

Abstract
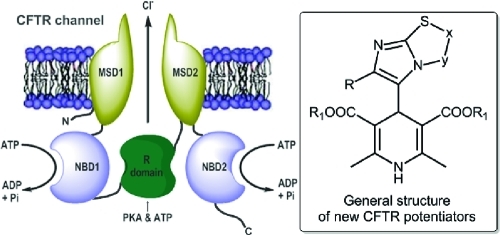
The pharmacology of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl– channel has attracted significant interest in recent years with the aim to search for rational new therapies for diseases caused by CFTR malfunction. Mutations that abolish the function of CFTR cause the life-threatening genetic disease cystic fibrosis (CF). The most common cause of CF is the deletion of phenylalanine 508 (ΔF508) in the CFTR chloride channel. Felodipine, nifedipine, and other antihypertensive 1,4-dihydropyridines (1,4-DHPs) that block L-type Ca2+ channels are also effective potentiators of CFTR gating, able to correct the defective activity of ΔF508 and other CFTR mutants (Mol. Pharmacol. 2005, 54, 1736.). For this purpose, we evaluated the ability of the previously and newly synthesized 4-imidazo[2,1-b]thiazoles-1,4-dihydropyridines without vascular activity and inotropic and/or chronotropic cardiac effects (J. Med. Chem. 2008, 54, 1592) to enhance the activity of ΔF508-CFTR. Our studies indicate compounds 17, 18, 20, 21, 38, and 39 as 1,4-DHPs with an interesting profile of activity.
Introduction
Cystic fibrosis (CF), the most common channelopathy, is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.1 The CFTR protein works as a cAMP-activated plasma membrane Cl– channel. As a matter of fact, in CF, the lack of CFTR Cl– channel function in the apical membranes of many epithelial cells prevents the normal secretion of Cl– onto the epithelial surface. There is also a loss of control of Na+ channels, resulting in increased fluid reabsorption. Consequently, there is a production of a thick and dehydrated mucus that is responsible for many of the most important manifestations of the disease in the respiratory tract. In particular, the impairment of mucociliary clearance in the airways, due to altered ion/fluid transport, favors the colonization of the airway surface by antibiotic-resistant bacteria. The presence of bacteria triggers in turn a severe inflammatory response that irreversibly damages the lung.2−4 The CFTR channel contains 12 transmembrane (TM) domains, with TM1, TM6 and TM12 contributing to the formation of the ion pore. In addition, there are two domains that bind cytoplasmic nucleotides (NBD1 and NBD2) and a regulatory domain. The channel is regulated by binding of ATP to NBDs and by the cAMP-dependent phosphorylation of serine residues in the regulatory domain. Deletion of phenylalanine (ΔF508) in NBD1 occurs in nearly 70% of patients with CF.5 ΔF508 causes two types of problems: an impaired trafficking of the protein and a defect in channel activity. The former defect is the most severe consequence of the mutation, consisting of the inability of the CFTR protein to exit the endoplasmic reticulum. The latter defect appears instead as a partial response to the cAMP activating stimulus. The two defects can be separately treated by pharmacological compounds called correctors and potentiators, respectively.6−9 Besides ΔF508, other CF mutations are also sensitive to potentiators.10
Antihypertensive 1,4-dihydropyridines (1,4-DHPs) are a well-known class of drugs that block voltage-dependent Ca2+ channels. Many DHPs are also effective potentiators of CFTR gating, able to correct the defective activity of ΔF508 and other CFTR mutants.8 Activation of mutant CFTR by 1,4-DHPs occurs through a mechanism that does not involve the inhibition of Ca2+ channels but possibly a direct interaction with the CFTR protein itself.9 Therefore, 1,4-DHPs represent an interesting family of compounds for the development of effective drugs for CF therapy. Indeed, they have been extensively studied, and a large amount of information is available on their medicinal chemistry properties.11 However, optimization of potency for CFTR versus Ca2+ channels is required to minimize possible side effects. In fact, felodipine, nifedipine, and other 1,4-DHPs used as drugs activate CFTR at concentrations that are considerably higher than those effective on Ca2+ channels. A better understanding of structural features required for CFTR activation may help in the development of more selective and effective DHP-based potentiators.7−9 In a recent study, we have published the synthesis, characterization, and functional in vitro assays in cardiac tissues and smooth muscle (vascular and nonvascular) of a number of 4-imidazo[2,1-b]thiazole-1,4-dihydropyridines.12 The binding properties for the novel compounds have been investigated, and the interaction with the binding site common to other 4-aryl-1,4-DHPs has been demonstrated. Interestingly, the novel 4-aryldihydropyridines are L-type calcium channel blockers with a peculiar pharmacological behavior. Indeed, the imidazo[2,1-b]thiazole system is found to confer to the 1,4-dihydropyridine scaffold an inotropic and/or chronotropic cardiovascular activity with a high selectivity toward the nonvascular tissue. With this information in mind, we designed and synthesized new imidazo[2,1-b]thiazole derivatives 16–26, and in an attempt to get major insights on structural features required for specific CFTR activation, we screened a set of newly (16–26) and previously synthesized 1,4-DHPs (27–47) (Chart 1) using cell-based assays to determine their activity as potentiator on ΔF508-CFTR. Moreover, we report functional studies to define the cardiovascular activity profile, the relaxant activity on K+-depolarized guinea pig ileum longitudinal smooth muscle, and the bronchodilatation effect on guinea pig trachea with the aim to evaluate potency for CFTR vs L-type calcium channels (LTCCs). In our analysis of cardiac and smooth muscle function, we have also included 1,4-DHPs [48 (DHP-256), 49 (DHP-194), 50 (DHP-229)]13 as reference compounds previously reported as CFTR potentiators with reduced potency on Ca2+ channels. Our results show that modifications of the 1,4-DHP scaffold, particularly in the 4-phenyl ring at C4, may lead to compounds with highly improved selectivity as CFTR potentiators.
Chart 1.

Chemistry
As shown in Scheme 1, the syntheses of the compounds 16–26 (Table 1) were accomplished starting from the bicyclic heterocycles obtained by cyclocondensation of the appropriate 2-aminothiazoles 1 and 2 and bromoketones 3 and 4. Imidazo[2,1-b]thiazoles 5–7 gave the corresponding aldehydes 8–10 by means of the Vilsmeier method. 1,4-DHPs 16–26 were synthesized by means of the well-known Hantzsch reaction with the appropriate aldehydes 8–15.12,14−16 All final products were confirmed with infrared spectra and 1H nuclear magnetic resonance.
Scheme 1.

Table 1. 1,4-Dihydropyridines 16–47.

