

Abstract
Myocarditis, often initiated by viral infection, may progress to autoimmune inflammatory heart disease, dilated cardiomyopathy and heart failure. Although cardiac myosin is a dominant autoantigen in animal models of myocarditis and is released from the heart during viral myocarditis, the characterization, role and significance of anti-cardiac myosin autoantibodies is poorly defined. In our study, we define the human cardiac myosin epitopes in human myocarditis and cardiomyopathies and establish a mechanism to explain how anti-cardiac myosin autoantibodies may contribute to heart disease. We show that autoantibodies to cardiac myosin in sera from myocarditis and dilated cardiomyopathies in humans targeted primarily epitopes in the S2 hinge region of cardiac myosin. In addition, anti-cardiac myosin antibodies in sera or purified IgG from myocarditis and cardiomyopathy targeted the beta-adrenergic receptor and induced antibody-mediated cAMP-dependent protein kinase A (PKA) cell signaling activity in heart cells. Antibody-mediated PKA activity in sera was abrogated by absorption with anti-human IgG. Antibody-mediated cell signaling of PKA was blocked by antigen-specific inhibition by human cardiac myosin or the beta-adrenergic receptor but not the alpha adrenergic receptor or bovine serum albumin. Propranolol, a beta blocker and inhibitor of the beta-adrenergic receptor pathway also blocked the antibody-mediated signaling of the beta-adrenergic receptor and PKA. The data suggest that IgG antibody against human cardiac myosin reacts with the beta-adrenergic receptor and triggers PKA signaling in heart cells. In-summary, we have identified a new class of crossreactive autoantibodies against human cardiac myosin and the beta-adrenergic receptor in the heart. In addition, we have defined disease specific peptide epitopes in the human cardiac myosin rod S2 region in human myocarditis and cardiomyopathy as well as a mechanistic role of autoantibody in the pathogenesis of disease.
Keywords: Myocarditis, cardiomyopathy, autoimmunity, myosin, beta-adrenergic receptor
Introduction
Myocarditis is an acute inflammatory disease of the heart and a precursor of dilated cardiomyopathy [1–8]. Dilated cardiomyopathy is a more chronic disease which is characterized by ventricular hypertrophy and which may be a direct result of myocarditis and lead to heart failure [1–3,9]. Myocarditis is often characterized by a cellular infiltrate, and if inflammation of the myocardium does not resolve during the acute stage, the heart may be compromised due to loss of myocytes [10], scarring from granulomatous inflammation [11,12], or fibrosis due to proliferation of fibroblasts and collagen deposition [13,14], Although TH1 or TH2 cell mediated immunity is blamed for disease, myocarditis has been reported to develop independently of TH1 and TH2 mechanisms involving local production of IL-17 and infiltration of neutrophils in acute inflammation [15]. In animal models of myocarditis, antibody deposition and antibody-mediated cell signaling of the beta-adrenergic receptor or the calcium channel may affect cardiomyocyte function and lead to cardiomyopathy or apoptosis in the myocardium [16–18]. Whichever type of disease occurs, the myocardium may become enlarged and functionally weakened due to the immune attack on the heart. In humans, little is known about the immune pathogenesis of antibody in heart disease.
Cardiac myosin has emerged as a dominant autoantigen in myocarditis in genetically susceptible animals [19–22], Transfer of anti-cardiac myosin monoclonal antibodies led to myocarditis in certain strains of mice [23–25], and the induction of circulating anti-cardiac myosin autoantibody and IgG deposition in myocardium was shown to be concomitant with the onset of myocarditis [25,26]. In the Lewis rat model of myocarditis, deposition of anti-cardiac myosin antibody occurred in damaged tissue where cardiac myosin was exposed, but anti-cardiac myosin antibodies recognized molecules expressed on the surface of cardiomyocytes and directly affected the heart [16]. In our recent study of the potential role and mechanism of autoantibodies against cardiac myosin in the heart, we demonstrated that autoantibody against cardiac myosin or its pathogenic peptide S2-16 directly targeted cardiac myocytes. Monoclonal anti-cardiac myosin antibodies from the Lewis rat model of myocarditis reacted with the cell surface of cardio-myocytes and crossreacted with the beta-adrenergic receptor [16]. The reaction with the beta-adrenergic receptor induced cell signaling and protein kinase A (PKA) induction which eventually may lead to apoptosis of heart cells [16].
