

Abstract
Recent gene expression studies on mouse models for retinal degeneration identified deregulation of Pituitary tumor transforming gene 1 (Pttg1) as a potential susceptibility factor involved in photoreceptor cell death. Pttg1 is a transcription regulatory protein involved in sister chromatid segregation, and Pttg1−/− mice exhibit testicular and splenic hypoplasia, thymic hyperplasia, aberrant cell cycle progression, chromosome instability, and impaired glucose homeostasis leading to diabetes, particularly in older males. Due to Pttg1 deregulation in dystrophic retinas, we characterized Pttg1−/− retinas using Hematoxylin and Eosin (H&E) staining, immunohistochemistry (IHC), and electroretinography (ERG). Seven month old Pttg1−/− mice were also examined for a diabetic retinopathy phenotype using Fluorescein Angiography (FA) to test for neovascularization. Our data reveal that up to 9 months of age, Pttg1−/− retinas have a healthy morphology and normal photoreceptor function. This study lays the groundwork for further investigation into the relevance of Pttg1 in retinal dystrophy.
Keywords: Pituitary tumor transforming gene 1 (Pttg1), Mouse knockout technology, Photoreceptor degeneration, Diabetic retinopathy, Fluorescein angiography, Susceptibility gene
Introduction
Pituitary tumor transforming gene 1 (Pttg1) was first identified in rat pituitary tumors, and according to sequence homology, was determined to be the vertebrate securin involved in sister chromatid segregation during mitosis [1]. Over expression of Pttg1 has been previously reported in a variety of endocrine-related tumors, including pituitary, thyroid, breast, ovarian, and uterine tumors, as well as non-endocrine-related cancers involving the central nervous system, pulmonary and gastrointestinal systems [1]. Pttg1 is considered a proto-oncogene for its role in NIH3T3 cell transformation and tumor formation, and correlation with tumor invasiveness and metastasis. Together with its function in cell replication, Pttg1 plays critical roles in DNA damage/repair, organ development and metabolism. More recently, Pttg1 was shown to mediate endothelial cell survival and angiogenesis through its regulation by phospho-Akt [2].
Despite its well characterized role in sister chromatid segregation and cell cycle progression, the murine Pttg1 knockout is surprisingly viable and fertile [1]. The phenotypic characteristics of these mice currently include testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, chromosome instability, and premature centromere division [1]. Pttg1−/− mice also have severely impaired glucose homeostasis and pancreatic beta cell proliferation leading to diabetes, particularly in males over 5 months of age [3].
The mouse Pttg1 gene shares 70% homology to human PTTG1; therefore, the Pttg1−/− mouse is an excellent model for the characterization of the protein in physiological functions with important implications to human disease. Upon comparing the gene expression retinal profiles of the Neural retinal leucine zipper knockout (Nrl−/−), which developmentally lack rods and express only cone photoreceptors, with the double knockout mice lacking G-protein coupled receptor kinase 1 (Nrl−/−Grk1−/−), we identified significant down regulation of Pttg1 in Nrl−/− mice but up regulation in the Nrl−/−Grk1−/− compared to wildtype mice [4]. Further studies into this transcription regulatory protein revealed its significant deregulation in other mouse models associated with pathological conditions related to defective vision, including Crumbs homology-1 (Crb1) and Ras protein specific guanine nucleotide releasing factor 1 (Ras-GRF1) null and mutant mice. Crb1−/− mice exhibit defects in retinal morphology, focal retinal disorganization, and humans with mutations in CRB1 develop Leber congenital amaurosis (LCA) [5]. The retinal phenotype in RasGRF1−/− mice includes severe light perception defects [6].
Due to the significant differential regulation of the Pttg1 transcripts in retinas with a known light-independent photoreceptor degeneration and other retinal dystrophies [5, 6], along with the characterized diabetes found in older males, we hypothesized that Pttg1 plays a critical role in the structure and function of the mouse retina. As a first step in studying the role of Pttg1 in the retina, characterization studies were performed using Hematoxylin and Eosin (H&E) staining, immunohistochemistry (IHC), and electroretinography (ERG). Furthermore, 7 month old male and female Pttg1−/− mice were examined for a diabetic retinopathy phenotype and neovascularization. This study examines the functional and morphologic characteristics associated with the loss of Pttg1 expression in the mouse retina to further understand its transcriptional variability and potential role in retinal dystrophy models.
Experimental Procedures
Experimental Animals
All mice were treated according to the guidelines established by the Institute for Laboratory Animal Research and the Association for Research in Vision and Ophthalmology (ARVO) Statement for Use of Animals in Ophthalmic and Vision Research. The IACUC of the University of Southern California approved all procedures involved in animal experiments. Pituitary tumor transforming gene 1 knockout mice (Pttg1−/−) were generously provided by Shlomo Melmed, M.D. (Cedars-Sinai Research Institute, University of California, Los Angeles) [1, 3]. The original Pttg1−/− mice were generated through the microinjection of 129/SvJ embryonic stem cells carrying the targeting vector into the blastocyst of a C57Bl/6J host. All Pttg1−/− mice were born and maintained in 12 h light:12 h dark cycles.