| compd | x–y | R | R1 |
|---|---|---|---|
| 16 | HC=CH | CF3 | C2H5 |
| 17 | HC=CH | 4-(CF3)-C6H4 | CH3 |
| 18 | HC=CH | 4-(CF3)-C6H4 | C2H5 |
| 19 | H3CC=CCH3 | 4-(CF3)-C6H4 | CH2CH=CH2 |
| 20 | HC=CH | 4-(OCF3)-C6H4 | CH3 |
| 21 | HC=CH | 4-(OCF3)-C6H4 | CH2CH=CH2 |
| 22 | H3CC=CH | C6H5 | C2H5 |
| 23 | H2C–CH2 | C6H5 | CH3 |
| 24 | HC=CH | 2,5-(OCH3)-C6H3 | CH2CH=CH2 |
| 25 | H3CC=CH | Cl | C2H5 |
| 26 | HC=CH | 2,3,4-Cl3C6H2 | C2H5 |
| 27a | HC=CH | CF3 | CH3 |
| 28a | H3CC=CH | CF3 | CH3 |
| 29a | ClC=CH | CH3 | CH3 |
| 30a | H3CC=CH | CH3 | CH3 |
| 31a | ClC=CH | Cl | CH3 |
| 32a | H3CC=CH | Cl | CH3 |
| 33a | HC=CCH3 | Cl | CH3 |
| 34a | H3CC=CCH3 | Cl | CH3 |
| 35a | HC=CH | 2-(OCH3)C6H4 | CH3 |
| 36a | HC=CH | 3-(OCH3)C6H4 | CH3 |
| 37a | HC=CH | 2-(OCF3)C6H4 | CH3 |
| 38a | HC=CH | 3-(OCF3)C6H4 | CH3 |
| 39a | HC=CH | 3-(CF3)C6H4 | CH3 |
| 40a | H3CC=CH | 2,5-(OCH3)2C6H3 | CH3 |
| 41a | ClC=CH | 2,5-(OCH3)2C6H3 | CH |
| 42a | H5C2C=CH | 2,5-(OCH3)2C6H3 | CH3 |
| 43a | HC=CH | 3,4-(OCH3)2C6H3 | CH3 |
| 44a | ClC=CH | 3,4,5-(OCH3)3C6H2 | CH3 |
| 45a | ClC=CH | 4-(NO2)-2,5-(OCH3)2C6H2 | CH3 |
| 46a | HC=CH | 6-(NO2)-2,5-(OCH3)2C6H2 | CH3 |
| 47a | HC=CH | 6-(NO2)-3,4-(OCH3)2C6H2 | CH3 |
Reference (12).
Pharmacology
CFTR Studies
The activity of 1,4-DHPs on ΔF508-CFTR was tested in FRT cells coexpressing the mutant CFTR protein and the halide-sensitive yellow fluorescent protein (HS-YFP) as previously described.8,13 Briefly, cells were previously incubated at 27 °C for 24 h to rescue the mutant protein from the endoplasmic reticulum8 and then acutely stimulated for 30–40 min with forskolin (20 μM) in the presence and absence of 1,4-DHPs at three different concentrations (0.2, 2, and 20 μM). CFTR activity was finally evaluated with a microplate reader by continuously measuring the fluorescence in each well before and after the addition of an I– rich solution (final concentration of 100 mM), as previously described.8,13 I– influx through the CFTR channel causes YFP quenching. Therefore, the rate of fluorescence quenching reflects the extent of CFTR activity. Accordingly, the readings from each well were normalized for the initial fluorescence value and the fluorescence decay following I– addition was fitted with an exponential function to derive the maximal quenching rate (dF/dt). The quenching rate (QR) was plotted against compound concentrations. The resulting dose–response relationships were then fitted with the Hill equation to estimate potency and maximal effect.
Functional Studies on Cardiac and Smooth Muscle Function
The pharmacological profile of compounds was derived in guinea pig isolated left and right atria to evaluate their inotropic and chronotropic effects, respectively, and in K+-depolarized (80 mM) guinea pig vascular (aortic strips) and nonvascular [ileum longitudinal smooth muscle (GPILSM)] to assess calcium antagonist activity. Compounds were checked at increasing doses to evaluate the percent decrease of developed tension in isolated left atrium driven at 1 Hz (negative inotropic activity), the percent decrease in atrial rate in spontaneously beating right atrium (negative chronotropic activity), and the percent inhibition of calcium-induced contraction in K+-depolarized aortic strips and GPILSM (vascular and nonvascular relaxant activity, respectively). These details are reported in the Supporting Information.
For some selected compounds (see Table 5) the characterization was extended to guinea pig tracheal ring preparations precontracted with carbachol (1 μM) to evaluate the induced inhibition.
Table 5. Bronchodilator Effect of Same Compounds on Guinea Pig Trachea.
| compd | mean activitya ± SEM | IC50 b (μM) | 95% CL (×10-6) |
|---|---|---|---|
| 16 | 77 ± 1.7c | 1.11 | 0.87–1.23 |
| 17 | 67 ± 1.3 | 0.061 | 0.043–0.084 |
| 18 | 57 ± 2.4 | 0.13 | 0.10–0.17 |
| 19 | 33 ± 1.1 | ||
| 20 | 26 ± 2.1 | ||
| 21 | 38 ± 2.2 | ||
| 25 | 92 ± 1.4 | 2.34 | 1.89–2.89 |
| 26 | 40 ± 2.1 | ||
| 28 | 64 ± 1.6d | 0.031 | 0.023–0.041 |
| 30 | 27 ± 1.3 | ||
| 31 | 87 ± 1.5c | 3.66 | 1.21–4.74 |
| 37 | 47 ± 1.1 | ||
| 38 | 64 ± 2.5c | 0.058 | 0.026–0.099 |
| 39 | 19 ± 0.7d | ||
| 45 | 35 ± 2.2c | ||
| nifedipine | 62 ± 2.3 | 0.096 | 0.070–0.13 |
| 48 | 30 ± 2.6 | ||
| 49 | 68 ± 2.2 | 0.00059 | 0.00042–0.00083 |
| 50 | 29 ± 1.7 | ||
| genistein | 14 ± 1.1 |
Percent inhibition of carbachol (1 μM) induced contraction on guinea pig trachea at 10–5 M. The 10–5 M concentration gave the maximum effect for most compounds.
Calculated from log concentration–response curves (Probit analysis by Litchfield and Wilcoxon17 with n = 6–7). When the maximum effect was <50%, the IC50 values were not calculated.
At 10–4 M.
At 10–7 M.
Data were analyzed using Student’s t-test and are presented as the mean ± SEM.17 Since the drugs were added in a cumulative manner, the difference between the control and the experimental values at each concentration was tested for P < 0.05. The potency of drugs defined as EC50 and IC50 was evaluated from log concentration–response curves (Probit analysis using Litchfield and Wilcoxon17 or GraphPad Prism18,19software) in the appropriate pharmacological preparations.
Results
A. CFTR Study
FRT cells expressing ΔF508-CFTR were incubated at 27 °C for 24 h to rescue the mutant protein from ER, thus improving its targeting to the plasma membrane. Subsequently the cells were stimulated with forskolin (20 μM) in the presence and absence of 1,4-DHPs at different concentrations. Under these conditions, the cAMP activating pathway is saturated by the high forskolin concentration. Therefore, any increase in ΔF508-CFTR activity caused by a compound above the level reached with forskolin alone can be attributed to a potentiator-like activity. We tested 4-imidazo[2,1-b]thiazole-1,4-dihydropyridines as well as 48, 49, and 50, identified as CFTR-selective potentiators from a previous study.13 We also included genistein as a non-1,4-DHP potentiator. Nifedipine, our reference compound, shows a maximal activity of 58 and Kd = 2.9 ± 0.4 μM. Compounds 48 [Kd = 0.37 ± 0.07 μM], 49 [Kd = 0.17 ± 0.03 μM], and 50 [Kd = 0.40 ± 0.05 μM] show efficacy comparable to that of nifedipine but, as reported previously,13 with greatly improved potency. In particular, 49 has 17-fold increased potency relative to nifedipine. Among 4-imidazo[2,1-b]thiazole-1,4-dihydropyridines, compounds 23, 24, 30, 32, 35, 37, 40–47 are inactive. In contrast, as shown in Figure 1, compounds 17, 20, 21, 38, and 39 effectively induce CFTR activity, with a maximal effect elicited at 20 μM comparable to that of nifedipine and 48–50. However, the 4-imidazo[2,1-b]thiazole-1,4-dihydropyridines appear significantly less potent given the small activity induced at 0.2 μM. There are compounds such as 18 with apparently high potency [Kd = 0.18 ± 0.03] but with partial activity (Emax = 42) (Table 2).