In human myocarditis, autoantibodies against cardiac myosin are elevated and associated with deterioration of cardiac function in patients with chronic myocarditis and cardiomyopathy [27–30]. Although anti-cardiac myosin antibodies are present in patients with cardiomyopathy and experimental animals with myocarditis, the mechanism of pathogenesis and significance of the anti-cardiac myosin autoantibodies is unknown. Immunoadsorption of circulating autoantibodies has been shown to improve cardiac function of patients with dilated cardiomyopathy [31,32] and certain subgroups of patients with dilated cardiomyopathy may benefit from immuno-suppressive treatment suggesting that antibodies may contribute to disease pathogenesis [32]. Furthermore, in human myocarditis, autoantibodies have been observed in myocardial biopsies [4,5] as well as a cellular infiltrate [33]. Our hypothesis is that anti-cardiac myosin antibodies may play a role in myocarditis and become pathogenic because they crossreact with epitopes present on the surface of heart cells where extracellular antigens trap antibodies and complement in tissues and target inflammation to the heart [34–37].
Evidence in our report demonstrates that anti-cardiac myosin IgG autoantibodies in sera from myocarditis and dilated cardiomyopathies recognize peptides within the S2 fragment of the human cardiac myosin rod and stimulate PKA activity in cardiomyocytes through the beta-adrenergic receptor. Autoantibodies against cardiac myosin and the beta-adrenergic receptor comprise a novel class of autoantibodies against the heart and explain how anti-cardiac myosin antibodies may lead to heart disease. Chronic over stimulation of the beta-adrenergic receptor is a potential pathogenic mechanism of crossreactive autoantibodies which may lead to dilated cardiomyopathy.
Research design and methods
Human sera
Sera were obtained from 14 myocarditis patients, 8 giant cell myocarditis patients, 13 dilated cardiomyopathy patients, 26 cardiomyopathy patients with diabetes, 103 diabetic patients (46 type 1, 57 type 2) without heart disease, and 27 control donors with no evidence of heart disease. All myocarditis and cardiomyopathy patients enrolled in the study presented with a depressed abnormal ejection fraction (18–58%) and no diagnosis of coronary artery disease or by Q waves and EKG. Normal subjects were defined as individuals without any symptom of heart disease, cardiomyopathy, autoimmune, metabolic or infectious diseases. Participants gave written informed consent, and the research protocols were approved by the University of Oklahoma Health Sciences Center, Mayo Clinic, VA Medical Center at Oklahoma City and the University of Colorado Health Sciences Center Institutional Review Boards.
Antigens
