

Abstract
Brief forebrain ischemia is a model of the delayed hippocampal neuronal loss seen in patients following cardiac arrest and resuscitation. Previous studies demonstrated that selective dysfunction of hippocampal CA1 subregion astrocytes occurs hours to days before delayed neuronal death. In this study we tested the strategy of directing protection to astrocytes to protect neighboring neurons from forebrain ischemia. Two well-studied protective proteins, heat shock protein 72 (Hsp72) or superoxide dismutase 2 (SOD2), were genetically targeted for expression in astrocytes using the astrocyte-specific human glial fibrillary acidic protein (GFAP) promoter. The expression constructs were injected stereotacticly immediately above the hippocampal CA1 region on one side of the rat brain two days prior to forebrain ischemia. Cell type specific expression was confirmed by double label immunohistochemistry. When the expression constructs were injected two days before transient forebrain ischemia, the loss of CA1 hippocampal neurons observed seven days later was significantly reduced on the injected side compared with controls. This neuroprotection was associated with significantly better preservation of astrocyte glutamate transporter-1 immunoreactivity at 5-h reperfusion and reduced oxidative stress. Improving the resistance of astrocytes to ischemic stress by targeting either the cytosolic or mitochondrial compartment was thus associated with preservation of CA1 neurons following forebrain ischemia. Targeting astrocytes is a promising strategy for neuronal preservation following cardiac arrest and resuscitation.
Keywords: global ischemia, hippocampus, heat shock protein, super-oxide dismutase, mitochondria
INTRODUCTION
Stroke and cerebral ischemia induced during cardiac arrest and resuscitation are major sources of neurological disability. In this country alone about 450,000 patients suffer a cardiac arrest each year, and neurological impairment is one of the most feared complications in those who are resuscitated. The only treatment shown to improve neurological outcome in these patients is hypothermia (Bernard et al., 2002; Group, 2002) Although most work on developing protective strategies has focused on neurons, the importance of astrocytes in neuronal survival of ischemic insults has been recognized for some time (Swanson et al., 2004; Takano et al., 2009), and lack of consideration of the role of astrocytes is thought to be a factor in the failure of potential neuroprotective strategies (Nedergaard and Dirnagl, 2005). Inhibition of astrocyte mitochondrial function (Voloboueva et al., 2007) or astrocyte glutamate uptake (Dugan et al., 1995; Rosenberg and Aizenman, 1989) impairs neuronal survival of excitotoxic injury. Excitotoxic neuronal injury is a common mechanism in both acute and chronic neurodegenerative diseases. Selective dysfunction of hippocampal CA1 astrocytes occurs at early reperfusion times, hours to days before the death of CA1 neurons following forebrain ischemia. In rats subjected to transient forebrain ischemia, CA1 astrocytes lose glutamate transport activity and immunoreactivity for glutamate transporter 1 (GLT-1) within a few hours of reperfusion and well before CA1 neuronal death (Chen et al., 2005; Ouyang et al., 2007; Yeh et al., 2005). In our previous study we demonstrated that increased generation of reactive oxygen species (ROS) and mitochondrial dysfunction in CA1 astrocytes contribute to ischemia-induced loss of GLT-1, and ultimately to delayed death of CA1 neurons (Ouyang et al., 2007). It is thus a plausible hypothesis that strategies aimed at maintaining astrocyte function would alleviate ischemia-induced loss of CA1 neurons. We tested this hypothesis by overexpressing protective genes selectively in hippocampal astrocytes in vivo.
Heat shock proteins (HSPs) are stress inducible molecular chaperones involved in the regulation of cellular protein homeostasis and cell survival (Guzhova and Margulis, 2006). Work from several laboratories including ours has demonstrated neuroprotection from ischemic brain injury with overexpression of chaperones and co-chaperones, both in animal stroke models and in cell culture (Giffard et al., 2008; Yenari et al., 1999). Mitochondrial dysfunction and oxidative stress are key determinants of outcome following cerebral ischemia (Chan, 2005), and the role of superoxide dismutase 2 (SOD2/manganese superoxide) is well established (Maier et al., 2006; Wang et al., 2005). SOD2 is a mitochondrial antioxidant enzyme, which scavenges superoxide radicals and overexpression provides direct neuroprotection, glioprotection and protection of the blood brain barrier (Jung et al., 2009; Maier et al., 2006; Wang et al., 2005). Reduction of SOD2 activity was shown to increase neuronal death induced by transient cerebral ischemia (Chan, 2005). In this study we tested the idea that targeting Hsp72 or SOD2 overexpression to hippocampal astrocytes would improve the survival of CA1 neurons after transient forebrain ischemia.
MATERIALS AND METHODS
GFAP Promoter Hsp72 and SOD2 Constructs
A plasmid containing the human GFAP promoter sequence, pGFAP-IkBa-SR, (Zhang et al., 2005) was a kind gift from Dr. M Schwaninger, and the human Hsp72 sequence, pcDNA3/Hsp72WT, was a kind gift from Dr. R. Ran (Ran et al., 2004). The 2.1 kb GFAP 5′ regulatory sequence (Besnard et al., 1991) was isolated from pGFAP-IkBa-SR using EcoRV and NheI. This fragment includes the 34 bp β-globin translation initiation sequence. pcDNA3/Hsp72WT was cut with NruI and NheI to remove the CMV promoter and the 6x-His tag, which were replaced with the GFAPp EcoRV-NheI fragment fused in-frame to the Hsp72 coding sequence to produce GFAPp-Hsp72. To place SOD2 downstream of the same sequence human SOD2 was polymerase chain reaction (PCR) amplified with primers: GCTAGCTTGAG CCGGGCAGTGT and AAGCTTACGTAGCGTGGTTTAC. These replaced the start codon with an NheI site and added a downstream HindIII site. The enzymes noted above were used to replace the Hsp72 coding sequence with that of SOD2 to create GFAPp-SOD2.