Genotype Analysis
Genomic DNA from Pttg1−/− mice was extracted using a mixture of Direct polymerase chain reaction (PCR) Lysis Reagent and Proteinase K (Viagen). A Taq PCR Core Kit (Qiagen) was used to verify the disruption of the Pttg1 gene using the following primers [3]:
+mPttg2:5′-GGTTTCAACGCCACGAGTCG-3′
−mPttg1:5′-CTGGCTTTTCAGTAACGCTGTTGAC-3′
Genomic DNA from each animal was also used to test for the presence of the Met450Leu RPE65 single nucleotide polymorphism (SNP) to ensure that the Pttg1−/− mice do not have this common susceptibility form associated with retinal degeneration [7]. Genomic DNA from C57Bl/6J mice were used as a control.
A common nonsense mutation screened for in mouse models is the presence of a DdeI restriction endonuclease enzyme site formed from a C to A transversion in codon 347 of exon 7 of the β subunit of the rod photoreceptor cGMP phosphodiesterase (PDE) gene found in retinal degeneration1 (rd1, RDLE) mice. The insertion of the DdeI site is a known marker for retinal degeneration susceptibility; therefore, the presence of the DdeI site in Pttg1−/− mice was screened using a restriction fragment length polymorphism (RFLP) pattern analysis. Genomic DNA from Pttg1−/− mice was used in the amplification of the potential DdeI insertion site using previously published primers [8]. Genomic DNA from rd1 mice was used as a positive control.
Electroretinography (ERG)
ERGs were recorded as previously described [9–12]. Pttg1−/− and C57Bl/6J mice born and maintained in ambient cyclic light (~5 lux) were examined at 5.5 months of age. At least 6 animals were recorded, consisting of 3 males and 3 female mice. Mice were dark-adapted overnight and their eyes were dilated with topical administration of phenylephrine (2.5%) and tropicamide (0.5%). Mice were anesthetized via an intraperitoneal injection of ketamine HCl (100 mg/kg body weight) and xylazine HCl (10 mg/kg body weight), and the cornea anesthetized with 0.5% topical tetracaine. Each mouse was placed in an aluminum foil-lined Faraday cage and a DLT fiber electrode was placed on the right cornea. A platinum reference electrode was placed on the lower eyelid and another ground electrode on the ipsilateral ear. Photopic stimuli of 10 μs duration of a maximum light intensity (log 2.01 cd s/m2) were delivered through one arm of a coaxial cable using a Grass PS22 xenon flash. The cable delivered both the flash 5 mm from the surface of the cornea and a background light (200 cd/m2), with spectral peaks at 485, 530, 543 nm and minimal transmission below 400 nm, was used. Dark-adapted maximum responses were measured using the non-attenuated light stimulus. The b-wave amplitude was measured from the trough of the a-wave to the peak of the b-wave. Average scotopic thresholds (log −4.59 cd.s/m2 no background light), scotopic maximum levels (log −1.01 cd.s/m2 no background light), mesopic a-wave and b-wave measurements (log 2.01 cd s/m2) were also recorded.
Eyecup Preparation
All mice were euthanized by overdose of carbon dioxide inhalation at mid-day under fluorescent lights. Retinas were dissected under room light using a dissecting microscope and eyes from each genotype and age examined were enucleated, marked for orientation and fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for at least 2 h. Each eyecup was washed twice in PBS for 5 min then dehydrated in a solution of 30% (wt/vol) sucrose in PBS overnight at 4°C. Each eyecup was then embedded in OCT media (Sakura Fineteck, Torrance, CA) [13] and frozen in liquid nitrogen. Retinal cryosections at 8 μm thickness were cut along the vertical meridian through the optic nerve and placed on poly (L-lycine)-coated glass slides.
Retinal Histology
Pttg1−/− and C57Bl/6J control mice of 1, 5, and 9 months of age were examined for changes in morphology associated with increasing age; including outer nuclear layer (ONL) thickness with H&E immunostaining. Briefly, retinal sections were pre-rinsed in double distilled water (ddH20) then dipped in Harris Hematoxylin for 45 s followed by a wash in ddH20. Slides were subsequently dipped in acid alcohol (70% ethanol 1% HCl) for 1 min, washed in ddH2O, then dipped 5 times in ammonia water (0.3% NH4OH) and washed. Slides were then dipped in Eosin-Phloxyine for 1 min, then dehydrated in a series of 95% ethanol and 100% ethanol followed by 6 min in xylene. Once the slides were dried, mounting medium was applied and slides were cover-slipped.