Human cardiac myosin was purified from human heart tissue according to Tobacman and Adelstein [38] with slight modifications as described previously [39]. Beta-adrenergic receptor 1 and 2 were purchased from Perkin Elmer Company (Waltham, MA, USA) alpha-2 adrenergic receptor (Rα2aAR) was purchased from Sigma Chemical Company (St Louis, MO, USA) and bovine serum albumin (BSA) was purchased from Fisher Scientific, Hanover park, IL, USA. Peptides of the S2 fragment of the human cardiac myosin rod were synthesized and purified by Genemed Synthesis, Inc. (San Francisco, CA, USA) as 25-mers with 11-amino acid overlap. Peptides of the light meromyosin (LMM)-fragment of human cardiac myosin beta-chain were synthesized as 18-mers with 5-amino acid overlap and were purified by high pressure liquid chromatography by the Molecular Biology Resource Facility at the University of Oklahoma Health Sciences Center (Oklahoma City, OK, USA). The complete amino acid sequence of human cardiac myosin was published by Vosberg et al. [40]. The amino acid sequences of the synthetic peptides comprising the human cardiac myosin S2 fragment are as follows and as described previously [16]. S2-1 SAEREKEMASMKEEFTRLK EALEKS, S2-2 FTRLKEALEKSEARRKELEEKMVSL, S2-3 RKELEEKMVSLLQEK NDLQLQVQAE, S2-4 KNDLQLQVQAEQDNLADAEERCDQL, S2-5 LADAEER CDQLIKNKIQLEAKVKEM, S2-6 KIQLEAKVKEMNERLEDEEEMNAEL, S2-7 LEDEEEMN AELTAKKRKLEDECSEL, 2-8 KRKLEDECSELKRDIDDLELTLAKV, S2-9 IDDL ELTLAKVE KEKHATENKVKNL, S2-10 KHATENKVKNLTEEMAG LDEIIAKL, S2-11 MAGLDE IIAKLTKEKKALQEAHQQA, S2-12 KKALQ EAHQQ ALDDLQAEEDKVNTL, S2-13 LQAEEDKVNTLTKAKVKLEQ QVDDL, S2-14 KVK LEQQ VDDLEGSLEQEKKVRMDL, S2-15 LEQEKKVRMDLER AKRKLEGDL KLT, S2-16 KRKLEGDLKLTQESIMDLENDKQQL, S2-17 IMDLENDKQ QLDERLKKKDFELNAL, S2-18 LKKKDFELNALNARIEDEQALGSQL, S2-19 IEDEQALGSQLQ KKLKELQARIEEL, S2-20 LKELQARIEELEEELESERTARAKV, S2-21 LESERTAR AKVEKLRSDLSR ELEEI, S2-22 RSDLSRELEEISERLEEAGGATSVQ, S2-23 LEEA GGATSVQIE MNKKREAEFQKM, S2-24 NKKREAEFQKMRR DLEEATLQ HEAT, S2-25 LEEATLQHEATAAALRKKHADSVAE, S2-26 LRKKHADSVAEL GEQIDNL QRVKQK, S2-27 QIDNLQRVKQKLEKEKSEFKLELDD, S2-28 EKSEFKLELD DVTSNMEQIIKAKAN, S2-29 NMEQIIKAKANLEKMCRTLEDQMNE, S2-30 MCRTLED QMN EHRSKAEETQRSVND, S2-31 KAEETQRSVNDLTSQRAKLQTENGE, S2-32 ETQR SVNDLTSQRAKLQTENGELSR. The amino acid sequence of the synthetic peptides comprising the human cardiac myosin LMM-fragment are as follows and as described previously [41]. LMM-1 KEALISSLTRGKLTYTQQ, LMM-2 TYTQQLED LKRQLEEEVK, LMM-3 EEEVKAKNALAHALQSAR, LMM-4 LQSARHDCDLLR EQYEEE, LMM-5 EQYEEETEAKAELQRVLSK, LMM-6 RVLSKANSEVAQWRT KYE, LMM-7 RTKYETDAIQRTEELEEA, LMM-8 ELEEAK KKLAQRLQEAEE, LMM-9 QEAEEAVEAVNAKCSSLE, LMM-10 CSSLEKT KH RLQNEIEDL, LMM-11 EIEDLMVDVERSNAAAAA, LMM-12 AAAAALDKKQRN FDKILA, LMM-13 DKILAEWKQKYEESQSEL, LMM-14 SQSELESSQKEARS LS TE, LMM-15 SLSTELFKLKNAYEESLE, LMM-16 EESLEHLETFKRENKNLQ, LMM-17 NKNLQEEISDLTEQLGSS, LMM-18 EQLGSSGKTIHELEKVRKQ, LMM-19 KVRKQLEAEKMELQSALE, LMM20 LQSALEEAEASLEHEEGKI, LMM-21 EEGKILRAQLEFNQIKAE, LMM-22 NQIKAEIERKLAEKDEEME, LMM-23 DEEMEQAKRNHLRWDSL, LMM-24 WDSLQTSLDAETRSRNE, LMM-25 RSRNEALRVKKKMEGDLN, LMM-2 6 EGDLNEMEIQLSHANRMA, LMM-27 ANRMAAEAQKQVKSLQSL, LMM-28 SLQSLLKDTQIQLDDAVR, LMM-28B DDAVRA-NDDLKENIAIVE, LMM-29 RANDDLKENIAIVERRNN, LMM-3 0 IAIVERRNNLLQAELEEL, LMM-31 ELEELRAVVEQTERSRKL, LMM-32 RSRKLAEQELIETSERVQ, LMM-33 SERVQLLHSQNTSLINQK, LMM-34 LINQKKKMDADLSQLQTE, LMM-35 TEVEEAVQESRNAEEKAKK, LMM-3 6 RNAEEKAKKAITDAAMMA, LMM-37 AAMMAEELKKEQDTSAHL, LMM-38 TSAHLERMKKNMEQTIKDL, LMM-39 TIKDLQHRLDEAEQIALK, LMM-40 EQIALKGGKKQLQKLEARV, LMM-41 LEARVRELENELEAEQKR, LMM-42 AEQKRNAESVKGMRKSER, LMM-43 RKSERRIKELTYQTEEDR, LMM-44 TEEDRKNLLRLQDLVDKL, LMM-45 LVDKLQLKVKAYKRQAEE, LMM-4 6 RQAEEAEEQANTNLSKFR, LMM-47 LSKFRKVQHELDEAEERA, LMM-48 AEERADIAESQVNKLRAK, LMM-49 KLRAKSRDIGTKGLNEE.