Transfection of Hippocampal Astrocytes to Overexpress Hsp72 or SOD2 In Vivo
All experiments using animals were performed in accordance with protocols approved by the Stanford University animal care and use committee, and in keeping with the National Institutes of Health guide. Recombinant DNA was used according to an approved biosafety protocol. DNA/lipid complex was prepared and injected stereotactically into brain as described previously (Sun et al., 2006; Xu et al., 2009). Briefly, adult male Sprague-Dawley rats (age, three months; 300–400 g) were anesthetized with isoflurane in 70% N2O and 30% O2 and DNA (10 μg) of expression plasmid encoding Hsp72, SOD2, or empty plasmid lacking the insert, mixed with the cationic lipid DOTAP (1:3 μg/μL, Roche Applied Science) was infused steretactically just outside the right hippocampus (from bregma −3.8 mm, M-L 2.0 mm, deep 2.5 mm) at 2 μL/min, maximal total volume 40 μL via a burr hole. Two days later rats were either sacrificed to assess expression, or subjected to forebrain ischemia.
Forebrain Ischemia
Forebrain ischemia was induced with bilateral carotid artery occlusion plus hypotension (Ouyang et al., 2007) according to an approved animal protocol. After induction of anesthesia a femoral artery catheter was placed for continuous blood pressure monitoring (Puritan-Bennett Corporation, Wilmington, MA). Hypotension (to 40 mm Hg) was induced by removing blood into heparinized sterile tubing after heparinization (Heparin 3 units per gram). Both carotid arteries were clamped for 10 min then unclamped and the shed blood reinfused. Rectal temperature was 37±1.0°C controlled by a Home-othermic blanket (Harvard Apparatus, Holliston, MA). Respiratory rate, oxygen saturation, and heart rate were monitored during surgery with a small animal Oximeter (STARR life sciences Corp., Allison Park, PA). Rats were sacrificed 5 h or seven days after ischemia under deep anesthesia with isoflurane. Brains were removed after perfusion with saline for protein collection or saline followed by ice-cold 4% phosphate buffered paraformaldehyde for immunohistochemistry.
Immunohistochemistry
Coronal brain sections, 50 μm, were made with a Vibratome (Leica VT 1000S, Heidelberger, Nussloch, Germany). Fluorescence immunohistochemistry was performed as previously described (Xu et al., 2009) using antibodies against Hsp 72 (C92F3A-5, Stressgen, Ann Arbor, MI), SOD2 (ab13534, Abcam, Cambridge, MA) GFAP (MAB 360, Cell Signaling, PO Box 3843, Boston, MA) and GLT-1 (Ab1782, Chemicon, Temecula, CA) or Map2 (Ab3418, Chemicon), or 4-hydroxy-2-nonenol (HNE) (#24327, OXIS, Beverly Hills, CA). Secondary antibodies were: Alexa Fluor 594, goat anti-mouse -IgG (Invitrogen, Carlsbad, CA 92008); Alexa fluor 488 goat anti rabbit IgG, (Invitrogen), or goat anti-guinea pig IgG HRP (Chemicon). To compare staining in different conditions brain sections were stained at the same time and photographed on the microscope with the same settings, followed by fluorescence densitometry to compare the levels of staining.
Assessment of CA1 Hippocampal Injury
Hippocampal injury was quantitated by Cresyl violet staining. Sections were mounted on gelatin-coated microscope slides, incubated in 1.0% cresyl violet acetate (Sigma, St. Louis, MO) for 5 min then dehydrated sequentially in 70, 90, 95, and 100% ethanol baths at room temperature. The density of cresyl violet staining was determined in a fixed area of CA1, and expressed as a percentage of the same area of CA1 in sham-operated animals.
Primary Cultures and Injury Paradigm
Primary astrocyte cultures were prepared from post-natal (Days 1–3) Swiss Webster mice (Simonsen, Gilroy, CA) as previously described (Dugan et al., 1995; Xu et al., 2001). Cultured cells were subjected to in vitro ischemia (oxygen glucose deprivation, OGD) in deoxygenated balanced salt solution lacking glucose in an anoxia chamber (Coy Laboratory Products Inc.), oxygen tension <0.02%, as previously described (Xu et al., 2009). Mitochondria were isolated as previously described (Xu et al., 2009) to measure mitochondrial complex activity, following 1 h OGD and 18 h recovery in the presence of oxygen and glucose. This duration of OGD is shorter than the duration needed to cause cell death. All experiments were repeated at least three times using cells isolated from different dissections.
Measurement of Mitochondrial Complex Activity
Rotenone-sensitive NADH-decylubiquinone oxidoreductase (complex I) activity was assayed at 340 nm using the acceptor coenzyme Q0 and nicotinamide adenine dinucleotide (NADH) as a donor as previously described (Xu et al., 2009). The assay was carried out in the presence and absence of 5 μmol/L rotenone to derive the rotenone sensitive complex-I activity, expressed as nmol NADH oxidized/min/mg protein (ε340 = 6.23/mmol/L/cm).
Cytochrome c oxidase (complex IV activity) was measured using a cytochrome c Oxidase Assay kit (CYTOX-OX1; Sigma), taking reduced cytochrome c as the donor. The initial rate of cytochrome c reduction was used for the calculation of activity. Proteins were measured with the Bio-Rad protein assay kit and activities were normalized to protein concentration and expressed as a ratio to normal control.
Western Blot Analysis
Western blots were performed as previously described (Xu et al., 2009) using hippocampi solubilized in lysis buffer. Equal amounts of protein, 50 μg/well, were separated by SDS-PAGE on a 4–20% polyacrylamide gel and electrotransferred onto Immobilon polyvinylidene fluoride membrane (Millipore Corp., Bedford, MA). Membranes were incubated overnight with 1:1,000 dilution of primary GLT-1 or SOD2 antibody and subsequently incubated with 1:2,000 HRP conjugated secondary antibody (Cell Signaling, Danvers, MA) for 90 min andvisualized with the enhanced chemiluminescence detection system (Amersham, Piscataway, NJ).
