism by which this occurs is yet to be determined, but may involve both cytokine dependent and independent pathways.(57) Indeed, our work suggests that direct cAMP transfer to infected cells and/or adenosine signaling are responsible, in part, for the phenotypic switch.42,58,59 Adenosine signaling has been shown in macrophages to reduce the production of pro-inflammatory cytokines including TNF-α, IL-12, MIP-1, and NO, as well as increase production of IL-10.60,61 Furthermore, adenosine signaling can increase intracellular cAMP and activate p38 pathways, consistent with our findings.(62) Thus, killing of HIV-1-infected BMM by Treg may be a result of both the antiviral response itself and the BMM activation state.
Regardless of the mechanism, a Treg directed phenotypic switch from M1 to M2 phenotype is advantageous for neuronal survival. The M1 phenotype is pro-inflammatory and is associated with neurodegeneration and increased viremia.63,64 An M2 phenotype however, is associated with tissue repair, anti-inflammatory cytokine production, and decreased ROS production.9,65,10 Thus an M2 phenotypic transformation in the setting of an inflammatory environment such as in HAND can contribute to resolution of inflammation and neuroprotection observed with Treg treatment.(9) Furthermore, some evidence suggests that M2 macrophages retain their phagocytic activity and have enhanced intracellular killing capability.(66)
As major CNS APCs and a source of cytokines and chemokines, perivascular macrophages and microglia strongly influence the activation state of astrocytes, and influence the trafficking of adaptive immune cells into the brain.(67) Astrocytes also can contribute to HIV-1 induced CNS damage as a potent source of CXCL10 production in HIV-1-infected individuals, especially with TNF-α and IFN-γ production by MPs. Importantly, Treg are able to decrease the production of these cytokines that could ultimately diminish astrocyte CXCL10 production.(9) Furthermore, HIV-1-infected macrophages induce astrocyte CXCL8 secretion, a chemokine that enhances chemotaxis of neutrophils and monocytes in HAD.(68) These findings illustrate the interaction between glia and immune cells that can contribute to neuroinflammation or, when influenced by Treg, resolution of inflammation. Recognition of the multifaceted functions of Treg is an important first step toward a complete understanding of the broad roles of adaptive immunity in the setting of HIV infection and particularly that of the CNS. This is certainly a vital part of the complex cellular and viral interplay present during HAND. Indeed, Treg also have the capacity to attenuate inflammation in a range of neurodegenerative diseases and may be harnessed for therapeuptic gain.(69)
Acknowledgments
We thank Ms. Robin Taylor for editorial support, Ms. Li Wu for providing mouse neurons, and James Talaska and Janice Taylor of the Confocal Laser Scanning Microscope Core Facility, University of Nebraska Medical Center, for providing technical assistance. We also thank Victoria B. Smith and Megan Michalak of the Cell Analysis Facility, University of Nebraska Medical Center, for providing assistance with flow cytometry. This work was supported by the National Key Basic Research Program of China 2006CB504201 (to Y.Z.) and National Institutes of Health United States of America Grants P20 RR15635, 1 P01 NS043985-01, 2R37 NS36126 and 5 P01 MH64570-03 (to H.E.G.).