Figure 1.

Compound efficacy on ΔF508-CFTR. The efficacy is obtained from dose–response relationships of YFP fluorescence experiments (mean ± SEM, n = 3).
Table 2. Activity of 1,4-DHPs and Genistein on ΔF508-CFTRa.
| compd | Emax (ms–1) | nH | mean Kd ± SEM (μM) |
|---|---|---|---|
| 16 | 60 | 0.66 | 19.4 ± 2.4 |
| 17 | 64 | 2.15 | 3.2 ± 0.5 |
| 18 | 42 | 1.69 | 0.18 ± 0.03 |
| 19 | 42 | 1.14 | 0.32 ± 0.06 |
| 20 | 64 | 1.8 | 3.5 ± 0.7 |
| 21 | 60 | 1.09 | 4.8 ± 0.7 |
| 22 | 60 | 0.93 | 14.8 ± 1.9 |
| 23 | inactiveb | ||
| 24 | inactiveb | ||
| 25 | 60 | 0.86 | 8.4 ± 0.8 |
| 26 | 46 | 0.44 | 3.2 ± 0.6 |
| 27 | 60 | 1.89 | 28.3 ± 2.5 |
| 28 | 60 | 0.88 | 12.0 ± 1.6 |
| 29 | 60 | 1.55 | 42.9 ± 4.9 |
| 30 | inactiveb | ||
| 31 | 60 | 1.04 | 6.8 ± 1.0 |
| 32 | inactiveb | ||
| 33 | 60 | 0.82 | 21.4 ± 2.4 |
| 34 | 60 | 1.32 | 10.9 ± 1.8 |
| 35 | inactiveb | ||
| 36 | 60 | 1.5 | 34.2 ± 4.4 |
| 37 | inactiveb | ||
| 38 | 64 | 0.81 | 1.4 ± 0.1 |
| 39 | 65 | 1.01 | 2.8 ± 0.4 |
| 40 | inactiveb | ||
| 41 | inactiveb | ||
| 42 | inactiveb | ||
| 43 | inactiveb | ||
| 44 | inactiveb | ||
| 45 | inactiveb | ||
| 46 | inactiveb | ||
| 47 | inactiveb | ||
| nifedipine | 58 | 2.58 | 2.9 ± 0.4 |
| 48 | 62 | 0.97 | 0.37 ± 0.07 |
| 49 | 56 | 3.58 | 0.17 ± 0.03 |
| 50 | 59 | 1.18 | 0.40 ± 0.05 |
| genistein | 61 | 1.3 | 9.4 ± 1.1 |
The table reports the activity of compounds on ΔF508-CFTR measured as I– influx (HS-YFP quenching rate). Dose–response relationships were generated, and the data were fitted with the Hill equation to obtain maximal effect on quenching rate (Emax), Hill coefficient (nH), and apparent dissociation constant (Kd). For compounds having low potency (i.e. 16, 22, 27, 29, 33, and 36) the fit was done with a forced Emax value of 60. Data were generated from three independent determinations.
Inactive up to 20 μM.
B. Functional Study in Cardiovascular System
All compounds were tested for their cardiovascular profile in guinea pig left atrium driven at 1 Hz and in spontaneously beating right atrium to evaluate their negative inotropic and/or chronotropic effects, respectively, and in 80 mM K+-depolarizing guinea pig aortic strips to assess their vasorelaxant activity. Data are collected in Table 3 for 16–26 and in Table S4 (Supporting Information) for 27–47, using nifedipine, genistein, and 48–50 as reference compounds. Nifedipine is active on all cardiovascular parameters considered with selectivity for vascular smooth muscle [IC50 = 0.009 μM (CL 0.003–0.02)]. In contrast, 48–50 have no negative chronotropic and vascular effects. These three compounds have selective negative inotropic activity with potency higher than those of nifedipine; in particular the potency of 49 is EC50 = 0.0022 μM (CL 0.0014–0.0036).
Table 3. Cardiovascular Activity of Tested Compounds.
| % decrease (mean ± SEM) |
EC50 of inotropic negative activity |
EC50 of chronotropic negative activity |
|||||
|---|---|---|---|---|---|---|---|
| compd | negative inotropic activitya | negative chronotropic activityb | EC50 c (μM) | 95% CL (×10-6) | EC50 c (μM) | 95% CL (×10-6) | mean vasorelaxant activitydactivity ± SEM |
| 16 | 93 ± 2.4 | 40 ± 1.4e | 1.07 | 0.76–1.49 | 48 ± 0.9 | ||
| 17 | 96 ± 3.7f | 33 ± 0.5g | 0.12 | 0.082–0.17 | 36 ± 2.1h | ||
| 18 | 90 ± 2.8g | 64 ± 1.1h | 0.033 | 0.026–0.044 | 0.15 | 0.071–0.67 | 6 ± 0.1 |
| 19 | 93 ± 2.2i | 52 ± 0.7 | 0.27 | 0.21–0.36 | 8.96 | 7.65–10.50 | 32 ± 1.9 |
| 20 | 89 ± 2.2 | 33 ± 1.6k | 2.33 | 1.90–2.66 | 60 ± 2.4e,j | ||
| 21 | 88 ± 0.1 | 77 ± 1.6k | 0.73 | 0.51–1.01 | 1.97 | 1.01–3.54 | 31 ± 2.8e |
| 22 | 44 ± 1.3f | 84 ± 1.6 | 7.15 | 4.30–10.01 | 5 ± 0.1f | ||
| 23 | 78 ± 2.2 | 90 ± 1.4k | 0.39 | 0.26–0.61 | 8.63 | 5.93–10.25 | 46 ± 3.1 |
| 24 | 81 ± 2.2l | 32 ± 1.1e | 0.34 | 0.23–0.48 | 31 ± 1.3 | ||
| 25 | 56 ± 1.7g | 36 ± 2.1l | 0.031 | 0.024–0.039 | 8 ± 0.2 | ||
| 26 | 90 ± 2.4e | 92 ± 2.7 | 0.43 | 0.31–0.57 | 0.61 | 0.41–0.89 | 17 ± 0.9 |
| nifedipine | 97 ± 2.0f | 85 ± 4.2g | 0.26 | 0.19–0.36 | 0.039 | 0.031–0.051 | 82 ± 1.3e,m |
| 48 | 80 ± 2.3e | 38 ± 0.4l | 0.029 | 0.021–0.040 | 17 ± 0.3f | ||
| 49 | 71 ± 1.1h | 5 ± 0.3 | 0.0022 | 0.0014–0.0036 | 12 ± 0.3f | ||
| 50 | 83 ± 1.4e | 6 ± 0.4 | 0.034 | 0.025–0.046 | 1 ± 0.01f | ||
| genistein | 71 ± 0.3l | 68 ± 2.1k | 0.028 | 0.021–0.037 | 44.48 | 34.95–56.61 | 22 ± 0.7i |
Decrease in developed tension on isolated guinea pig left atrium at 10–4 M, expressed as percent changes from the control (n = 5–6). The left atria were driven at 1 Hz.