Immunohistochemistry
The mouse anti-Arrestin1 (Arr1) mouse monoclonal antibody (D9F2) and anti-Arrestin4 (Arr4) rabbit polyclonal antibody (mCAR-LUMIJ) were used to immunologically identify rod and cone photoreceptors, respectively. Pttg1−/− and C57Bl/6J mice of 1.5 and 9 months were examined using our established protocol with minor modifications [13]. Frozen retinal sections of C57Bl/6J and Pttg1−/− mice were blocked in IHC blocking buffer (10% normal goat serum, 1% bovine serum albumin [BSA] and 0.2% Triton X-100 in PBS) for 30 min, then incubated with mCAR-LUMIJ (1:1,000) or D9F2 (1:20,000) for 1 h at room temperature. After three 5 min washes in PBS, the slides were incubated in fluorescein anti-rabbit IgG (1:200; Vector Laboratories) for Arr4 immunostaining, and Alexa Fluor 568 anti-mouse IgG (1:400; Invitrogen) for Arr1 immunostaining for 1 h at room temperature. Slides were either mounted with Vectashield Mounting Medium for fluorescence with DAPI (Vector Laboratories) or incubated with the monomeric cyanine nucleic acid stain To-Pro®3 iodide (1:2,000 Invitrogen), mounted with Vectashield Mounting Medium and cover-slipped. Animals were examined at 1.5 and 9 months to detect changes in photoreceptor number with increasing age. Representative images are from the superior region of the retina from male mice.
Fluorescein Angiography (FA)
Retinas from 7 month old male and female Pttg1−/− mice along with age matched C57Bl/6J controls were examined for retinal blood vessel leakage using Fluorescein Angiography (FA) as previously described [4]. Prior to anesthesia, pupils were dilated with topical phenylephrine HCl (2.5%) and tropicamide (0.5%). Following dilation, the mice were anesthetized with an intraperitoneal injection of ketamine HCl (100 mg/kg body weight) and xylazine HCl (10 mg/kg body weight) and kept on a heating pad for the duration of the experiment. The mice were placed in a lateral recumbence with the visual axis of the eye upward. The focal axis of the hand held Kowa Genesis Small Animal Camera was aligned with the visual axis of the mouse. An intraperitoneal injection of 0.05 ml of 10% sodium fluorescein dye solution (Akorn, Buffalo Grove, IL) was administered at the same time the dataphot (internal time) on the Kowa Vk02 2.11 Digital Imaging for Genesis program was initiated. Time lapse digital photos were taken of the center, top, bottom, left, and right quadrants for every time point at approximately 1, 3, 5 and 7 min post injection. Pictures were taken of the central, inferior and superior regions of the retina for comparison.
Results
Pttg1−/− Mice are Met450Met for Rpe65 and Lack the Recessive Mutation for rd1
Tails from the 6 Pttg1−/− mice used for establishing our breeding colony were clipped and genomic DNA extracted for genotype analysis to verify the disruption of Pttg1. A C57Bl/6J mouse was used as the positive control (PC). Figure 1a represents the results of the genotype analysis, which confirm that the animals were knockouts for Pttg1.
Fig. 1.

Genotype Analysis of the Pttg1−/−. a The six mice used for breeding were confirmed as Pttg1 knockouts. The positive control (PC) used was the genomic DNA of a C57Bl/6J mouse. b Genotype analysis for the Rpe65 SNP indicates that all 6 mice are Met450Met. c Digestion with the DdeI restriction endonuclease enzyme shows a lack of the DdeI transversion mutation found in rd1 (RDLE) mice. Genomic DNA from C57Bl/6J and RDLE mice were used as negative and positive controls for the DdeI mutation, respectively
To verify that the Pttg1−/− mice all have the same variant of the Rpe65 gene product at amino acid residue 450, Met450 specific and Leu450 specific primers were used. Figure 1b demonstrates the results of the PCR product of both primer sets, alongside appropriate positive controls. Accordingly, all of the Pttg1−/− mice were Met450Met for Rpe65 and are therefore not susceptible to light damage [7].
To rule out another inherited form of retinal degeneration, Pttg1−/− mice were screened for the presence of the nonsense mutation in the β subunit of the phosphodiesterase gene (β-PDE) containing two DdeI sites. Following PCR amplification of the nucleotide region containing the potential DdeI site, the PCR products were subjected to DdeI digestion and electrophoresed. Figure 1c demonstrates that the DdeI sites are absent from the 6 Pttg1−/− mice. Genomic DNA from rd1 mice were used as a positive control. The absence of a digested PCR product in these Pttg1−/− animals represents no insertion of a DdeI site [8].