Detection of ROS
ROS production in brain parenchyma following ischemia was assessed with hydroethidine (HEt) which is oxidized by ROS to ethidium. HEt (Molecular Probes, Eugene, OR) 0.5 mg in 200 μL saline was administered via tail vein 1 h prior to sacrifice at 5 h following fore-brain ischemia. Brains were perfused with ice cold saline, then 4% paraformaldehyde, removed and postfixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 48 h. Ethidium fluorescence in CA1 was identified using a Zeiss axiovert epifluorescence microscope. In addition, lipid peroxidation products were detected with antibody to HNE as described above in immunohistochemistry.
Statistics
All assay data represent at least three independent experiments. Protection from forebrain ischemia was tested once with the indicated numbers of animals per group. Data are reported as mean ± SEM. Statistical differences were determined using ANOVA followed by Scheffes post hoc test with P < 0.05 considered significant (Sigmastat, Ashburn, VA) or by t-test when only two groups were compared.
RESULTS
Astrocyte Specific Expression of Hsp72
After stereotactic injection just above the hippocampus the GFAPp-Hsp72 construct directed expression of Hsp72 specifically in astrocytes. Single immunostaining showed widespread expression of Hsp72 in the CA1 region (Fig. 1A left). Injection of control DNA results in almost no immunoreactivity for Hsp72 (Fig. 1A, right). Double immunostaining showed Hsp 72 colocalized with the astrocyte specific marker GFAP in the CA1 region (Fig. 1B, left) but not with the neuronal marker Map2 (Fig. 1B, right).
Fig. 1.

GFAPp-Hsp72 causes overexpression of Hsp72 selectively in astrocytes in vivo. Immunostained sections of brains harvested two days after stereotaxic infusion of GFAPp-Hsp72 or control DNA are shown. Sections were stained with antibody to Hsp72 (green) and then counterstained with PI (red) to show nuclei (A). The CA1 neuronal cell body level is indicated on the right. (B) Sections were double immunostained to identify astrocytes (GFAP in red, left panel) or neurons (Map2 in red, right panel). Colocalization was observed with GFAP but not Map2. Not all astrocytes were transfected, as cells staining for GFAP but not Hsp72 can be observed.
Selective Overexpression of Hsp72 in Astrocytes Reduces Death of CA1 Neurons
Selective loss of CA1 hippocampal neurons (white arrows) was clear in control injected brains (Fig. 2A middle panel) observed seven days after transient fore-brain ischemia. When GFAPp-Hsp72 was injected above the hippocampus on one side (black arrow for microinjection track, Fig. 2A, lower panel), the loss of CA1 neurons was significantly reduced compared with the noninjected side or control plasmid injected hippocampi. Quantitation of cresyl violet staining showed significantly greater survival of CA1 neurons in the GFAPp-Hsp72 group compared with the control plasmid injected group at seven days survival (Fig. 2B, P < 0.05).
Fig. 2.
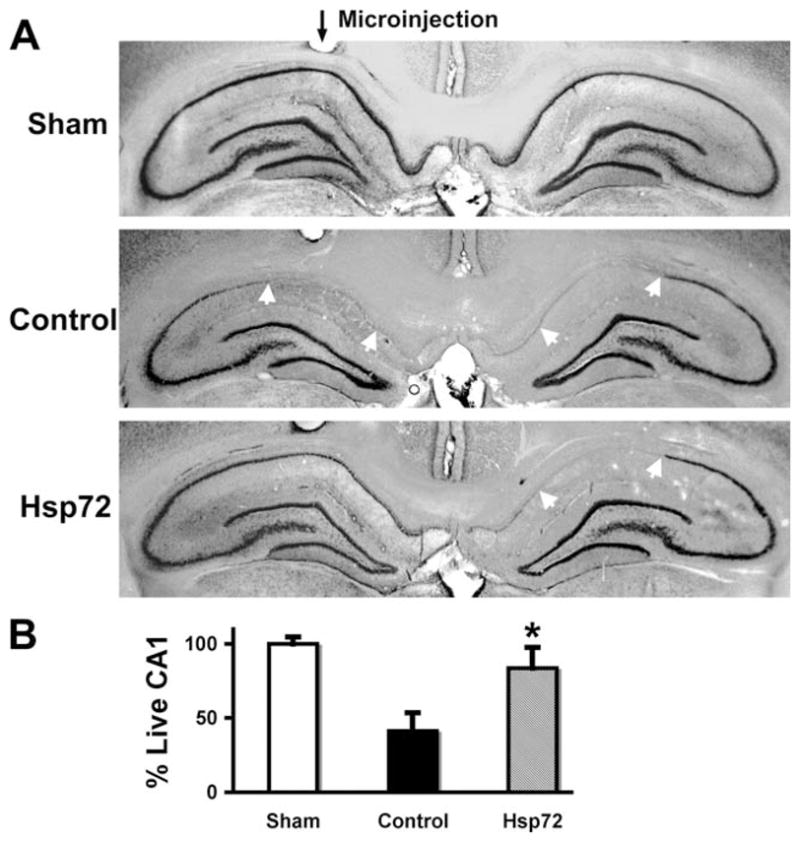
Targeted overexpression of Hsp72 in astrocytes reduces vulnerability of CA1 neurons to forebrain ischemia. A: GFAPp-Hsp72 or control DNA was injected stereotaxicly just above CA1 on one side (black arrow for microinjection track) two days before forebrain ischemia. Selective loss of CA1 hippocampal neurons (between white arrows in middle and lower panels in (A) was observed at seven days reperfusion by cresyl violet staining. The loss of CA1 hippocampal neurons was significantly reduced on the Hsp72 injected side compared with the noninjected side (lower panel) or the control DNA injected hippocampus (middle panel). B: Loss of CA1 neurons was quantified by cresyl violet staining density. There was significantly greater preservation of CA1 neurons ipsilateral to injection in the GFAPp-Hsp72 group compared with the control plasmid injected group (n = 5, *P < 0.05).