Decrease on atrial rate on guinea pig spontaneously beating isolated right atrium at 10–5 M, expressed as percent changes from the control (n = 7–8). Pretreatment heart rate ranged from 165 to 190 beats/min.
Calculated from log concentration–response curves (Probit analysis by Litchfield and Wilcoxon17 with n = 6–7). When the maximum effect was <50%, the EC50 inotropic, EC50 chronotropic, and IC50 values were not calculated.
Percent inhibition of calcium-induced contraction on K+-depolarized guinea pig aortic strip at 10–4 M (n = 5–6).
At 10–6 M.
At 10–5 M.
At 10–7 M.
At 5 × 10–7 M.
At 5 × 10–5 M.
IC50 = 0.016 μM (CL 0.012–0.025).
At 10–4 M.
At 5 × 10–6 M.
IC50 = 0.009 μM (CL 0.003–0.020).
In isolated vascular smooth muscle depolarized with 80 mM K+, genistein is inactive. In cardiac isolated tissues genistein induced negative inotropic and chronotropic activities with about 1588-fold selectivity for negative inotropic potency [EC50 = 0.028 μM (CL 0.021–0.037)] respective to negative chronotropic potency [EC50 = 44.48 μM (CL 34.98–56.61)]. As previously described, genistein up to 10 μM induced positive inotropic activity.20
All other compounds, except 20, are devoid of vascular effects. The potency of 20 was not significantly different from that of nifedipine [IC50 = 0.016 μM (CL 0012–0025), IC50 = 0.009 μM (CL 0003–0020), respectively] even if the intrinsic activity is significantly lower.
Compounds 28, 30, and 44 are also devoid of cardiac effects. In fact, their intrinsic activity on cardiac parameters is less than 50% for the inotropic and 40% for the chronotropic effect at the highest concentration studied.
A significant group of compounds (11 of 32) expressed negative inotropic selectivity: 16, 17, 24, 25, 29, 31–34, 37, and 46, with potency between 0.039 and 1.96 μM. Among them, nine compounds (17, 25, 29, 31–34, 37, and 46) were more potent than nifedipine [EC50 = 0.26 μM (CL 0.19–0.36)]. Derivatives 17, 29, 31, and 33 have negative inotropic effect about 2.6 times greater than that of nifedipine. However, all these compounds have no intrinsic activity that exceeds 80%. Therefore, the intrinsic activity is significantly lower than that of nifedipine considering the concentration needed to obtain the maximal intrinsic activity with the exception of 16 and 17.
Most of the tested compounds (18, 19, 21, 23, 26, 27, 35, 36, 38–43, and 47) showed both negative inotropic and chronotropic properties with different selectivity. In particular, unlike the reference nifedipine, none of these compounds showed negative chronotropic selectivity. Compounds 19, 23, 27, 39, and 47, different from the reference nifedipine, showed a negative inotropic over chronotropic selectivity. In particular in 19, 23, 39, and 47 are predominantly negative inotropic agents with a selectively ratio of about 33, 22, 12, and 9, respectively. Derivatives 35, 38, and 40 (Table S4) have only a weak negative inotropic selectivity.
Only compounds 22 and 45 have negative chronotropic selectivity [EC50 = 7.15 μM (CL 4.30–10.01) and EC50 = 18.52 μM (CL 14.19–24.16) respectively] even though 286 and 446 times less than that of nifedipine.
C. Functional Study in Guinea Pig Longitudinal Smooth Muscle
It is well-known that calcium entry blockers such as nifedipine have relevant inhibitory effects on nonvascular smooth muscle.21 All compounds were also tested in K+-depolarized (80 mM) guinea pig ileum longitudinal smooth muscle to evaluate both the effects on nonvascular smooth muscle and the selective action by comparison with the cardiovascular system. These data are collected in Table 4. For compounds 32, 36, 37, 44, and 46, these data are reported in the Supporting Information (Table S5). As previously mentioned, among the cardiovascular system, nifedipine showed a clear-cut vascular potency and selectivity [EC50 = 0.009 μM (CL 0.003–0.02)]. However, with regard to the nonvascular smooth muscle, the potency of nifedipine increases 6 times if compared to that measured in aortic strips [EC50 = 0.0015 μM (CL 0.0011–0.0022)].
Table 4. Relaxant Activity of Tested Compounds on K+-Depolarized Guinea Pig Ileum Longitudinal Smooth Muscle.
| compd | mean activitya ± SEM | IC50 b (μM) | 95% CL (×10-6) |
|---|---|---|---|
| 16 | 90 ± 1.4c | 0.014 | 0.010–0.018 |
| 17 | 69 ± 3.6d | 0.0046 | 0.0036–0.0058 |
| 18 | 83 ± 1.3e | 0.00088 | 0.00021–0.0017 |
| 19 | 72 ± 2.3 | 0.46 | 0.36–0.61 |
| 20 | 73 ± 3.2d | 0.0033 | 0.0025–0.0042 |
| 21 | 90 ± 3.4f | 0.0053 | 0.0038–0.0073 |
| 22 | 83 ± 2.4 | 0.25 | 0.19–0.32 |
| 23 | 80 ± 1.1 | 0.26 | 0.20–0.33 |
| 24 | 96 ± 2.5g | 0.32 | 0.24–0.42 |
| 25 | 94 ± 1.4g | 0.057 | 0.043–0.076 |
| 26 | 77 ± 0.2 | 0.038 | 0.029–0.050 |
| 27 | 70 ± 2.3h | 0.21 | 0.14–0.31 |
| 28 | 92 ± 1.4 | 0.095 | 0.072–0.13 |
| 29 | 96 ± 3.3g | 0.55 | 0.19–0.93 |
| 30 | 70 ± 1.3i | 8.83 | 5.53–10.41 |
| 31 | 60 ± 1.4 | 0.68 | 0.55–0.84 |
| 33 | 97 ± 0.6g | 0.51 | 0.39–0.62 |
| 34 | 78 ± 1.2 | 0.32 | 0.24–0.41 |
| 35 | 88 ± 2.2g | 0.86 | 0.64–1.02 |
| 38 | 76 ± 1.3f | 0.018 | 0.010–0.097 |
| 39 | 96 ± 1.0j | 0.036 | 0.028–0.046 |
| 40 | 74 ± 1.4i | 4.89 | 3.74–6.40 |
| 41 | 91 ± 2.3k | 1.14 | 0.73–1.85 |
| 42 | 92 ± 1.1i | 2.16 | 1.03–2.94 |
| 43 | 90 ± 2.3 | 0.45 | 0.10–0.91 |
| 45 | 92 ± 2.2j | 0.074 | 0.057–0.097 |
| 47 | 86 ± 1.3k | 11.43 | 8.78–14.87 |
| nifedipine | 70 ± 0.36e | 0.0015 | 0.0011–0.0022 |
| 48 | 83 ± 2.6i | 0.23 | 0.17–0.31 |
| 49 | 52 ± 0.6i | 1.85 | 1.31–2.03 |
| 50 | 68 ± 1.5i | 0.94 | 0.64–1.38 |
| genistein | 84 ± 1.6k | 9.95 | 8.01–12.35 |
Percent inhibition of calcium-induced contraction on K+-depolarized (80 mM) guinea pig longitudinal smooth muscle at 10–6 M.