Enzyme-linked-immunosorbent assay
Immulon 4 (Dynatech) microtiter plates were coated at 4°C overnight with either human cardiac myosin beta-chain, LMM peptides or S2 peptides at 10 μg/ml in 0.1 M carbonate–bicarbonate coating buffer (pH 9.6). Plates were washed with PBS containing 0.05% Tween 20, blocked with 1% BSA in PBS buffer for 1 h at 37°C, and washed with PBS/Tween 20. For the peptide enzyme-linked-immunosorbent assay (ELISA), 50 μl serum (1/100 serum dilution in 1% BSA in PBS buffer) was added to microtiter wells and incubated overnight at 4°C. Plates were washed with PBS/Tween 20, and 50 μl goat anti-human IgG (u-chain specific, Sigma, St Louis, MO, USA) conjugated with alkaline phosphatase (1/250 dilution) was added and incubated at 37°C for 1 h. Plates were washed with PBS/Tween 20, and 50 μl of substrate para-nitrophenyl-phosphate 104 (Sigma) in 0.1 M diethanolamine buffer (pH 9.8) was added. Optical density (OD) was measured at 405 nm in an ELISA plate reader (Dynex, Chantilly, VA, USA). For the peptide ELISA, the ELISA was read when a well characterized sera with high reactivity to a known peptide was positive and reached the same OD (1.0) in every assay performed. A positive OD of 1.0 at 405 nm in the ELISA as described above was used as the standard. Tests were performed separately three times and each of these tests had triplicate samples. The background OD was determined for each serum and subtracted from total serum reactivity for each individual peptide. OD values used for bar graphs were averages obtained from three separate assays. We collected our data from assays performed on different days using the same sera and found no significant difference in reactivity with the peptides. The peptide ELISA was highly reproducible from day to day with a positive and negative peptide control and the average mean difference between the tests performed in different days was ± 0.053 OD. Controls used included serum alone, secondary antibody conjugate alone, and 1% BSA alone. Standardized positive and negative control sera were used in each assay. Positive control sera were sera from patients with inflammatory heart disease and strong antibody responses against cardiac myosin (>6400) and its peptides. Negative control sera was obtained from healthy subjects whose myosin antibody titer range was < 100–200.
To determine the human cardiac myosin ELISA antibody titer, sera were diluted 1/100 in 1% BSA in PBS buffer and thereafter diluted two-fold, and the ELISA was performed as described above. For the ELISA titer, goat anti-human IgG (gamma-chain specific, Cappel, Aurora, OH, USA) conjugated with alkaline phosphatase (1/1000 dilution in 1% BSA in PBS buffer) was used. For the human cardiac myosin ELISA antibody titers, tests were performed separately three times and in each assay the titration was done in duplicate. Titers were determined as the highest dilution with OD value of 0.10 at approximately 60 min. Anti-human cardiac myosin antibody titers in the ELISA were controlled with negative and positive control sera. Positive and negative control sera were as described above. Anti-human cardiac myosin antibody titers were highly reproducible (+0.05). A similar ELISA was performed when using the beta-adrenergic receptor as antigen.