Supporting Information Available
Supplementary tables and figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Robertson K. R.; Smurzynski M.; Parsons T. D.; Wu K.; Bosch R. J.; Wu J.; McArthur J. C.; Collier A. C.; Evans S. R.; Ellis R. J. The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS 2007, 21 (14), 1915–21. [DOI] [PubMed] [Google Scholar]
- Antinori A.; Arendt G.; Becker J. T.; Brew B. J.; Byrd D. A.; Cherner M.; Clifford D. B.; Cinque P.; Epstein L. G.; Goodkin K.; Gisslen M.; Grant I.; Heaton R. K.; Joseph J.; Marder K.; Marra C. M.; McArthur J. C.; Nunn M.; Price R. W.; Pulliam L.; Robertson K. R.; Sacktor N.; Valcour V.; Wojna V. E. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69 (18), 1789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur J. C.; Brew B. J.; Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005, 4 (9), 543–55. [DOI] [PubMed] [Google Scholar]
- Kraft-Terry S. D.; Buch S. J.; Fox H. S.; Gendelman H. E. A coat of many colors: neuroimmune crosstalk in human immunodeficiency virus infection. Neuron 2009, 64 (1), 133–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everall I. P.; Hansen L. A.; Masliah E. The shifting patterns of HIV encephalitis neuropathology. Neurotox. Res. 2005, 8 (1−2), 51–61. [DOI] [PubMed] [Google Scholar]
- Price R. W.; Epstein L. G.; Becker J. T.; Cinque P.; Gisslen M.; Pulliam L.; McArthur J. C. Biomarkers of HIV-1 CNS infection and injury. Neurology 2007, 69 (18), 1781–8. [DOI] [PubMed] [Google Scholar]
- Reynolds A. D.; Stone D. K.; Hutter J. A.; Benner E. J.; Mosley R. L.; Gendelman H. E. Regulatory T cells attenuate th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J. Immunol. 2010, 184 (5), 2261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A. D.; Stone D. K.; Mosley R. L.; Gendelman H. E. Proteomic studies of nitrated alpha-synuclein microglia regulation by CD4+CD25+ T cells. J. Proteome Res. 2009, 8 (7), 3497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Gong N.; Huang X.; Reynolds A. D.; Mosley R. L.; Gendelman H. E. Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. J. Immunol. 2009, 182 (6), 3855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A. D.; Stone D. K.; Mosley R. L.; Gendelman H. E. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J. Immunol. 2009, 182 (7), 4137–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A. D.; Kadiu I.; Garg S. K.; Glanzer J. G.; Nordgren T.; Ciborowski P.; Banerjee R.; Gendelman H. E. Nitrated alpha-synuclein and microglial neuroregulatory activities. J. Neuroimmune Pharmacol. 2008, 3 (2), 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone D. K.; Reynolds A. D.; Mosley R. L.; Gendelman H. E. Innate and adaptive immunity for the pathobiology of Parkinson’s disease. Antioxid. Redox Signal. 2009, 11 (9), 2151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Zhang Y.; Wu M.; Li J.; Zhou W.; Li G.; Li X.; Xiao B.; Christadoss P. Suppression of ongoing experimental autoimmune myasthenia gravis by transfer of RelB-silenced bone marrow dentritic cells is associated with a change from a T helper Th17/Th1 to a Th2 and FoxP3+ regulatory T-cell profile. Inflamm. Res. 2010, 59 (3), 197–205. [DOI] [PubMed] [Google Scholar]
- Macatangay B. J.; Szajnik M. E.; Whiteside T. L.; Riddler S. A.; Rinaldo C. R. Regulatory T cell suppression of Gag-specific CD8 T cell polyfunctional response after therapeutic vaccination of HIV-1-infected patients on ART. PLoS One 2010, 5 (3), e9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorborn G.; Pomeroy L.; Isohanni H.; Perry M.; Peters B.; Vyakarnam A. Increased sensitivity of CD4+ T-effector cells to CD4+CD25+ Treg suppression compensates for reduced Treg number in asymptomatic HIV-1 infection. PLoS One 2010, 5 (2), e9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antons A. K.; Wang R.; Oswald-Richter K.; Tseng M.; Arendt C. W.; Kalams S. A.; Unutmaz D. Naive precursors of human regulatory T cells require FoxP3 for suppression and are susceptible to HIV infection. J. Immunol. 2008, 180 (2), 764–73. [DOI] [PubMed] [Google Scholar]
- Hu H.; Fernando K.; Ni H.; Weissman D. HIV envelope suppresses CD4+ T cell activation independent of T regulatory cells. J. Immunol. 2008, 180 (8), 5593–600. [DOI] [PubMed] [Google Scholar]