Hsp72 Overexpression Reduces GLT-1 Loss, ROS Levels, and Preserves Mitochondrial Complex Activity
We previously demonstrated reduced transport activity and protein levels of GLT-1 at 5-h reperfusion following forebrain ischemia (Ouyang et al., 2007). GLT-1 immunoreactivity at 5 h reperfusion is significantly preserved when GFAPp-Hsp72 is injected prior to ischemia. This is seen by immunostaining (Fig. 3A) and by Western (Fig. 3B), in GFAPp-Hsp72 injected compared with control DNA injected hippocampi (n = 3; P < 0.05). There was no significant difference in GLT-1 levels between GFAPp-Hsp72 and control DNA injected sham-operated animals, or between noninjected and control DNA injected animals (data not shown). Since we previously found increased ROS correlated with reduced levels of GLT-1 we assessed ROS using HEt. Brains removed after 5 h reperfusion showed greater ethidium fluorescence in control plasmid injected hippocampi compared with the GFAPp-Hsp72 injected hippocampi (Fig. 3C).
Fig. 3.
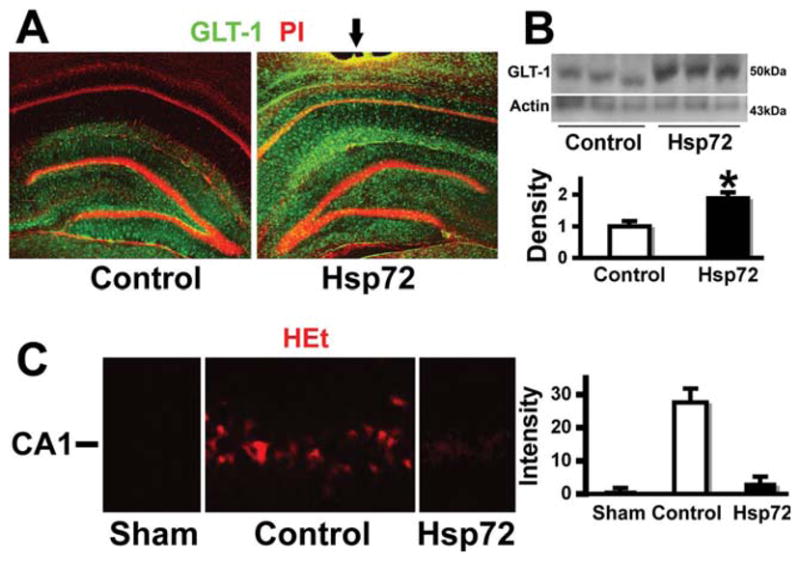
Overexpressing Hsp72 preserves GLT-1 staining and reduces ROS. A: GLT-1 immunoreactivity was significantly greater after GFAPp-Hsp72 injection compared with control (control DNA injected) after 10 min ischemia and 5-h reperfusion. B: Western blot also shows better preservation of GLT-1 protein on the GFAPp-Hsp72 injected side following ischemia compared with control injected (n = 3; P = 0.03). C: HEt shows greater ROS and ethidium fluorescence in control injected hippocampus compared with GFAPp-Hsp72 injected hippocampus or sham (n = 8; *P = 0.001).
Previously we showed that primary astrocytes from the CA1 subregion lost GLT-1 staining and increased ROS levels after 1 h OGD (Ouyang et al., 2007). Here we assessed mitochondrial Complex I and IV activity following 1 hr OGD and 18 h of in vitro reperfusion in isolated mitochondria. The activities of Complexes I and IV were decreased significantly at 18 hr recovery after 1 hr OGD in astrocytes whether transfected with a control DNA plasmid or not transfected (see Fig. 4). Overexpressing Hsp72 prior to OGD markedly reduced this loss of activity for both complexes (Fig. 4A,B).
Fig. 4.
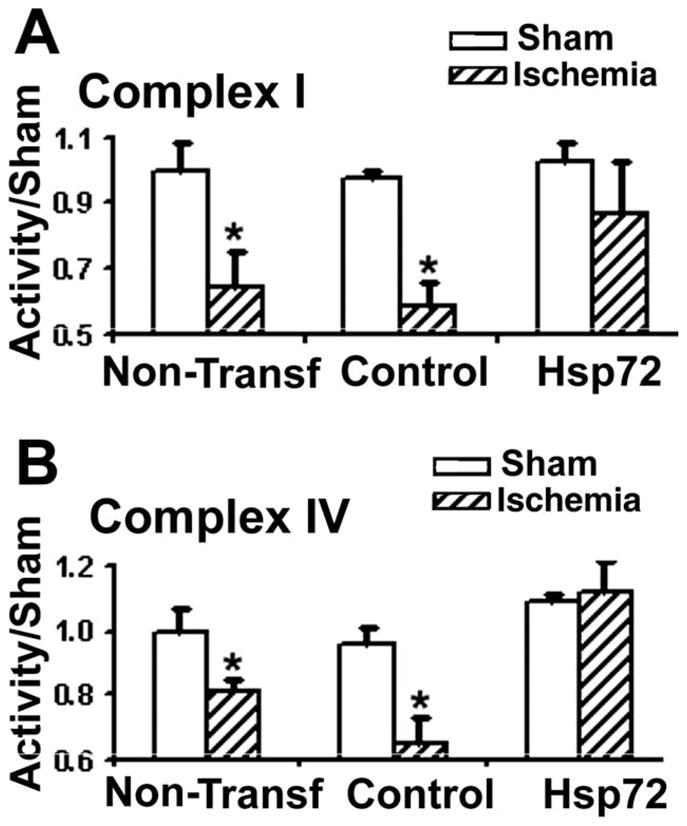
Overexpression of Hsp72 preserves electron transport chain complex activity in astrocytes subjected to 1 h OGD and 18 h reperfusion. A: Complex I activity was reduced in cultures with or without transfection of control plasmid relative to incubation in the presence of oxygen and glucose. Hsp72 overexpression resulted in preservation of complex I activity. B: Complex IV activity was also significantly reduced, and this reduction was prevented with Hsp72 overexpression (n = 3; *P < 0.05).