Competitive inhibition ELISA
Competitive inhibition ELISA was performed in triplicate as described [42]. Inhibitors were prepared in PBS and mixed with an equal volume of antibody and incubated at 37°C for 1 h and overnight at 4°C. Fifty microlitre of serum antibody–antigen mixture was added to wells coated with human cardiac myosin or the beta-adrenergic receptor (10 μg/ml). The remainder of the assay was performed as described above. Percent inhibition was calculated as follows: 100 × (1 − [A410 inhibitor + antibody/A410 PBS + antibody]). Maximal (100%) reactivity was determined by incubating antibody with PBS without inhibitor.
Western immunoblots
Human cardiac myosin was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as described by Laemmli [43]. After proteins were separated by SDS-PAGE using the SE 280 11-cm vertical slab gel unit (Hoeffer Scientific Instruments, San Francisco, CA, USA) the gel was removed and overlayed with a sheet of Immobilon-P (Millipore, Bedford, MA, USA), and transferred overnight at 50 mA current in the transfer unit TE (Hoeffer Scientific Instruments). Efficiency of the protein transfer was determined by staining a portion of the blot with amido black. Blot strips were blocked with PBS containing 3% non-fat milk for 1 h at 37°, washed five times with 0.05% Tween-20 in PBS, incubated overnight at 37° with 1:100 dilution of sera from patients in each of the test groups. After washing, a horseradish peroxidase-labeled goat anti-human IgG (gamma-chain specific, Sigma) was incubated with the blots. Blots were then developed with H2O2 substrate and 4-chloro-l-naphthol as previously described [39]. Positive and negative control sera were used for standardization in each blot assay. Values were reported as 0 = no reactivity, 1 + = detectable reactivity, 2 + = moderate reactivity, 3 + = strong reactivity and 4 + = very strong reactivity.
Purification of IgG from human sera
The IgG fraction from the patient serum was purified using the Melon gel IgG purification kit (Pierce, Rockford, IL, USA) according to the manufacturer's instructions. Serum was diluted 1:10 with Melon gel purification buffer and loaded onto a gravity-flow column. The IgG was eluted with the purification buffer, and absorbance of the fractions was measured at 280 nm.
Protein kinase assays
As previously described [16], 1 × 107 H9c2 primary rat heart cells (ATCC) were plated in T75 cell culture flasks over night under standard cell culture conditions. Sera (1:100 dilution) or purified IgG from positive and negative sera were incubated with cells in a final volume of 15 ml of medium for 1 h before reactions were stopped by ice-cold PBS rinsing. The cells were mechanically dislodged from the flasks, centrifuged, and solublized in 0.35 ml protein extraction buffer before homogenization. PKA activity of H9c2 cells was measured using SignaTECT cAMP-dependant protein kinase assay system (Promega Corporation, Madison, WI, USA) according to the manufacturer's instruction. The specific activity of the enzyme in pmol per min per ng for each sample was calculated and results were presented as percentage above the basal PKA rate. For inhibition of antibody-induced PKA activity, beta-blocker propranolol (10 μM), betal-blocker CGP 20712A (3 μM) and beta 2-blocker ICI 188.551 (2 μM) were added into reaction mixtures. For competitive inhibition of antibody reactivity, 500 μg/ml of inhibitor human cardiac myosin or BSA (negative control antigen) was mixed with the serum dilution and incubated as described in competitive inhibition ELISA above. The antibodies and inhibitor mixtures were added into reaction with the H9c2 cells for the PKA assay performed as previously described above.
Statistics
Reactivity of serum groups in the ELISA were compared by the non parametric Mann-Whitney U-test, the Student unpaired two-tailed t-test or the one way ANOVA to determine significance (p values <0.05) between the groups.
Results
Autoantibodies against human cardiac myosin
Although evidence has shown that cardiac myosin plays a role in myocarditis in animal models, there have been fewer studies of human disease. Here, we confirm the presence of autoantibodies against human cardiac myosin in myocarditis and dilated cardiomyopathies in Figures 1 and 2. Figure 1 illustrates that the most significant anti-human cardiac myosin titers (IgG) were observed in myocarditis (p ≤ 0.0001), and titers decreased as the disease progressed to cardio-myopathy. Sera titers against human cardiac myosin ranged from 200 to 51,200 in myocarditis and from 100 to 25,600 in cardiomyopathies. Normal healthy control serum titers ranged from < 100 to 800 with the mean titer below 400.
Figure 1.