Calculated from log concentration–response curves (Probit analysis by Litchfield and Wilcoxon17 with n = 6–7). When the maximum effect was <50%, the IC50 values were not calculated.
At 10–7 M.
At 10–8 M.
At 5 × 10–9 M.
At 5 × 10–8 M.
At 5 × 10–6 M.
At 3 × 10–7 M.
At 10–5 M.
At 5 × 10–7 M.
At 5 × 10–5 M.
All three 1,4-DHPs previously described (48–50)13 are able to relax the intestinal smooth muscle but with a potency significantly lower than that of nifedipine. In the same preparation genistein induced relaxation with intrinsic activity similar to that of 48 but with lower potency [IC50 = 9.95 μM (CL 8.01–12.35); IC50 = 0.0015 μM (CL 0.0011–0.0022); respectively].
All the 1,4-DHPs bearing a imidazo[2,1-b]thiazole system at C4 are able to relax the guinea pig longitudinal smooth muscle of ileum. They show an increased activity in nonvascular smooth muscle with respect to vascular smooth muscle with the exception of 18, which has a potency comparable to that of nifedipine [IC50 = 0.00088 μM (CL 0.00021–0.0017); IC50 = 0.0015 μM (CL 0.0011–0.0022); respectively]. All other compounds are on the whole less potent than the reference nifedipine. In particular, compounds 17, 20, and 21 are about 3 times less potent than nifedipine, whereas 16, 25, 26, 28, 36, 38, 39, and 45 are about 65 times less potent. Compound 28 showed a selective activity for the GPILSM, since it was ineffective on the cardiovascular parameters studied.
D. Functional Study in Trachea
Compounds showing an interesting activity profile as CFTR potentiators combined with minor effects on cardiovascular parameters were also tested on isolated tracheal rings to measure bronchodilatation (data of selected compounds and references are collected in Table 5 with those of reference compounds). The compounds studied have a different potency profile in guinea pig trachea smooth muscle.
49 has a strong selectivity for the trachea compared to nifedipine. Indeed, 49 is 163 times more potent than nifedipine. 48 and 50 have intrinsic activity ≤30%, while genistein has intrinsic activity lover then 20%.
The new 1,4-DHPs studied showed a panel of various activities. Among the compounds studied, 19–21, 26, 30, 37, 39, and 45 have intrinsic activity less than 50%. 17 and 18 have the same intrinsic activity of nifedipine, while 38 reaches the same intrinsic activity at a concentration 10 times higher. However, the calculated potencies for 17, 18, and 38 were not significantly different from that of nifedipine [EC50 = 0.061 μM (CL 0.043–0.084), EC50 = 0.13 μM (CL 0.10–0.17), EC50 = 0.058 μM (CL 0.026–0.099), and EC50 = 0.096 μM (CL 0.070–0.13), respectively]. The relaxant potencies of 16, 25, and 31 are lower than that of nifedipine (11-, 24-, and 38-fold, respectively).
Interestingly, compound 28, which has no effect on cardiovascular parameters, relaxes both intestinal and tracheal smooth muscles.
Discussion
Our study had the main objective of identifying 1,4-DHPs with optimal activity as potentiators of the CFTR Cl– channel and negligible effects on voltage-dependent Ca2+ channels (VDCCs). In a previous paper, it was shown that these two activities might be separated by modifying the substituents of 1,4-DHP scaffold.13 Indeed a cell-based functional assay approach was used to identify compounds, such as 48–50, with high potency and efficacy on mutant CFTR and dramatically reduced activity on VDCCs.13 In the present study, we have proved these results by testing these compounds on physiologically relevant functions associated with Ca2+ channel activity in heart, aorta smooth muscle strips, and ileum. Our results in aortic strips (Figure 2) demonstrate that the CFTR-selective 1,4-DHPs previously identified13 indeed have low potency and efficacy, compared to an antihypertensive drug such as nifedipine, on the contractility of smooth muscle of blood vessels. These compounds, namely, 48–50, also have negligible negative chronotropic activity on the guinea pig right atrium. In contrast, these 1,4-DHPs are similar to nifedipine as negative inotropic agents and also as relaxant agents on intestinal smooth muscle (Tables 3 and 4). These results may reflect a different expression of Ca2+ channel subtypes in tissues used for functional assays. It is known that the activities of the cardiac calcium channel blockers are related to their interaction with the α1-subunits of VDCCs. In particular, the negative inotropic effect is due to interaction with the Cav1.2 isoform while the negative chronotropic effect seems to be related to interaction with the Cav1.3 isoform.22 Furthermore, effects of Ca2+ channel blockers such as nifedipine on vascular and nonvascular smooth muscle cells appear to be linked to the interaction with the Cav1.2 isoform.23 Moreover, the discovery that CFTR is expressed not only in epithelial cells but also in the smooth muscle, where it may modulate contractility, complicates the interpretation of results.24 To address this point, we have also tested a classical non-1,4-DHP CFTR potentiator such as genistein. The finding that genistein has, at least in part, a profile similar to that of CFTR-selective 1,4-DHPs (i.e., low activity on aorta and high relaxant effect on ileum) suggests the possibility that CFTR contributes to smooth muscle function in some organs (Figure 2).
Figure 2.
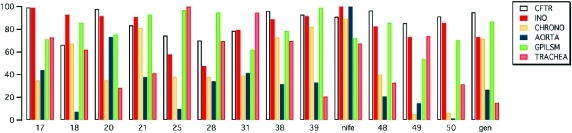
Evaluation of selected compounds on CFTR, negative inotropic activity (INO), negative chronotropic activity (CHRONO), vasorelaxation of guinea pig aortic strips (AORTA), ileum longitudinal smooth muscle (GPILSM), and tracheal smooth muscle (TRACHEA). Data are normalized for the most effective compound (100%) in each assay.
Here we have tested 4-imidazo[2,1-b]thiazole-1,4-dihydropyridines as a possible new class of CFTR potentiators. Some of these compounds are indeed highly effective on ΔF508-CFTR and have a low activity on vascular smooth muscle cells similar to 48–50. However, they have a relatively low potency as CFTR potentiators (Kd in the micromolar range).
The evaluation of the screening tests on CFTR, on cardiac tissue, and on vascular and nonvascular tissues for identification of the most promising molecules of the small library of the imidazo[2,1-b]thiazole-1,4-DHP allows us to define some emerging relationships.