These results suggest that targeting astrocytes is effective to protect neighboring neurons from forebrain ischemia. However, to strengthen this conclusion we tested a second protective gene, SOD2. While Hsp72 overexpression protects mitochondria, Hsp72 is present in the cytosolic compartment, so the protection is likely indirect. SOD2 was therefore chosen as it is a direct mitochondrial antioxidant, and therefore might be expected to directly protect mitochondria, reduce injury, and protect essential oxidation sensitive astrocyte proteins, if mitochondrial oxidative stress is important.
Astrocyte Selective Overexpression of SOD2
As with GFAPp-Hsp72 we found that the GFAPp-SOD2 construct directed overexpression in the injected hippocampus in astrocytes but not neurons by double immunostaining for SOD2 and GFAP for astrocytes or Map2 for neurons (Fig. 5A,B). Levels of SOD2 were significantly increased by injection of GFAPp-SOD2 into hippocampus when assessed by Western blot and densitometry of injected and control injected hippocampi (Fig. 5C).
Fig. 5.
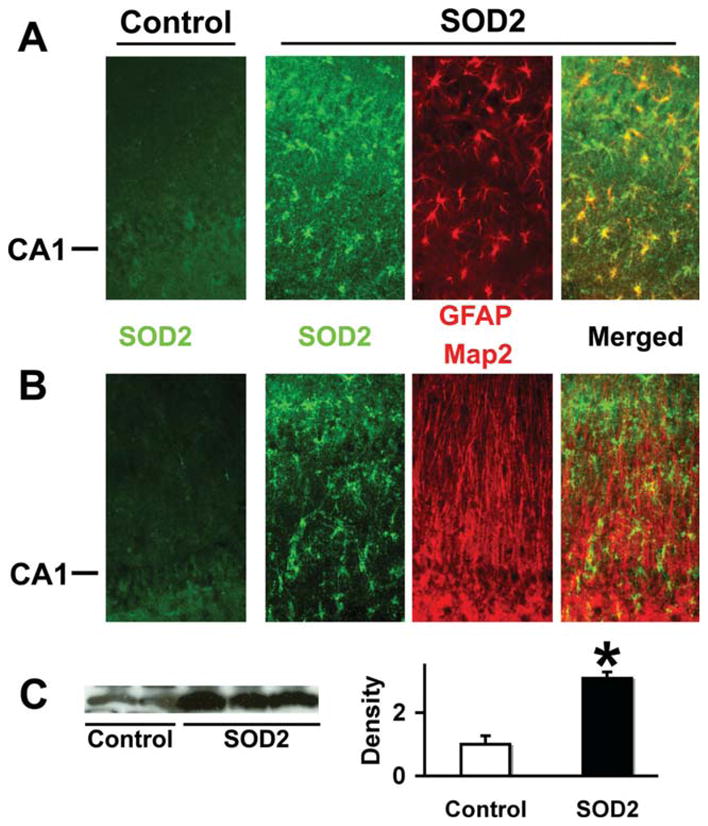
SOD2 overexpression colocalizes with the astrocyte marker GFAP but not Map2. Immunostaining of control DNA injected hippocampi demonstrated low levels of SOD2 staining (left -Control panels in A and B). SOD2 staining was significantly increased in CA1 astrocytes after stereotactic injection of GFAPp-SOD2. Double immunostaining showed SOD2 colocalized with the astrocyte marker GFAP (A) but not with neuron marker Map2 (B). The CA1 neuronal cell body layer is indicated at left. C: Western blot shows increased SOD2 protein levels in injected hippocampi compared with control plasmid injected hippocampi by densitometry (n = 3; P < 0.05).
SOD2 Overexpression in Astrocytes Reduces Neuron Death, ROS production, and GLT-1 Loss
Overexpression of SOD2 in astrocytes by microinjection of GFAPp-SOD2 reduced the extent of CA1 neuronal death caused by forebrain ischemia, compared with control DNA injected hippocampi (Fig. 6A,B) assessed 7 days after forebrain ischemia (P < 0.05).
Fig. 6.
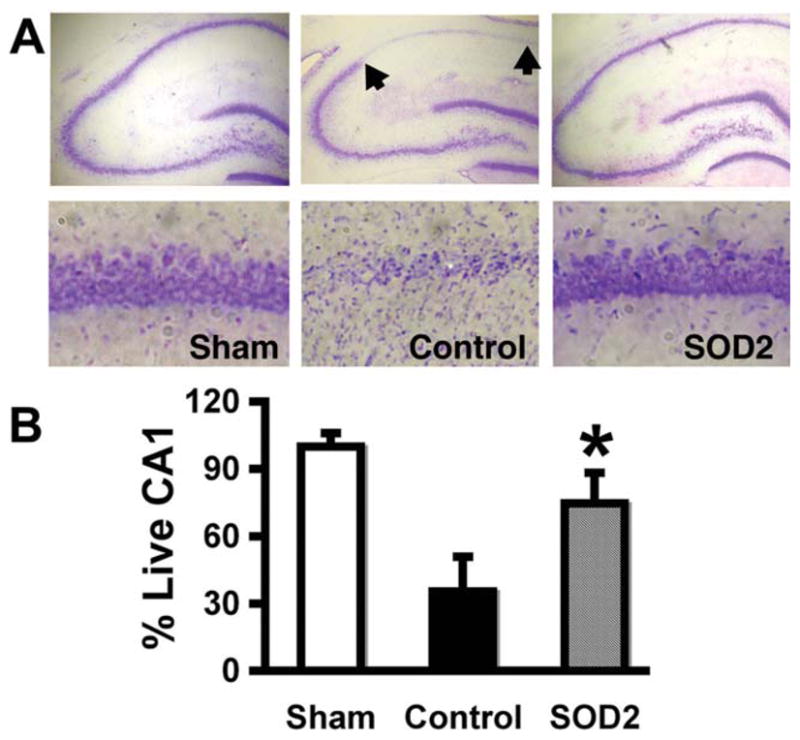
Overexpression of SOD2 in astrocytes reduces vulnerability of CA1 neurons. GFAPp-SOD2 or control DNA was injected unilaterally above the hippocampus two days prior to forebrain ischemia. A: Cresyl violet stained brain sections show that selective CA1 hippocampal neuronal loss is significantly lower in GFAPp-SOD2 injected brain than in control seven days after forebrain ischemia. B: Quantitation of cresyl violet staining showed significant preservation of CA1 neurons (n = 3; P < 0.05).