Anti-human cardiac myosin titers (IgG) in sera from myocarditis (p ≤ 0.0001) were significantly higher compared with cardiomyopathy and normal control sera in the ELISA.
Figure 2.
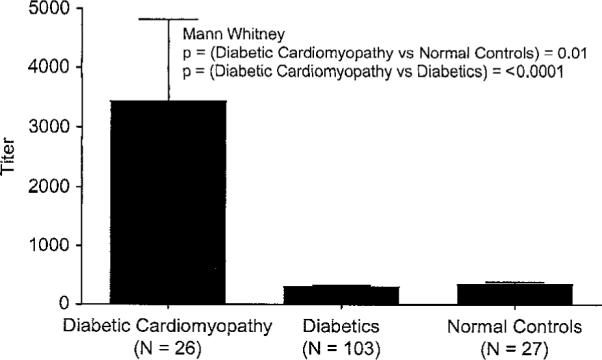
Anti-human cardiac myosin titers (IgG) of diabetic cardiomyopathy sera compared with diabetic sera and normal control donors in the ELISA. Diabetic cardiomyopathy sera titers were significantly higher than in diabetic (p ≤ 0.0001) and control (p = 0.01) sera groups.
Although the mechanisms of cardiomyopathy are different in the setting of diabetes, our data indicates that immune responses against cardiac myosin were present in diabetic cardiomyopathy as well as in myocarditis. Figure 2 illustrates the anti-human cardiac myosin titers (IgG) of the diabetic cardiomyopathy group compared with uncomplicated diabetics and control donors. In cardiomyopathy with diabetes, the mean serum titer was approximately 3500 while diabetic and normal healthy control serum titer means were < 400. The reactivity of the diabetic cardiomyopathy group of sera was significantly different from both diabetic sera (p ≤ 0.0001) and normal healthy control subjects (p = 0.01). IgM against cardiac myosin was found to be much lower than IgG against cardiac myosin in myocarditis and cardiomyopathy sera. We studied many of our sera in the Western immunoblots and found that the cardiomyopathy serum IgG did often react with the 200 kDa band of purified human cardiac myosin (data not shown), even in the absence of ELISA detectable antibody, while 20 normal sera did not react on the blot or ELISA as has been previously described [41]. Therefore, IgG autoantibodies against human cardiac myosin heavy chain were strongly associated with myocarditis and decreased with disease progression to cardiomyopathy as previously reported by Caforio [1,44–49]. The increased reactivity of the myocarditis and cardiomyopathy groups with human cardiac myosin in either the ELISA or the Western immunoblot indicated the appearance of autoantibodies which recognized both conformational or denatured epitopes on the cardiac myosin heavy chain.
Localization of epitopes in human cardiac myosin
Although antibodies in myocarditis recognized the intact cardiac myosin molecule, we would expect that epitopes within the molecule would be recognized by antibodies as the molecule was broken down and presented to the immune system. We found that in myocarditis and cardiomyopathies, cardiac myosin epitopes were targeted by IgG antibodies.
To study human cardiac myosin epitopes, we generated a group of synthetic peptides spanning the rod region including the S2 hinge region and the LMM regions of the rod. Peptides were reacted in an ELISA with sera (1:100 dilution) from myocarditis and cardiomyopathy to determine the regions associated with human disease. Human cardiac myosin peptide epitopes recognized by IgG in myocarditis and cardiomyopathy were significantly different from normal reactivity and are illustrated in bar graphs shown in Figure 3(A), (B). In Figure 3(A), human cardiac myosin peptides significantly recognized in myocarditis were S2-1, S2-9, S2-10, S2-17, S2-26, S2-28 and S2-30; in dilated cardiomyopathy, significant peptide epitopes were S2-1, S2-6, S2-9, S2-10. S2-12, S2-17, S2-19, S2-26, S2-28 and S2-30; in giant cell myocarditis, significant peptide epitopes were S2-1, S2-10, S2-17, S2-19 and S2-26. Baseline reactivity of normal sera illustrates the presence of low levels of autoantibody in normal healthy subjects. Human LMM peptides were not significantly recognized. Although the sera from giant cell myocarditis and dilated cardiomyopathy did not appear to have high levels of anti-cardiac myosin antibodies by ELJSA, they did recognize human cardiac myosin peptides and were often positive by immunoblots (not shown).