- Hryniewicz A.; Boasso A.; Edghill-Smith Y.; Vaccari M.; Fuchs D.; Venzon D.; Nacsa J.; Betts M. R.; Tsai W. P.; Heraud J. M.; Beer B.; Blanset D.; Chougnet C.; Lowy I.; Shearer G. M.; Franchini G. CTLA-4 blockade decreases TGF-beta, IDO, and viral RNA expression in tissues of SIVmac251-infected macaques. Blood 2006, 108 (12), 3834–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boasso A.; Vaccari M.; Nilsson J.; Shearer G. M.; Andersson J.; Cecchinato V.; Chougnet C.; Franchini G. Do regulatory T-cells play a role in AIDS pathogenesis. AIDS Rev. 2006, 8 (3), 141–7. [PubMed] [Google Scholar]
- Del Pozo-Balado Mdel M.; Leal M.; Mendez-Lagares G.; Pacheco Y. M. CD4(+)CD25(+/hi)CD127(lo) phenotype does not accurately identify regulatory T cells in all populations of HIV-infected persons. J. Infect. Dis. 2010, 201 (3), 331–5. [DOI] [PubMed] [Google Scholar]
- Owen R. E.; Heitman J. W.; Hirschkorn D. F.; Lanteri M. C.; Biswas H. H.; Martin J. N.; Krone M. R.; Deeks S. G.; Norris P. J. HIV+ elite controllers have low HIV-specific T-cell activation yet maintain strong, polyfunctional T-cell responses. AIDS 2010, 24 (8), 1095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast A.; Prado J. G.; Kang Y. H.; Chen F.; Riddell L. A.; Luzzi G.; Goulder P.; Klenerman P. HIV-1 infection is characterized by profound depletion of CD161+ Th17 cells and gradual decline in regulatory T cells. AIDS 2010, 24 (4), 491–502. [DOI] [PubMed] [Google Scholar]
- Mendez Lagares G.; Leal Noval M.; Del Pozo Balado M. D.; Leon J. A.; Pacheco Y. M. Is it age or HIV that drives the regulatory T-cells expansion that occurs in older HIV-infected persons. Clin. Immunol. 2010, 136 (1), 157–9. [DOI] [PubMed] [Google Scholar]
- Pereira L. E.; Villinger F.; Onlamoon N.; Bryan P.; Cardona A.; Pattanapanysat K.; Mori K.; Hagen S.; Picker L.; Ansari A. A. Simian immunodeficiency virus (SIV) infection influences the level and function of regulatory T cells in SIV-infected rhesus macaques but not SIV-infected sooty mangabeys. J. Virol. 2007, 81 (9), 4445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase A. J.; Yang H. C.; Zhang H.; Blankson J. N.; Siliciano R. F. Preservation of FoxP3+ regulatory T cells in the peripheral blood of human immunodeficiency virus type 1-infected elite suppressors correlates with low CD4+ T-cell activation. J. Virol. 2008, 82 (17), 8307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinter A.; McNally J.; Riggin L.; Jackson R.; Roby G.; Fauci A. S. Suppression of HIV-specific T cell activity by lymph node CD25+ regulatory T cells from HIV-infected individuals. Proc. Natl. Acad. Sci. U.S.A. 2007, 104 (9), 3390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canki M.; Thai J. N.; Chao W.; Ghorpade A.; Potash M. J.; Volsky D. J. Highly productive infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) indicates no intracellular restrictions to HIV-1 replication in primary human astrocytes. J. Virol. 2001, 75 (17), 7925–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A. D.; Banerjee R.; Liu J.; Gendelman H. E.; Mosley R. L. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J. Leukoc. Biol. 2007, 82 (5), 1083–94. [DOI] [PubMed] [Google Scholar]
- Banerjee R.; Mosley R. L.; Reynolds A. D.; Dhar A.; Jackson-Lewis V.; Gordon P. H.; Przedborski S.; Gendelman H. E. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS One 2008, 3 (7), e2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendelman H. E.; Orenstein J. M.; Martin M. A.; Ferrua C.; Mitra R.; Phipps T.; Wahl L. A.; Lane H. C.; Fauci A. S.; Burke D. S.; et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 1988, 167 (4), 1428–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkina S. K.; Zhang Y.; Nelsestuen G. L.; Oetting W. S.; Ibrahlm H. N. Temporal stability of the urinary proteome after kidney transplant: more sensitive than protein composition. J. Proteome Res. 2009, 8 (1), 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberg A. L.; Mahoney D. W.; Eckel-Passow J. E.; Malone C. J.; Wolfinger R. D.; Hill E. G.; Cooper L. T.; Onuma O. K.; Spiro C.; Therneau T. M.; Bergen H. R. 3rd. Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 2008, 7 (1), 225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y.; Drai D.; Elmer G.; Kafkafi N.; Golani I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125 (1−2), 279–84. [DOI] [PubMed] [Google Scholar]