GLT-1 immunoreactivity was evaluated and immunostaining demonstrated that GLT-1 was relatively well preserved in the CA1 region following forebrain ischemia after injection of GFAPp-SOD2 compared with injection of control DNA at 5-h reperfusion after 10 min transient forebrain ischemia (Fig. 7A lower right compared with lower left). To confirm that ROS production was reduced we assessed oxidative damage by staining for the lipid peroxidation product HNE. As seen in Fig. 7B, the level of immunostaining is reduced when SOD2 is overexpressed, demonstrating effective antioxidant protection.
Fig. 7.

Overexpressing SOD2 in astrocytes preserves GLT-1 and reduces HNE staining in the hippocampal CA1 region. A: GLT-1 immunoreactivity (green) in the hippocampal CA1 region was markedly reduced at 5-h reperfusion after 10 min ischemia, lower left panel, unlike dentate gyrus where staining is preserved. In contrast, GFAPp-SOD2 injected CA1 shows preserved GLT-1 immunoreactivity after forebrain ischemia in control, lower right panel. B: Immunostaining and quantitation of HNE fluorescence reveals marked reduction of oxidative stress in GFAPp-SOD2 injected ischemic animals compared with control DNA injected (n = 3; P = 0.002).
DISCUSSION
We have demonstrated protection of CA1 neurons with a strategy of selectively targeting protective genes to astrocytes. Astrocyte specific overexpression of Hsp72 or SOD2 was achieved using the GFAP promoter. This is the first demonstration of neuroprotection from ischemia in vivo via astrocyte protection, to the best of our knowledge. In both cases protection was accompanied by preservation of the astrocytic glutamate transporter GLT-1, and reduced evidence of oxidative stress in the CA1 region, emphasizing the importance of reducing oxidative stress and preserving GLT-1 for survival of CA1 neurons following forebrain ischemia.
While prior work focused on CA1 neuronal vulnerability to ischemia, attributing selective vulnerability to neuronal connectivity, this has not led to effective protective strategies. Although astrocytes survive more severe ischemia than neurons in some paradigms (Xu et al., 2001), not only astrocyte survival but also the level of astrocyte function is critical to neuronal survival, as demonstrated here. Thus one important component of selective CA1 neuronal vulnerability to forebrain ischemia is CA1 astrocyte vulnerability.
Preservation of astrocytic GLT-1 likely mitigates excitotoxicity following forebrain ischemia, as astrocyte glutamate uptake has long been appreciated to protect neurons (Dugan et al., 1995; Rosenberg and Aizenman, 1989; Swanson et al., 2004). Preservation of astrocyte function(s) is likely the primary mechanism of protection of CA1 neurons when the protective genes are selectively overexpressed in astrocytes, since little astrocyte death occurs in this paradigm (Ouyang et al., 2007). Of the many important astrocyte functions, preservation of GLT-1 appears central. These results support the idea that astrocyte function in the hours following forebrain ischemia is a critical determinant of neuronal survival days later. GLT-1 is susceptible to oxidative damage (Lauderback et al., 2001), and both strategies employed here were associated with GLT-1 preservation.
Numerous observations indicate that glial cells interact with and support neighboring neurons (Kettenmann and Ransom, 2005; Nedergaard et al., 2003; Ransom et al., 2003; Swanson et al., 2004; Takano et al., 2009). Under stress conditions astrocytes may release neuroprotective molecules including metabolic substrates, antioxidants, trophic factors, and possibly heat shock proteins (Nedergaard and Dirnagl, 2005; Swanson et al., 2004; Tytell, 2005). Some neurons are relatively deficient in Hsp72 induction (Hatayama et al., 1997; Mathur et al., 1994; Satoh et al., 1994), and addition of extracellular Hsp70 can enhance neuronal tolerance to heat shock and apoptosis (Guzhova et al., 2001). Lower Hsp72 and reduced induction may be linked to enhanced susceptibility of neurons to pathological conditions including ischemia or excitotoxicity (Planas et al., 1997). In this case, Hsp72 released from CA1 astrocytes may supplement a deficient neuronal response. Further study is needed to determine whether Hsp72 release contributes to protection. The heat shock or stress response is now appreciated to occur not only in a cell autonomous fashion, but also to be regulated by endocrine and neuronal signaling (Prahlad and Morimoto, 2009). Hsp72 may play a role in the interaction between astrocytes and neurons that occurs in response to ischemic stress.
Previous work suggests that astrocyte mitochondria are an important target of ischemic brain injury (Bambrick et al., 2004). The mitochondrial respiratory chain is the major site of ROS production (Lin and Beal, 2006). Overexpression of Hsp72 in astrocytes better sustained function and possibly permitted more rapid return to normal function after ischemia, improving neuronal support. SOD2 directly reduces mitochondrial ROS. This protects mitochondria and likely reduces oxidative stress for the rest of the cell. In contrast Hsp72 is a cytosolic protein with no evidence for translocation into mitochondria. The reduction of oxidative stress and mitochondrial dysfunction with Hsp72 may be both indirect, due to reduced stress in the cytoplasmic compartment resulting in less mitochondrial stress, as well as direct, due to involvement of Hsp70 family members in import of essential, nuclear encoded mitochondrial proteins (Stuart et al., 1996; Young et al., 2003). We previously found that Hsp72 overexpression preserved mitochondrial membrane potential, and moderated the increase in state IV respiration (Ouyang et al., 2006). Here we further find preservation of respiratory complexes I and IV activities with Hsp72 overexpression.