Normal Photoreceptor Function and Structure Observed in Pttg1−/− Mice
In order to determine the possible relevance of Pttg1 in the murine retina, the morphology of Pttg1−/− retinas were examined with increasing age for changes in outer nuclear layer (ONL) thickness using H&E staining. As shown in Fig. 2, representative images of the ONL thickness in Pttg1−/− mice at 1, 5, and 9 months of age demonstrate that they have a similar ONL thickness to C57Bl/6J controls of the same age. The only age where this appeared to be slightly less is in 1 month old animals. Also, the outer segments of 1 month old Pttg1−/− retinas appeared slightly shorter when compared to controls; however, due to limited sampling in this study we cannot definitely state that this difference is significant. Despite this observation, the retinas from the 5 and 9 month old mice appear to have a similar ONL thickness and outer segment length. This lack of an obvious thinning of the ONL with increasing age similar to the Nrl−/−Grk1−/− retina [4], or morphological deformities observed in Crb1−/− retinas [5], which both exhibit deregulation of Pttg1 gene expression, confirms that the absence of Pttg1 is not essential for maintaining photoreceptor number and viability in these mice up to 9 months.
Fig. 2.
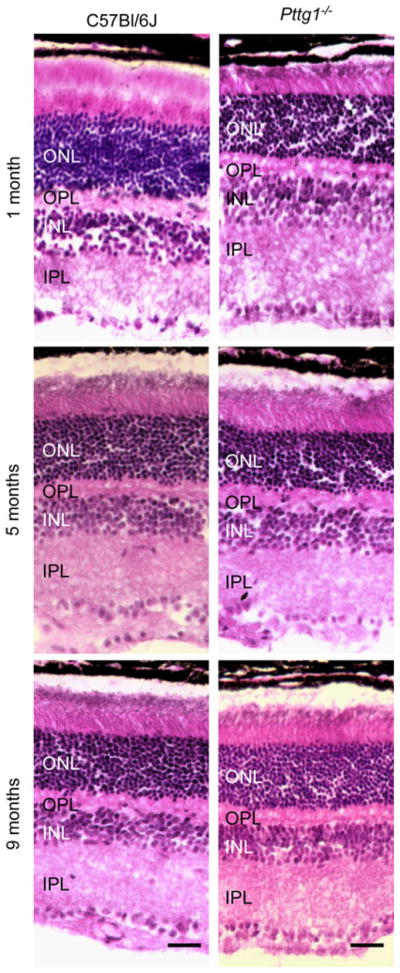
Hematoxylin and Eosin (H&E) Histological Staining of Retinal Morphology and Outer Nuclear Layer (ONL) Thickness in Pttg1−/− and C57Bl/6J. All mice were reared in cycling light and light adapted for 1 h. The retinal morphology and ONL thickness are similar between the Pttg1−/− and C57Bl/6J and 1, 5 and 9 months of age. Digital images were taken of the central inferior region of the retina. The scale bar represents a length of 20 μm
Alteration to rod and cone cell function was analyzed in Pttg1−/− mice in comparison to age matched C57Bl/6J controls in both males and females at 7 months of age. According to the scotopic threshold and maximum b-wave amplitudes plotted in Fig. 3a, there is no statistically significant difference (2 way ANOVA) in rod function in Pttg1−/− mice when compared to controls. A similar observation was made when examining photopic ERGs for cone function in the same animals during 13 min of exposure to background illumination (Fig. 3b). Both mouse strains showed the same light-adaptation of b-wave amplitude. Statistically similar (2 way ANOVA) mesopic a- and b-wave maximum amplitudes (Fig. 3a) also demonstrate that the expression of Pttg1 is not essential for normal rod or cone function in the mouse.
Fig. 3.

Amplitude of Dark-Adapted Scotopic and Mesopic and Light-Adapted Photopic ERG Responses in C57Bl/6J and Pttg1−/− Mice. Seven-month old mice were dark-adapted overnight before ERG recording. a Scotopic threshold (log −4.59 cd s/m2 ) and maximum (log −1.01 cd s/m2) responses showed no statistical difference (2 way ANOVA) in rod function comparing knockout and control mice. Mesopic a- and b-wave maximum amplitudes confirmed no statistical difference (2 way ANOVA) in combined rod and cone function. b Pure cone function was also the same in photopic ERG responses (log 2.01 cd s/m2) during 13 min of background illumination (200 cd/m2)
To further examine the potential importance of Pttg1 for murine cone photoreceptors, Arrestin 4 (Arr4) immunohistochemistry (IHC) staining was conducted in 1.5 and 9 month old animals to examine changes associated with increasing age. Representative images in Fig. 4 reveal similar cone numbers in both Pttg1−/− and C57Bl/6J mice at 9 months. Although the fluorescent staining appears slightly lighter in the 1.5 month old Pttg1−/− mice, this is perhaps due to the plane of focus that was adjusted to view both the synaptic and inner segment staining of the cones. Similar to the H&E stained section, the 1.5 month old Pttg1−/− retina appears to have an outer segment that is slightly shorter than the C57Bl/6J mice (Fig. 2 and Fig. 4).
Fig. 4.