- Okumura A.; Lu G.; Pitha-Rowe I.; Pitha P. M. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (5), 1440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Paul W. E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010, 20 (1), 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman W. J.; Verbsky J. W.; Barchet W.; Colonna M.; Atkinson J. P.; Ley T. J. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004, 21 (4), 589–601. [DOI] [PubMed] [Google Scholar]
- Dwyer K. M.; Deaglio S.; Gao W.; Friedman D.; Strom T. B.; Robson S. C. CD39 and control of cellular immune responses. Purinergic Signal. 2007, 3 (1−2), 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinvalet D.; Thiery J.; Chowdhury D. Granzymes and cell death. Methods Enzymol. 2008, 442, 213–30. [DOI] [PubMed] [Google Scholar]
- Martinvalet D.; Dykxhoorn D. M.; Ferrini R.; Lieberman J. Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 2008, 133 (4), 681–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrin P.; Surprenant A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. EMBO J. 2009, 28 (14), 2114–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johann A. M.; Barra V.; Kuhn A. M.; Weigert A.; von Knethen A.; Brune B. Apoptotic cells induce arginase II in macrophages, thereby attenuating NO production. FASEB J. 2007, 21 (11), 2704–12. [DOI] [PubMed] [Google Scholar]
- Bopp T.; Becker C.; Klein M.; Klein-Hessling S.; Palmetshofer A.; Serfling E.; Heib V.; Becker M.; Kubach J.; Schmitt S.; Stoll S.; Schild H.; Staege M. S.; Stassen M.; Jonuleit H.; Schmitt E. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 2007, 204 (6), 1303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur J. C.; Steiner J.; Sacktor N.; Nath A. Human immunodeficiency virus-associated neurocognitive disorders: Mind the gap. Ann. Neurol. 2010, 67 (6), 699–714. [DOI] [PubMed] [Google Scholar]
- Kraft-Terry S. D.; Stothert A. R.; Buch S.; Gendelman H. E. HIV-1 neuroimmunity in the era of antiretroviral therapy. Neurobiol. Dis. 37 (3), 542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loures F. V.; Pina A.; Felonato M.; Araujo E. F.; Leite K. R.; Calich V. L. Toll-like receptor 4 signaling leads to severe fungal infection associated with enhanced proinflammatory immunity and impaired expansion of regulatory T cells. Infect. Immun. 2010, 78 (3), 1078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali D. A.; Collison L. W.; Workman C. J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8 (7), 523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvas S.; Rouse B. T. Treg control of antimicrobial T cell responses. Curr. Opin. Immunol. 2006, 18 (3), 344–8. [DOI] [PubMed] [Google Scholar]
- Lim A.; French M. A.; Price P. CD4+ and CD8+ T cells expressing FoxP3 in HIV-infected patients are phenotypically distinct and influenced by disease severity and antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2009, 51 (3), 248–57. [DOI] [PubMed] [Google Scholar]
- Crowe S. M.; Westhorpe C. L.; Mukhamedova N.; Jaworowski A.; Sviridov D.; Bukrinsky M. The macrophage: the intersection between HIV infection and atherosclerosis. J. Leukoc. Biol. 2010, 87 (4), 589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre D.; Speranza F.; Martegani R. Impact of highly active antiretroviral therapy on organ-specific manifestations of HIV-1 infection. HIV Med. 2005, 6 (2), 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi S.; Iwasaki T.; Imado T.; Higasa S.; Kakishita E.; Shirasaka T.; Sano H. Relationship of CD4+CD25+ regulatory T cells to immune status in HIV-infected patients. AIDS 2005, 19 (9), 879–86. [DOI] [PubMed] [Google Scholar]
- Cao W.; Jamieson B. D.; Hultin L. E.; Hultin P. M.; Detels R. Regulatory T cell expansion and immune activation during untreated HIV type 1 infection are associated with disease progression. AIDS Res. Hum. Retroviruses 2009, 25 (2), 183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes S. S.; Borges I. K.; Lima J. E.; Milanez Pde A.; Conchon-Costa I.; Felipe I.; Saridakis H. O.; Watanabe M. A. Involvement of regulatory T cells in HIV immunopathogenesis. Curr. HIV Res. 2010, 8 (4), 340–6. [DOI] [PubMed] [Google Scholar]