As previously demonstrated, imidazo[2,1-b]thiazole substituents at C4 are able to change not only the cardiovascular parameters12 but also the CFTR activity (Figure 2). From a quantitative standpoint, compounds bearing a 3- or 4-trifluoromethylphenyl group or a 3- or 4-trifluoromethoxyphenyl group at the 6-position of the imidazo[2,1-b]thiazole (17, 18, 20, 21, 38, 39) are the most effective on ΔF508-CFTR. A shift of the trifluoromethoxyphenyl group at position 2 of the phenyl ring 37 or a replacement of the trifluoromethoxyphenyl group with a methoxy group (35, 36) has a detrimental effect on ΔF508-CFTR. Also the introduction of more than one methoxy group or a nitro group on the phenyl ring drastically decreases activity on ΔF508-CFTR (24, 40–47). All compounds with a trifluoromethyl group or a methyl group or chlorine in position 6 are less CFTR-selective (16, 25, 27–34). The 2- and 3-positions of the imidazo[2,1-b]thiazole ring moderately affect the ΔF508-CFTR activity more than the cardiovascular profile. In both cases, the aromaticity of the system must be kept as demonstrated by compound 23 which is inactive in the assays. A comparison of the ΔF508-CFTR activities of compounds 27, 28, 33, and 34 seems to indicate that a methyl in position 2 of the heterocycle can positively influence the effect. This influence is greater if the L-type calcium channel blocker activity is considered (27 vs 28). The results in the functional assay point out that esterification of the carboxylic functions in positions 3 and 5 of the 1,4-DHP ring with bulky and more hydrophobic groups such as allylic or ethyl groups decreases potency on CFTR while two methyl groups are better for CFTR potentiators13 (see compounds 21 and 18 vs 20 and 17). On the contrary, the introduction of ester, ethyl, or allyl chains enhances L-type calcium channel blocker activity.
Conclusions
Our study confirms the possibility in the development of 1,4-DHPs with increased activity and selectivity as potentiators of mutant CFTR and, at the same time, reduced activity as inhibitors of voltage-dependent Ca2+ channels of the cardiovascular system and smooth muscle of nonvascular tissues (see Figure 3). This is highly desirable for developing drugs able to correct the basic defect in cystic fibrosis while having minimal side effects. However, the 1,4-DHPs evaluated in this study still keep some activity in nonepithelial cells. This may be caused by residual activity on some types of Ca2+ channels and/or expression of CFTR in cardiac and smooth muscle cells. To further minimize undesired effects, it is possible to generate novel 1,4-DHPs based on SAR obtained from the present and previous studies. In general the results suggest that the core imidazo[2,1-b]thiazole is confirmed as a suitable heterocycle to explore new potentiators of mutant CFTR. Of further interest is the effect of the imidazo[2,1-b]thiazole substitution pattern on the profile of biological action. CFTR-selective potentiators might be better achieved by a phenyl ring with appropriate substituents at the 6-position of the imidazo[2,1-b]thiazole, while the substituents at the 2-position can influence cardiovascular parameters to the same extent. In addition, systemic effects may be further minimized by considering delivery of 1,4-DHP potentiators via aerosol.
Figure 3.

(a) Comparison of potencies of different biological parameters for compounds with the best, interesting profile. (b) Reference compounds. Data are expressed as −log potency ± CL.
Experimental Section
A. Chemistry
All the compounds prepared have a purity of at least 95% as determined by combustion analysis. The melting points are uncorrected. Analyses (C, H, N) were within 0.4% of the theoretical values. TLC was performed on Bakerflex plates (silica gel IB2-F). The eluent was a mixture of petroleum ether 60–80 °C/acetone in various proportions. Kieselgel 60 (Merck) was used for flash chromatography. The IR spectra were recorded in Nujol on a Nicolet Avatar 320 E.S.P.; αmax is expressed in cm–1. The 1H NMR spectra were recorded on a Varian Gemini (300 MHz); the chemical shift (referenced to solvent signal) is expressed in δ (ppm) and J in Hz. Abbreviations are as follows: th = thiazole, im = imidazole, ar = aromatic, py = pyridine, ex = H linked to N that exchanged with D2O.
All solvents and reagents, unless otherwise stated, were supplied by Sigma-Aldrich Chemical Co. Ltd. and were used as supplied. For a new starting compound, see Table S1 in Supporting Information.
General Procedure for the Synthesis of the 1,4-Dihydropyridines 16–26
Methyl acetoacetate or ethyl acetoacetate or allyl acetoacetate (2 mM) and 30% NH4OH (4 mM) were added to a stirred solution of the appropriate aldehyde 8–15 (1 mM) dissolved in isopropyl alcohol (50 mL). The reaction mixture was refluxed for 1–4 days (according to a TLC test acetone/petroleum ether 55–85 °C, 1:9 v/v, 2:8 v/v), and to it were added methyl acetoacetate or ethyl acetoacetate or allyl acetoacetate (4 mM) and 30% NH4OH (2 mM) every 12 h. After cooling, the mixture was evaporated to dryness under reduced pressure. All the derivatives were purified by flash chromatography on silica gel (acetone/petroleum ether 40–60 °C from 1.9 to 4.6, v/v) to provide the pure DHPs as a pale yellow syrup. The resulting oily residue was diluted with cold acetone to afford a white solid collected by filtration for 16, 19, 21–26. For 18 the oily residue was diluted with diethyl ether. For 17 and 20 resulting oils were taken with acetone–petroleum ether 1/1 v/v to obtain a white solid collected by filtration.
Diethyl 2,6-Dimethyl-4-(6-(trifluoromethyl)imidazo[2,1-b]thiazol-5-yl)-1,4-dihydropyridine-3,5-dicarboxylate (16)
25% yield. 1H NMR: 0.96 (6H, t, COOCH2CH3, J = 7.1), 2.23 (6H, s, CH3), 3.90 (4H, q, COOCH2CH3, J = 7.1), 5.52 (1H, s, py), 7.44 (1H, d, th, J = 4.6), 7.63 (1H, d, th, J = 4.6), 9.06 (1H, s, NH, ex D2O). Mp = 185–190 °C. MW = 443.4435. Anal. (C19H20F3N3O4S) C, H, N.
Dimethyl 2,6-Dimethyl-4-(6-(4-(trifluoromethyl)phenyl)imidazo[2,1-b]thiazol-5-yl)-1,4-dihydropyridine-3,5-dicarboxylate (17)
25% yield. 1H NMR: 2.20 (6H, s, CH3), 3.18 (6H, s, COOCH3), 5,72 (1H, s, py), 7.27 (1H, m, th), 7.33 (1H, m, th), 7.79 (2H, d, ar, J = 5.2), 8.09 (2H, d, ar, J = 5.2), 9.13 (1H, s, NH, ex D2O). Mp = 227. MW = 491.1126. Anal. (C23H20F3N3O4S) C, H, N.
Diethyl 2,6-Dimethyl-4-(6-(4-(trifluoromethyl)phenyl)imidazo[2,1-b]thiazol-5-yl)-1,4-dihydropyridine-3,5-dicarboxylate (18)
30% yield. 1H NMR: 0.81 (6H, t, COOCH2CH3, J = 7.1), 2.15 (6H, s, CH3), 3.75 (4H, m, COOCH2CH3), 5.65 (1H, s, py), 7.32 (1H, d, th, J = 4.4), 7.43 (1H, d, th, J = 4.4), 7.70 (2H, d, ar, J = 8.4), 8.04 (2H, d, ar, J = 8.4), 8.98 (1H, s, NH, ex D2O). Mp = 175–180. MW = 519.5400. Anal. (C25H24F3N3O4S) C, H, N.