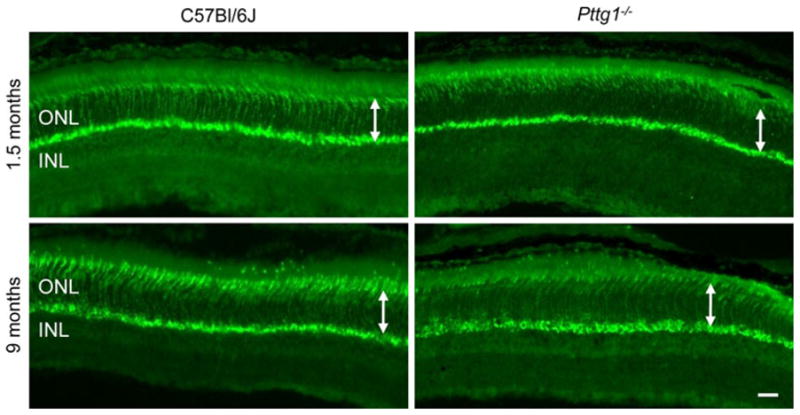
Mouse Cone Arrestin4 Immunohistochemical Staining in the Superior Region of Retinas from Pttg1−/− and C57Bl/6J Mice. IHC staining patterns with PAb mCAR-LUMIJ indicate a similar number of cone cells between Pttg1−/− mice and C57Bl/6J controls at both 1.5 and 9 months of age. White arrows demonstrate no change in ONL thickness. Scale bar, 10 μm
The immunohistological staining pattern for rod plus cone photoreceptors was performed using Arrestin 1 (Arr1) in Pttg1−/− and C57Bl/6J retinas at 1.5 and 9 months (Fig. 5). Qualitatively, the retinas appear to be similar in rod cell number at both ages examined. Rod photoreceptor structure and distribution of Arr1 appears to be normal in Pttg1−/− retinas. Interestingly, in 1.5 month old mice, the outer segment appears to be slightly shorter than those of C57Bl/6J mice; a similar observation made in 1.5 month old Pttg1−/− mice when stained with mCAR-LUMIJ (Fig. 4) and H&E (Fig. 2). Despite this, the structure, orientation, ONL thickness, and morphology all appear to be similar between Pttg1−/− and C57Bl/6J mice. It is important to note that the IHC analysis performed here is merely qualitative. To verify this initial observation in the Pttg1−/− retinas compared to controls, additional quantitative studies with more animals and a spider plot analysis would be necessary.
Fig. 5.
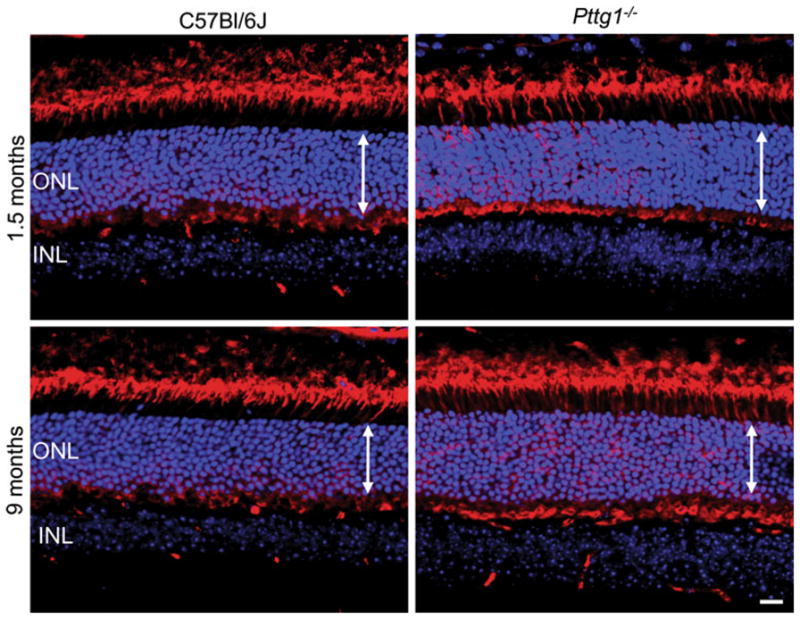
Mouse Arrestin1 (Arr1) Immunohistochemical Staining in the Superior Region of Retinas from Pttg1−/− and C57Bl/6J Mice. IHC staining patterns with MAb D9F2 for Arr1 (red) indicate a similar number of rod photoreceptor cells between Pttg1−/− mice and C57Bl/6J controls at both 1.5 and 9 months of age. Propidium Iodide (TroPro3) was used to stain nuclei (blue). White arrows demonstrate no change in ONL thickness. Scale bar,10 μm
Pttg1−/− Male and Female Mice have Normal Fluorescein Angiography Staining Patterns
The disruption of Pttg1 in mice has been shown to severely impair glucose homeostasis leading to diabetes in late adulthood, especially in males associated with nonautoimmune insulinopenia and reversed alpha/beta cell ratio [3]. Pttg1−/− animals therefore exhibit sexually dimorphic diabetes mellitus [3]. Based on these observations, we hypothesized that male Pttg1−/− mice over 5 months of age had defects in vasculature similar to diabetic retinopathy. Diabetic retinopathy is caused by damage to retinal blood vessels through the formation of microaneurysms, retinal hemorrhage, or retinal neovascularization [16].