The postischemic mitochondrial redox state is characterized by increased oxidation of respiratory chain components (Perez-Pinzon et al., 1998; Rosenthal et al., 1995). This hyperoxidation may result from loss of electron carriers, such as cytochrome c and NADH, after ischemia (Perez-Pinzon et al., 1999). The loss of cytochrome c from mitochondria might affect respiratory chain activity or trigger apoptosis (Charriaut-Marlangue et al., 1996; Nitatori et al., 1995). SOD2 overexpression likely mitigates hyperoxidation. In septic liver dysfunction, prior Hsp72 induction was associated with preservation of Complex IV and mitochondrial morphology (Chen et al., 2004).
There are GFAP expressing progenitor cells in the sub-ventricular and subgranular zone throughout life. Our GFAP promoter constructs could be expressed if taken up by such precursor cells. Two factors suggest that expression in precursor cells might not contribute markedly to the protection observed here. First we evaluated histological outcome on Day 7, and second the extent of protection is quite large. Prior work showed increased neurogenesis following forebrain ischemia (Sharp et al., 2002), but with little increase on Day 6, and peak proliferation about Day 11. The time needed to differentiate into cells expressing NeuN was about one month after the ischemic event. There is little evidence that the cells born in the subgranular zone repopulate CA1, rather the majority of newborn neurons are found in the granular layer following forebrain ischemia (Sharp et al., 2002; Yamashima et al., 2007). We observed a doubling in the survival of CA1 neurons, comprising nearly 40% of CA1 neurons. It is very unlikely that replacement of CA1 neurons from any source would be of this magnitude over this brief time period. Thus any contribution to protection from precursors is likely to be small.
In conclusion, enhancing astrocyte resistance to ischemic stress by overexpressing either the cytosolic protein Hsp72 or the mitochondrial protein SOD2 resulted in improved survival of CA1 neurons following forebrain ischemia. Preservation of astrocytic GLT-1 appears to play a key role. This suggests that strategies to protect astrocytes may prove effective in reducing neurological injury following cardiac arrest and resuscitation.
Acknowledgments
Grant sponsor: NIH; Grant numbers: NS053898, GM49831.
References
- Bambrick L, Kristian T, Fiskum G. Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem Res. 2004;29:601–608. doi: 10.1023/b:nere.0000014830.06376.e6. [DOI] [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Besnard F, Brenner M, Nakatani Y, Chao R, Purohit HJ, Freese E. Multiple interacting sites regulate astrocyte-specific transcription of the human gene for glial fibrillary acidic protein. J Biol Chem. 1991;266:18877–18883. [PubMed] [Google Scholar]
- Chan PH. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann NY Acad Sci. 2005;1042:203–209. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- Charriaut-Marlangue C, Aggoun-Zouaoui D, Represa A, Ben-Ari Y. Apoptotic features of selective neuronal death in ischemia, epilepsy and gp 120 toxicity. TrendsNeurosci. 1996;19:109–114. doi: 10.1016/s0166-2236(96)80039-7. [DOI] [PubMed] [Google Scholar]
- Chen HW, Kuo HT, Lu TS, Wang SJ, Yang RC. Cytochrome c oxidase as the target of the heat shock protective effect in septic liver. Int J Exp Pathol. 2004;85:249–256. doi: 10.1111/j.0959-9673.2004.00393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Hsu-Chou H, Lu JL, Chiang YC, Huang HM, Wang HL, Wu T, Liao JJ, Yeh TS. Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology. 2005;49:703–714. doi: 10.1016/j.neuropharm.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Bruno VM, Amagasu SM, Giffard RG. Glia modulate the response of murine cortical neurons to excitotoxicity: Glia exacerbate AMPA neurotoxicity. J Neurosci. 1995;15:4545–4555. doi: 10.1523/JNEUROSCI.15-06-04545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giffard RG, Han RQ, Emery JF, Duan M, Pittet JF. Regulation of apoptotic and inflammatory cell signaling in cerebral ischemia: The complex roles of heat shock protein 70. Anesthesiology. 2008;109:339–348. doi: 10.1097/ALN.0b013e31817f4ce0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group THACAS. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Guzhova I, Kislyakova K, Moskaliova O, Fridlanskaya I, Tytell M, Cheetham M, Margulis B. In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res. 2001;914(1–2):66–73. doi: 10.1016/s0006-8993(01)02774-3. [DOI] [PubMed] [Google Scholar]
- Guzhova I, Margulis B. Hsp70 chaperone as a survival factor in cell pathology. Int Rev Cytol. 2006;254:101–149. doi: 10.1016/S0074-7696(06)54003-3. [DOI] [PubMed] [Google Scholar]
- Hatayama T, Takahashi H, Yamagishi N. Reduced induction of HSP70 in PC12 cells during neuronal differentiation. J Biochem. 1997;122:904–910. doi: 10.1093/oxfordjournals.jbchem.a021851. [DOI] [PubMed] [Google Scholar]
- Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. 2009;29:7003–7014. doi: 10.1523/JNEUROSCI.1110-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Ransom B. Neuroglia. New York: Oxford University Press; 2005. [Google Scholar]
- Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: The role of Abeta1–42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH. Evaluating therapeutic targets for reperfusion-related brain hemorrhage. Ann Neurol. 2006;59:929–938. doi: 10.1002/ana.20850. [DOI] [PubMed] [Google Scholar]