Diallyl 4-(2,3-Dimethyl-6-(4-(trifluoromethyl)phenyl)imidazo[2,1-b]thiazol-5-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (19)
10% yield. 1H NMR: 1.88 (6H, s, CH3), 2.31 (3H, s, CH3), 2.64 (3H, s, CH3), 4.31 (2H, dd, J = 5.45, J = 13.65, CH2CH=CH2), 4.50 (2H, dd, J = 5.45, J = 13.65, CH2CH=CH2), 4.88 (2H, dd, J = 1.55, J = 17.2, CH2CH=CH2), 5.00 (2H, dd, J = 1.55, J = 10.5, CH2CH=CH2), 5,43 (1H, s, py), 5.58 (2H, m, CH2CH=CH2), 7.30 (2H, d, ar, J = 8.2), 7.68 (2H, d, ar, J = 8.2), 8.18 (1H, s, NH, ex D2O). Mp = 165–170. MW = 571.6149. Anal. (C29H28F3N3O4S) C, H, N.
Dimethyl 2,6-Dimethyl-4-(6-(4-(trifluoromethoxy)phenyl)imidazo[2,1-b]thiazol-5-yl)-1,4-dihydropyridine-3,5-dicarboxylate (20)
20% yield. 1H NMR: 2.20 (6H, s, CH3), 3.21 (6H, s, COOCH3), 5.67 (1H, s, py), 7.27 (1H, d, th, J = 3.1),7.32 (1H, d, th, J = 3.1), 7.42 (2H, d, ar, J = 5.7), 7.96 (2H, d, ar, J = 5.7), 9.08 (1H, s, NH, ex D2O). Mp = 217. MW = 507.1075. Anal. (C23H20F3N3O5S) C, H, N.
Diallyl 2,6-Dimethyl-4-(6-(4-(trifluoromethoxy)phenyl)imidazo[2,1-b]thiazol-5-yl)-1,4-dihydropyridine-3,5-dicarboxylate (21)
10% yield. 1H NMR: 2.16 (6H, s, CH3), 4.15 (2H, dd, J = 5.55, J = 13.5, CH2CH=CH2), 4.33 (2H, dd, J = 5.55, J = 13.5, CH2CH=CH2), 4.91 (2H, dd, J = 1.5, J = 10.35, CH2CH=CH2), 4.98 (2H, dd, J = 1.5, J = 10.35, CH2CH=CH2), 5.54 (2H, m, CH2CH=CH2), 5.62 (1H, s, py), 7.28 (1H, d, th, J = 4.35), 7.36 (2H, d, ar, J = 8.7), 7.38 (1H, d, th, J = 4.35), 7.86 (2H, d, ar, J = 8.7), 9.00 (1H, s, NH, ex D2O). Oil. MW = 559.1388. Anal. (C27H24F3N3O5S) C, H, N.
Diethyl 2,6-Dimethyl-4-(2-methyl-6-phenylimidazo[2,1-b]thiazol-5-yl)-1,4-dihydropyridine-3,5-dicarboxylate (22)
20% yield. 1H NMR: 0.85 (6H, t, COOCH2CH3, J = 7.05), 2.12 (6H, s, CH3), 2.42 (3H, s, CH3), 3.71 (2H, q, COOCH2CH3, J = 7.05), 3.81 (2H, q, COOCH2CH3, J = 7.05), 5.54 (1H, s, py), 7.25 (3H, m, th + ar), 7.36 (1H, t, ar, J = 6.95), 7.72 (2H, d, ar, J = 6.95), 8.80 (1H, s, NH, ex D2O). Mp = 205. MW = 465.1722. Anal. (C25H27N3O4S) C, H, N.
Dimethyl 2,6-Dimethyl-4-(6-phenyl-2,3-dihydroimidazo[2,1-b]thiazol-5-yl)-1,4-dihydropyridine-3,5-dicarboxylate (23)
30% yield. 1H NMR: 2.16 (6H, s, CH3), 3.23 (6H, s, COOCH3), 3.89 (2H, d, thn, J = 6.2), 3.98 (2H, d, thn, J = 6.2), 5.44 (1H, s, py), 7.23 (1H, t, ar, J = 7.3),7.35 (2H, t, ar, J = 7.3), 7.70 (2H, d, ar, J = 7.3), 8.82 (1H, s, NH, ex D2O). Mp = 245. MW = 424.4966. Anal. (C22H22N3O4S) C, H, N.
Diallyl 4-(6-(2,5-Dimethoxyphenyl)imidazo[2,1-b]thiazol-5-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (24)
15% yield. 1H NMR: 1.92 (6H, s, CH3), 3.46 (3H, s, OCH3), 3.69 (3H, s, OCH3), 4.35 (2H, dd, J = 5.15, J = 13.95, CH2CH=CH2), 4.48 (2H, dd, J = 5.15, J = 13.95, CH2CH=CH2), 4.86 (2H, dd, J = 1.4, J = 17.2, CH2CH=CH2), 4.98 (2H, dd, J = 1.4, J = 10.6, CH2CH=CH2), 5.09 (1H, s, py), 5.69 (2H, m, CH2CH=CH2), 6.44 (1H, s, ar), 6.86 (1H, s, ar), 7.11 (1H, d, th, J = 4.6), 7.74 (1H, d, th, J = 4.6), 8.14 (1H, s, NH, ex D2O). Mp = 200–205. MW = 535.6157. Anal. (C28H29N3O6S) C, H, N.
Diethyl 4-(6-Chloro-2-methylimidazo[2,1-b]thiazol-5-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (25)
75% yield. 1HNMR: 0.99 (6H, t, COOCH2CH3, J = 7.2), 2.25 (6H, s, CH3), 2.42 (3H, s, CH3), 3.90 (4H, m, COOCH2CH3), 5.12 (1H, s, py), 7.58 (1H, s, th), 9.04 (1H, s, NH, ex D2O). Mp = 175. MW = 423.1019. Anal. (C19H22ClN3O4S) C, H, N.
Diethyl 2,6-Dimethyl-4-(6-(2,3,4-trichlorophenyl)imidazo[2,1-b]thiazol-5-yl)-1,4-dihydropyridine-3,5-dicarboxylate (26)
20% yield. 1H NMR: 0.93 (6H, t, COOCH2CH3, J = 7.1), 1.94 (6H, s, CH3), 3.91 (4H, m, COOCH2CH3), 5.07 (1H, s, py), 7.05 (1H, d, ar, J = 8.4), 7.26 (1H, d, th, J = 4.4), 7.66 (1H, d, ar, J = 8.4), 7.95 (1H, d, th, J = 4.4), 8.21 (1H, s, NH, ex D2O). Mp = 230–235. MW = 554.8765. Anal. (C24H22Cl3N3O4S) C, H, N.
B. CFTR Assays
For details, see Supporting Information, section S10.
C. Functional Studies
For details, see Supporting Information, section S11.
Acknowledgments
This work was supported by grants from University of Bologna, Italy, from Telethon Italy (Grant GGP10026), from Fondazione Italiana per la Fibrosi Cistica (Grant FFC2/2009), and from the Cystic Fibrosis Foundation Therapeutics. The authors thank Alessandro Casoni for animal care.