In order to examine changes in retinal vasculature associated with sexually dimorphic Pttg1−/− mediated diabetes, fluorescein angiography (FA) staining was performed on 7 month old Pttg1−/− male and female mice. C57Bl/6J mice at both ages were used for comparison. The central, superior and inferior regions of the retina are provided in Fig. 6. The images show no apparent changes in vasculature associated with leakage or breaks. This led us to conclude that the diabetic phenotype observed in older male Pttg1−/− mice does not affect the retina and these animals did not exhibit a phenotype similar to diabetic retinopathy up to 7 months of age. Although the diabetic phenotype presents at 5 months of age, the diabetic retinopathy phenotype may be apparent at an older age, such as 1 year. This possibility warrants further investigation.
Fig. 6.
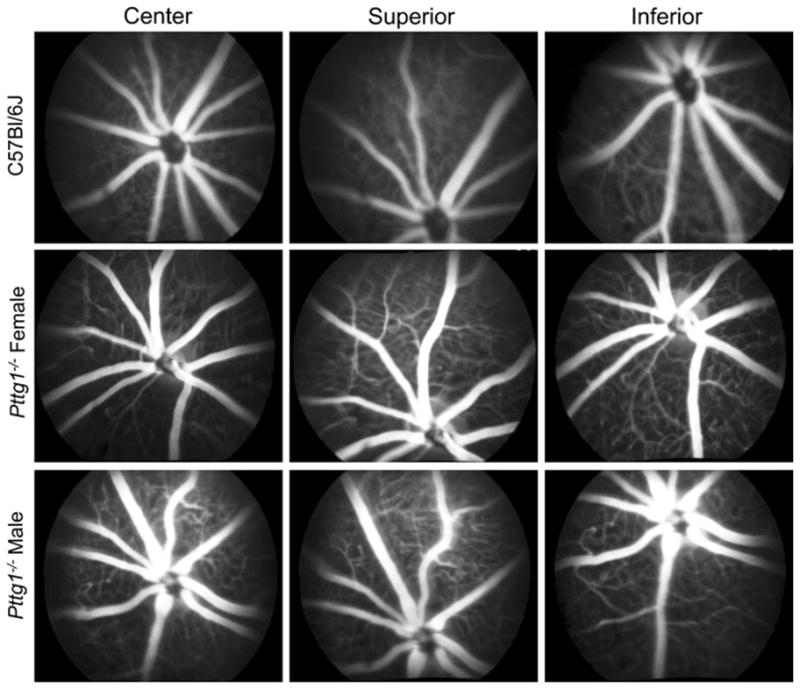
Fluorescein Angiography (FA) of Retinal Vasculature from Pttg1−/− and C57Bl/6J Mice. Representative images of Fluorescein Angiography (FA) from the central, superior and inferior regions of the retinas from 7 month old animals. Both male and female Pttg1−/− mice were compared to a male C57Bl/6J healthy control [4]. No blood leakage was found in retinas from all animals examined
Discussion
Pttg1 is a well established cell-cycle regulatory protein and proto-oncogene with critical roles involving sister chromatid segregation to metabolism. In addition to its established functions, Pttg1 deregulation has recently been observed in several mouse models for retinal degeneration. The Pttg1 gene is differentially regulated in the Nrl−/−, Nrl−/−Grk1−/−, RasGRF1−/− and Crb1−/− knockout mice [4–6]. While some of the knockout mice listed have significant over-expression of Pttg1, others down-regulate the gene by significant margins. Retinas of Pttg1 null mice were therefore examined to further elucidate the structural and functional relevance of Pttg1 in the retina.
According to our retinal morphology studies, Pttg1−/− mice are comparable to C57Bl/6J controls, with a slightly smaller ONL thickness at 1 month. However by 5 months, the ONL thickness is similar to controls and maintains this thickness up to 9 months of age. By rearing the mice in light:dark cycles, we were able to test for the presence of light mediated photoreceptor cell death. Because the ONL thickness of these mice were stable up to 9 months, it is clear that the photoreceptor cells are viable and not susceptible to light mediated degeneration.
Nrl−/− mice have retinas lacking rods but express photoreceptors that share morphological, molecular and electrophysiological characteristics with cones [14]. In the initial DNA microarray studies performed on Nrl−/− mice, Pttg1 was identified as significantly down regulated by 10.3 fold when compared to wildtype controls [15]. We therefore postulated that the loss of Pttg1 influenced cone cell function and number with age. Using previously established ERG and IHC techniques [9], our examination of cone functional ERGs in parallel to IHC of cone Arr4, we observed that Pttg1−/− mice have photopic b-wave amplitudes and light-adaptation similar to controls and healthy cone cell numbers (Fig. 4) that persist up to 9 months.