- Card C. M.; McLaren P. J.; Wachihi C.; Kimani J.; Plummer F. A.; Fowke K. R. Decreased immune activation in resistance to HIV-1 infection is associated with an elevated frequency of CD4(+)CD25(+)FOXP3(+) regulatory T cells. J. Infect. Dis. 2009, 199 (9), 1318–22. [DOI] [PubMed] [Google Scholar]
- Gong N.; Liu J.; Reynolds A. D.; Gorantla S.; Mosley R. L.; Gendelman H. E. Brain ingress of regulatory T cells in a murine model of HIV-1 encephalitis. J. Neuroimmunol, available online September 16, 2010, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrisekoop N.; van Gent R.; de Boer A. B.; Otto S. A.; Borleffs J. C.; Steingrover R.; Prins J. M.; Kuijpers T. W.; Wolfs T. F.; Geelen S. P.; Vulto I.; Lansdorp P.; Tesselaar K.; Borghans J. A.; Miedema F. Restoration of the CD4 T cell compartment after long-term highly active antiretroviral therapy without phenotypical signs of accelerated immunological aging. J. Immunol. 2008, 181 (2), 1573–81. [DOI] [PubMed] [Google Scholar]
- Tiemessen M. M.; Jagger A. L.; Evans H. G.; van Herwijnen M. J.; John S.; Taams L. S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. U.S.A. 2007, 104 (49), 19446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Golden M. Putting on the brakes: cyclic AMP as a multipronged controller of macrophage function. Sci. Signal. 2009, 2 (75), pe37. [DOI] [PubMed] [Google Scholar]
- Barnholt K. E.; Kota R. S.; Aung H. H.; Rutledge J. C. Adenosine blocks IFN-gamma-induced phosphorylation of STAT1 on serine 727 to reduce macrophage activation. J. Immunol. 2009, 183 (10), 6767–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G.; Szabo C.; Nemeth Z. H.; Kvetan V.; Pastores S. M.; Vizi E. S. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J. Immunol. 1996, 157 (10), 4634–40. [PubMed] [Google Scholar]
- Hasko G.; Kuhel D. G.; Chen J. F.; Schwarzschild M. A.; Deitch E. A.; Mabley J. G.; Marton A.; Szabo C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J. 2000, 14 (13), 2065–74. [DOI] [PubMed] [Google Scholar]
- Nemeth Z. H.; Leibovich S. J.; Deitch E. A.; Sperlagh B.; Virag L.; Vizi E. S.; Szabo C.; Hasko G. Adenosine stimulates CREB activation in macrophages via a p38 MAPK-mediated mechanism. Biochem. Biophys. Res. Commun. 2003, 312 (4), 883–8. [DOI] [PubMed] [Google Scholar]
- Lane J. H.; Sasseville V. G.; Smith M. O.; Vogel P.; Pauley D. R.; Heyes M. P.; Lackner A. A. Neuroinvasion by simian immunodeficiency virus coincides with increased numbers of perivascular macrophages/microglia and intrathecal immune activation. J. Neurovirol. 1996, 2 (6), 423–32. [DOI] [PubMed] [Google Scholar]
- Regis E. G.; Barreto-de-Souza V.; Morgado M. G.; Bozza M. T.; Leng L.; Bucala R.; Bou-Habib D. C. Elevated levels of macrophage migration inhibitory factor (MIF) in the plasma of HIV-1-infected patients and in HIV-1-infected cell cultures: a relevant role on viral replication. Virology 399 (1), 31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbein G.; Varin A. The macrophage in HIV-1 infection: from activation to deactivation. Retrovirology 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groesdonk H. V.; Schlottmann S.; Richter F.; Georgieff M.; Senftleben U. Escherichia coli prevents phagocytosis-induced death of macrophages via classical NF-kappaB signaling, a link to T-cell activation. Infect. Immun. 2006, 74 (10), 5989–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadiu I.; Glanzer J. G.; Kipnis J.; Gendelman H. E.; Thomas M. P. Mononuclear phagocytes in the pathogenesis of neurodegenerative diseases. Neurotox. Res. 2005, 8 (1−2), 25–50. [DOI] [PubMed] [Google Scholar]
- Zheng J. C.; Huang Y.; Tang K.; Cui M.; Niemann D.; Lopez A.; Morgello S.; Chen S. HIV-1-infected and/or immune-activated macrophages regulate astrocyte CXCL8 production through IL-1beta and TNF-alpha: involvement of mitogen-activated protein kinases and protein kinase R. J. Neuroimmunol. 2008, 200 (1−2), 100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosloski L. M.; Ha D. M.; Stone D. K.; Hutter J. A.; Pichler M. R.; Reynolds A. D.; Gendelman H. E.; Lee Mosley R. Adaptive Immune Regulation of Glial Homeostasis as an Immunization Strategy for Neurodegenerative Diseases. J. Neurochem. 2010, 114 (5), 1261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.