Glossary
Abbreviations Used
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- TM
transmembrane
- LTCC
L-type calcium channel
- 1,4-DHP
1,4-dihydropyridine
- GPILSM
guinea pig ileum longitudinal smooth muscle
- NMR
nuclear magnetic resonance
- IR
infrared
- DMSO
dimethylsulfoxide
- DMF
N,N-dimethylformamide
- SEM
standard error of the mean
- ER
endoplasmic reticulum
- CL
confidence limit
Supporting Information Available
Chemistry details; analytical data for compounds 5–26; IR data for compounds 16–26; CFTR and functional assays; functional data for compounds previously described12 (cardiovascular data for compounds 27–47 and relaxant activity on GPLSM for compounds 32, 36, 37, 44, and 46). This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Riordan J. R.; Rommens J. M.; Kerem B.; Alon N.; Rozmahel R.; Grzelczak Z.; Zielenski J.; Lok S.; Plavsic N.; Chou J. L.; et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989, 245, 1066–1973. [DOI] [PubMed] [Google Scholar]
- Wilschanski M.; Durie P. R. Patterns of GI disease in adulthood associated with mutations in the CFTR gene. Gut 2007, 56, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilewski J. M.; Frizzell R. A. Role of CFTR in airway disease. Physiol. Rew. 1999, 79, S215–S255. [DOI] [PubMed] [Google Scholar]
- Mitchell I.; Corey M.; Woenne R.; Krastins I. R.; Levison H. Bronchial hyperreactivity in cystic fibrosis and asthma. J. Pediatr. 1978, 93, 744–748. [DOI] [PubMed] [Google Scholar]
- Ackerman M. J.; Clapham D. E. Ion channels—basic science and clinical disease. N. Engl. J. Med. 1997, 336, 1575–1586. [DOI] [PubMed] [Google Scholar]
- Van Goor F.; Straley K. S.; Cao D.; Gonzalez J.; Hadida S.; Hazlewood A.; Joubran J.; Knapp T.; Makings L. R. V.; Miller M.; et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. 2006, 290, L1117–1130. [DOI] [PubMed] [Google Scholar]
- Pedemonte N.; Lukacs G. L.; Du K.; Caci E.; Zegarra-Moran O.; Galietta L. J. V.; Verkman A. S. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005, 115, 2564–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedemonte N.; Diena T.; Caci E.; Nieddu E.; Mazzei M.; Ravazzolo R.; Zegarra-Moran O.; Galietta L. J. Antihypertensive 1,4-dihydropyridines as correctors of the cystic fibrosis transmembrane conductance regulator channel gating defect caused by cystic fibrosis mutations. Mol. Pharmacol. 2005, 68, 1736–1746. [DOI] [PubMed] [Google Scholar]
- Pedemonte N.; Sonawane N. D.; Taddei A.; Hu J.; Zegarra-Moran O.; Suen Y. F.; Robins L. I.; Dicus C. W.; Willenbring D.; Nantz M. H.; Kurth M. J.; Galietta L. J.; Verkman A. S. Phenylglycine and sulfonamide correctors of defective delta F508 and G551D cystic fibrosis transmembrane conductance regulator chloride-channel gating. Mol .Pharmacol. 2005, 67, 1797–1807. [DOI] [PubMed] [Google Scholar]
- Caputo A.; Hinzpeter A.; Caci E.; Pedemonte N.; Arous N.; Di Duca M.; Zegarra-Moran O.; Fanen P.; Galietta L. J. Mutation-specific potency and efficacy of cystic fibrosis transmembrane conductance regulator chloride channel potentiators. J Pharmacol Exp Ther. 2009, 330, 783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrold M. W.Angiotensin Converting Enzyme Inhibitors, Antagonists and Calcium Blockers. In Foye’s Principles of Medicinal Chemistry, 5th ed.; Williams D. A., Lemke T. L., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2002; pp 551–556. [Google Scholar]
- Budriesi R.; Ioan P.; Locatelli A.; Cosconati S.; Leoni A.; Ugenti M. P.; Andreani A.; Di Toro R.; Bedini A.; Spampinato S.; Marinelli L.; Novellino E.; Chiarini A. Imidazo[2,1-b]thiazole system: a scaffold endowing dihydropyridines with selective cardiodepressant activity. J. Med. Chem. 2008, 51, 1592–1600. [DOI] [PubMed] [Google Scholar]
- Pedemonte N.; Boido D.; Moran O.; Giampieri M.; Mazzei M.; Ravazzolo R.; Galietta L. J. Structure–activity relationship of 1,4-dihydropyridines as potentiators of the cystic fibrosis transmembrane conductance regulator chloride channel. Mol. Pharmacol. 2007, 72, 197–207. [DOI] [PubMed] [Google Scholar]
- Andreani A.; Rambaldi M.; Bonazzi D Compounds with antitumor activity. V. Hydrazone derivatives of 5-formylimidazo(2,1-b)thiazoles and 6-substituted 2,3-dihydro-5-formylimidazo(2,1-b)thiazoles. Farmaco 1980, 35, 573–580. [DOI] [PubMed] [Google Scholar]
- Andreani A.; Rambaldi M.; Locatelli A.; Bossa R.; Galatulas I.; Ninci M. Synthesis and cardiotonic activity of 2,5-dimethoxyphenylimidazo[2,1-b]thiazoles. Eur. J. Med. Chem. 1992, 27, 431–433. [Google Scholar]
- Andreani A.; Rambaldi M.; Carloni P.; Greci L.; Stipa P. Imidazo[2,1-b]thiazole carbamates and acylureas as potential insect control agents. J. Heterocycl. Chem. 1989, 26, 525–529. [Google Scholar]
- Tallarida R. J.; Murray R. B.. Manual of Pharmacologic Calculations with Computer Programs, 2nd ed.; Springer-Verlag: New York, 1987. [Google Scholar]
- Motulsky H., Christopoulos A.. Fitting Models to Biological Data Using Linear and Non Linear Regression. A Practical Guide to Curve Fitting; GraphPad Software Inc.: San Diego, CA, 2003; www.graphpad.com. [Google Scholar]
- Motulsky H. J.Prism 5 Statistics Guide; GraphPad Software Inc.: San Diego, CA, 2007; www.graphpad.com. [Google Scholar]
- Li H.; Zhang Y.; Tian Z.; Qiu X.; Gu J.; Wu J. Genistein stimulates myocardial contractility in guinea pigs by different subcellular mechanisms. Eur. J. Pharmacol. 2008, 597, 70–74. [DOI] [PubMed] [Google Scholar]
- Bolger G. T.; Genco P.; Klockowski R.; Luchowski E.; Siegel H.; Janis R. A.; Triggle A. M.; Triggle D. J. Characterization of binding of the Ca++ channel antagonist, [3H]nitrendipine, to guinea-pig ileal smooth muscle. J. Pharmacol. Exp. Ther. 1983, 225, 291–309. [PubMed] [Google Scholar]
- Hetzenauer A.; Sinnegger-Brauns M. J.; Striessnig J.; Singewald N. Brain activation pattern induced by stimulation of L-type Ca2+-channels: contribution of Ca(V)1.3 and Ca(V)1.2 isoforms. Neuroscience 2006, 139, 1005–1015. [DOI] [PubMed] [Google Scholar]
- Hirota S.; Janssen L. J. Store-refilling involves both L-type Ca2+ channels and reverse mode Na+/Ca2+ exchange. Eur. Respir. J. 2007, 30, 269–278. [DOI] [PubMed] [Google Scholar]
- Vandebrouck C.; Melin P.; Norez C.; Robert R.; Guibert C.; Mettey Y.; Becq F. Evidence that CFTR is expressed in rat tracheal smooth muscle cells and contributes to bronchodilation. Respir. Res. 2006, 7, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.