Rod photoreceptor staining using the Arr1 antibody, D9F2, demonstrates qualitatively that there is no apparent difference in the number of rod photoreceptors at either 1.5 or 9 months of age. However, a slight difference in the thickness of the outer segment layer was observed in retinas at 1.5 months of age (Fig. 5), but it is not apparent if this developmental difference plays a physiologically relevant role. One possibility is the delayed development of the outer segments of these photoreceptors due to the loss of Pttg1, which is important in cell-cycle progression. Further statistical measurements would quantitatively answer this question.
Retina function at 7 months of age was examined using scotopic, mesopic and photopic ERG analyses. Statistical analysis of maximum a- and b-wave amplitudes along with threshold values demonstrate that the retinas of Pttg1−/− mice function normally at 7 months. The slight differences observed at 1.5 months in ONL thickness, as well as outer segment thinning, was not examined by ERG analysis; yet, it is apparent that they do not have a long term detrimental loss of either rod or cone function.
We performed FA staining on older male and female mice to detect signs of diabetic retinopathy at an age where animals were previously described to exhibit characteristics of diabetes mellitus [3]. However, in our FA experiments in both male and female mice at 7 months, we observed normal immunological staining patterns and the absence of any leakage or damage to blood vessels (Fig. 6). These FA experiments were performed in parallel with other knockout mice that did exhibit leagage and damaged blood vessels [4]. In conclusion, Pttg1−/− male mice over 5 months exhibit diabetes due to their inefficient pancreatic beta cell turnover [3]; conversely, their retinas appear to function normally, have a healthy morphology at the ages we examined, and do not exhibit a neovascular phenotype similar to diabetic retinopathy.
To further elucidate the factors that maintain the maturation and daily metabolism of a healthy retina, we identified Pttg1 as a novel target in the homeostasis of retinal structure and function. The results presented here demonstrate that the loss of Pttg1 does not manifest into a retinal degenerative phenotype in mouse and its expression is not essential in the maintenance of normal retinal function up to 9 months of age. Despite the previous observations that Pttg1 leads to diabetes mellitus in male mice, the animals examined here lacked neovascularization or a diabetic retinopathy phenotype, although this does not rule out the possibility of metabolic deficits in still older mice. Furthermore, deregulation of Pttg1 must not be minimized because the over expression of the gene transcript may potentially lead to retinal neovascularization and photoreceptor dystrophy.
Future studies on retinal disease models with enhanced Pttg1 expression crossed with the Pttg1−/− mouse may help to identify Pttg1 as an important susceptibility modifier or regulator involved in retinal degeneration. Based on our experiments, Pttg1 is not a primary insult leading to photoreceptor cell death; however, it may contribute secondarily to the functional loss of other essential proteins for normal retinal function. For example, Pttg1 has been shown to stimulate the production of angiogenic growth factors such as basic fibroblast growth factor (bFGF) and enhance the stability of endothelial cells in solid tumors [3]. Although further examination is required, the observed enhanced expression of Pttg1 may be a secondary effect of the retinal neovascularization phenotype in the Nrl−/−Grk1−/− retina [2, 4, 16]. Another possibility is that the expression of Pttg1 is sensitive to changes in the expression of functionally relevant proteins, such as Nrl and Crb1, and may be an indicator for retinal stress, but not demonstrate a causative effect. Whether or not the variability in Pttg1 expression is a consequence of or a contributor to retinal degeneration has yet to be determined. Although numerous mechanisms of action have been identified for Pttg1, these studies provide the foundation for further studies to examine the relevance of this multi-functional protein in the murine retina.
Acknowledgments
Cheryl M. Craft, Ph.D. is the Mary D. Allen Chair in Vision Research, Doheny Eye Institute and a Research to Prevent Blindness Senior Scientific Investigator. Supported by NIH EY015851 (CMC), National Eye Institute Core Grant EY03040 (Doheny Eye Institute), Research to Prevent Blindness (DEI), the Foundation Fighting Blindness (DEI), Mary D. Allen Foundation (Richard Newton Lolley Memorial Scholarship (RMY), William Hansen Sandberg Memorial Foundation (RMY), Tony Gray Foundation, Dorie Miller, and Dr. Paul R. Burton. We sincerely appreciate the generous continued support for our vision research program by Mrs. Mary D. Allen. We also thank Dr. Shlomo Melmed for the Pttg1−/− mice and Dr. Larry A. Donoso for the Arr1 D9F2 antibody. The authors acknowledge the expert technical support of Bruce Brown, Alexander Wang for his help with the experiments, Lawrence Rife for ERG analysis, Fernando Gallardo for FA, and Ernesto Barron for figures. Also, we thank Dr. Shun-Ping Huang, Dr. Freddi Zuniga and Leng-Ying Chen for their scientific contributions.