- Mathur SK, Sistonen L, Brown IR, Murphy SP, Sarge KD, Morimoto RI. Deficient induction of human hsp70 heat shock gene transcription in Y79 retinoblastoma cells despite activation of heat shock factor 1. Proc Natl Acad Sci USA. 1994;91:8695–8699. doi: 10.1073/pnas.91.18.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. 2005;50:281–286. doi: 10.1002/glia.20205. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. JNeurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Xu LJ, Sun YJ, Giffard RG. Overexpression of inducible heat shock protein 70 and its mutants in astrocytes is associated with maintenance of mitochondrial physiology during glucose deprivation stress. Cell Stress Chaperones. 2006;11:180–186. doi: 10.1379/CSC-182R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Mumford PL, Sick TJ. Prolonged anoxic depolarization exacerbates NADH hyperoxidation and promotes poor electrical recovery after anoxia in hippocampal slices. Brain Res. 1998;786(1–2):165–170. doi: 10.1016/s0006-8993(97)01438-8. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Xu GP, Born J, Lorenzo J, Busto R, Rosenthal M, Sick TJ. Cytochrome C is released from mitochondria into the cytosol after cerebral anoxia or ischemia. J Cereb Blood Flow Metab. 1999;19:39–43. doi: 10.1097/00004647-199901000-00004. [DOI] [PubMed] [Google Scholar]
- Planas AM, Soriano MA, Estrada A, Sanz O, Martin F, Ferrer I. The heat shock stress response after brain lesions: Induction of 72 kDa heat shock protein (cell types involved, axonal transport, transcriptional regulation) and protein synthesis inhibition. Prog Neurobiol. 1997;51:607–636. doi: 10.1016/s0301-0082(97)00004-x. [DOI] [PubMed] [Google Scholar]
- Prahlad V, Morimoto RI. Integrating the stress response: Lessons for neurodegenerative diseases from C. elegans. Trends Cell Biol. 2009;19:52–61. doi: 10.1016/j.tcb.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran R, Lu A, Zhang L, Tang Y, Zhu H, Xu H, Feng Y, Han C, Zhou G, Rigby AC, Sharp FR. Hsp70 promotes TNF-mediated apoptosis by binding IKK gamma and impairing NF-kappa B survival signaling. Genes Dev. 2004;18:1466–1481. doi: 10.1101/gad.1188204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom B, Behar T, Nedergaard M. New roles for astrocytes (stars at last) Trends Neurosci. 2003;26:520–522. doi: 10.1016/j.tins.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex [published erratum appears in Neurosci Lett 1990 Aug 24;116(3):399] Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- Rosenthal M, Feng ZC, Raffin CN, Harrison M, Sick TJ. Mitochondrial hyperoxidation signals residual intracellular dysfunction after global ischemia in rat neocortex. J Cereb Blood Flow Metab. 1995;15:655–665. doi: 10.1038/jcbfm.1995.81. [DOI] [PubMed] [Google Scholar]
- Satoh J, Tabira T, Yamamura T, Kim SU. HSP72 induction by heat stress is not universal in mammalian neural cell lines. J Neurosci Res. 1994;37:44–53. doi: 10.1002/jnr.490370107. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Liu J, Bernabeu R. Neurogenesis following brain ischemia. Brain Res Dev Brain Res. 2002;134(1–2):23–30. doi: 10.1016/s0165-3806(01)00286-3. [DOI] [PubMed] [Google Scholar]
- Stuart RA, Ono H, Langer T, Neupert W. Mechanisms of protein import into mitochondria. Cell Struct Funct. 1996;21:403–406. doi: 10.1247/csf.21.403. [DOI] [PubMed] [Google Scholar]
- Sun Y, Ouyang YB, Xu L, Chow AM, Anderson R, Hecker JG, Giffard RG. The carboxyl-terminal domain of inducible Hsp70 protects from ischemic injury in vivo and in vitro. J Cereb Blood Flow Metab. 2006;26:937–950. doi: 10.1038/sj.jcbfm.9600246. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- Takano T, Oberheim N, Cotrina ML, Nedergaard M. Astrocytes and ischemic injury. Stroke. 2009;40(3 Suppl):S8–S12. doi: 10.1161/STROKEAHA.108.533166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytell M. Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int J Hyperthermia. 2005;21:445–455. doi: 10.1080/02656730500041921. [DOI] [PubMed] [Google Scholar]
- Voloboueva LA, Suh SW, Swanson RA, Giffard RG. Inhibition of mitochondrial function in astrocytes: Implications for neuroprotection. J Neurochem. 2007;102:1383–1394. doi: 10.1111/j.1471-4159.2007.4634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ma JH, Giffard RG. Overexpression of copper/zinc super-oxide dismutase decreases ischemia-like astrocyte injury. Free Radic Biol Med. 2005;38:1112–1118. doi: 10.1016/j.freeradbiomed.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Xu L, Sapolsky RM, Giffard RG. Differential sensitivity of murine astrocytes and neurons from different brain regions to injury. Exp Neurol. 2001;169:416–424. doi: 10.1006/exnr.2001.7678. [DOI] [PubMed] [Google Scholar]
- Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab. 2009;29:365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashima T, Tonchev AB, Borlongan CV. Differential response to ischemia in adjacent hippocampalsectors: Neuronal death in CA1 versus neurogenesis in dentate gyrus. Biotechnol J. 2007;2:596–607. doi: 10.1002/biot.200600219. [DOI] [PubMed] [Google Scholar]
- Yeh TH, Hwang HM, Chen JJ, Wu T, Li AH, Wang HL. Glutamate transporter function of rat hippocampal astrocytes is impaired following the global ischemia. Neurobiol Dis. 2005;18:476–483. doi: 10.1016/j.nbd.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Yenari MA, Giffard RG, Sapolsky RM, Steinberg GK. The neuroprotective potential of heat shock protein 70 (HSP70) Mol Med Today. 1999;5:525–531. doi: 10.1016/s1357-4310(99)01599-3. [DOI] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- Zhang W, Potrovita I, Tarabin V, Herrmann O, Beer V, Weih F, Schneider A, Schwaninger M. Neuronal activation of NF-kb contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:30–40. doi: 10.1038/sj.jcbfm.9600004. [DOI] [PubMed] [Google Scholar]