Contributor Information
Rosanne M. Yetemian, Division of Retinal Molecular Biology, Department of Ophthalmology, The Mary D. Allen Laboratory for Vision Research, Doheny Eye Institute, Los Angeles, CA 90033, USA
Cheryl M. Craft, Email: ccraft@usc.edu, Division of Retinal Molecular Biology, Department of Ophthalmology, The Mary D. Allen Laboratory for Vision Research, Doheny Eye Institute, Los Angeles, CA 90033, USA. Department of Cell & Neurobiology, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA. Department of Ophthalmology, Keck School of Medicine of the University of Southern California, 1355 San Pablo Street, DVRC 405, Los Angeles, CA 90033-9224, USA
References
- 1.Wang Z, Yu R, Melmed S. Mice lacking pituitary tumor transforming gene show testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, and premature centromere division. Mol Endocrinol. 2001;15:1870–1879. doi: 10.1210/mend.15.11.0729. [DOI] [PubMed] [Google Scholar]
- 2.Caporali S, Levati L, Starace G, et al. AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol Pharmacol. 2008;74:173–183. doi: 10.1124/mol.107.044743. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Moro E, Kovacs K, et al. Pituitary tumor transforming gene-null male mice exhibit impaired pancreatic beta cell proliferation and diabetes. Proc Natl Acad Sci USA. 2003;100:3428–3432. doi: 10.1073/pnas.0638052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yetemian RM, Brown BM, Craft CM. Loss of G-protein coupled receptor kinase 1 in Nrl−/− mice leads to neovascularization, enhanced inflammatory response, and age-related cone dystrophy. Invest Ophthalmol Vis Sci. 2010;51:6196–6206. doi: 10.1167/iovs.10-5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van de Pavert SA, Sanz AS, Aartsen WM, et al. Crb1 is a determinant of retinal apical Muller glia cell features. Glia. 2007;55:1486–1497. doi: 10.1002/glia.20561. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez-Medarde A, Barhoum R, Riquelme R, et al. RasGRF1 disruption causes retinal photoreception defects and associated transcriptomic alterations. J Neurochem. 2009;110:641–652. doi: 10.1111/j.1471-4159.2009.06162.x. [DOI] [PubMed] [Google Scholar]
- 7.Kim SR, Fishkin N, Kong J, et al. Rpe65 Leu450Met variant is associated with reduced levels of the retinal pigment epithelium lipofuscin fluorophores A2E and iso-A2E. Proc Natl Acad Sci USA. 2004;101:11668–11672. doi: 10.1073/pnas.0403499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pittler SJ, Baehr W. Identification of a nonsence mutation in the rod photoreceptor cGMP phosphodiesterase β-subunit gene of the rd mouse. Proc Natl Acad Science USA. 1991;88:8322–8326. doi: 10.1073/pnas.88.19.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown BM, Ramirez T, Rife L, Craft CM. Visual arrestin 1 contributes to cone photoreceptor survival and light-adaptation. Invest Ophthalmol Vis Sci. 2010;51:2372–2380. doi: 10.1167/iovs.09-4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu X, Brown B, Rife L, Craft CM. Slowed photoresponse recovery and age-related degeneration in cones lacking G protein-coupled receptor kinase 1. In: Hollyfield JG, Anderson RE, LaVail MM, editors. Advances in experimental medicine and biology. Vol. 572. Springer; 2006. pp. 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu X, Li A, Brown B, Weiss ER, et al. Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol Vis. 2002;8:462–471. [PubMed] [Google Scholar]
- 12.Zhu X, Wu K, Rife L, et al. Carboxypeptidase E is required for normal synaptic transmission from photoreceptors to the inner retina. J Neurochem. 2005;95:1351–1362. doi: 10.1111/j.1471-4159.2005.03460.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhu X, Craft CM. Modulation of CRX transactivation activity by phosducin isoforms. Mol Cell Biol. 2000;20:5216–5226. doi: 10.1128/mcb.20.14.5216-5226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daniele LL, Lillo C, Lyubarsky AL, et al. Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Invest Ophthalmol Vis Sci. 2005;46:2156–2167. doi: 10.1167/iovs.04-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh EC, Cheng H, Hao H, et al. Rod differentiation factor NRL activates the expression of nuclear receptor NR2E3 to suppress the development of cone photoreceptors. Brain Res. 2008;1236:16–29. doi: 10.1016/j.brainres.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caporali A, Pani E, Horrevoets AJ, et al. Neurotrophin p75 receptor (p75NTR) promotes endothelial cell apoptosis and inhibits angiogenesis: implications for diabetes-induced impaired neovascularization in ischemic limb muscles. Circ Res. 2008;103:e15–e26. doi: 10.1161/CIRCRESAHA.108.177386. [DOI] [PMC free article] [PubMed] [Google Scholar]