Figure 3.


(A) Comparison of sera from myocarditis, dilated cardiomyopathy, giant cell myocarditis and normal control patients in their reactivity with 82 synthetic peptides spanning the S2 (25-mers) hinge region and LMM (18-mers) tail region of the human cardiac myosin rod in the ELISA (sera @ 1/100 dilution). Significant reactivity (OD) compared to normal controls for the peptides is indicated with p values and grey bars. (B) Comparison diabetic, diabetic cardiomyopathy, and normal sera reactivity with S2 and LMM synthetic peptides. Sera from the control donors rarely reacted with S2 peptides at an OD >0.2. Significant reactivity compared to normal controls is indicated by p values and grey bars.
In Figure 3(B), human cardiac myosin peptides significantly recognized in diabetic cardiomyopathy were S2-4, S2-6, S2-8, S2-10 and S2-28 as well as LMM 7, LMM 20, LMM 45, LMM 46 and LMM 47 when compared to sera from diabetic and normal healthy subjects. Although sera from uncomplicated diabetics was not completely unormal, the data overall suggested that there was increased significant recognition of a specific group of human cardiac myosin epitopes in diabetic cardiomyopathy which was clearly different from diabetes without cardiomyopathy. The data suggest that in diabetics the cardiac myosin molecule is presented to the immune system, and increased recognition of cardiac myosin peptides occurs in diabetic cardiomyopathy. Taken together, the S2 hinge region of cardiac myosin was the target of autoimmune responses against the heart in myocarditis and cardiomyopathies. In diabetic cardiomyopathy, both the S2 hinge and LMM regions of the human cardiac myosin rod were recognized. The total number of human cardiac myosin peptides or epitopes recognized by sera from cardiomyopathies were increased in dilated cardiomyopathy ≥ diabetic cardiomyopathy > myocarditis > giant cell myocarditis > diabetic > normal healthy subjects.
Crossreactive autoantibodies against cardiac myosin and beta-adrenergic receptors in human cardiomyopathies
Our recent finding that anti-cardiac myosin antibody in the rat model of myocarditis crossreacted with the beta-adrenergic receptor [16] prompted us to look at human myocarditis and cardiomyopathies for antibodies against the beta-adrenergic receptor and determine if it was crossreactive with human cardiac myosin. To study potential crossreactivity of antibodies with cardiac myosin and the beta-adrenergic receptor in humans, purified IgG from a selected patient sera was inhibited from binding to human cardiac myosin by the beta-adrenergic receptor and vice versa. Dose response inhibition curves (500-1 μg/ml inhibitor) shown in Figure 4 indicates that the reactivity of purified IgG with the beta receptor or human cardiac myosin could be blocked with human cardiac myosin or the beta-adrenergic receptor. The reaction of the sera was markedly inhibited when the beta-adrenergic receptor or cardiac myosin was used as the inhibitor, whereas BSA or the Rα2aAR were not inhibitory to the serum or IgG ELISA reactivity. These data further suggest that in human sera, populations of antibodies exist which are crossreactive with human cardiac myosin and the beta-adrenergic receptors. Rat [16] and human mAbs (data not shown) support the hypothesis that crossreactive antibodies against cardiac myosin and the beta-adrenergic receptor are present in disease.
Figure 4.
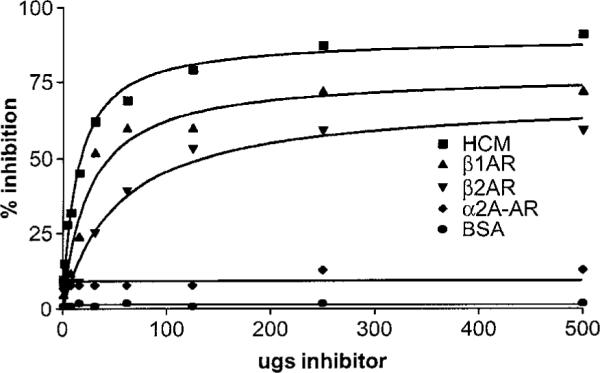
Dose response curves demonstrate antigen-specific inhibition of IgG reactivity with the beta-1 adrenergic receptor by human cardiac myosin and B1 or B2 adrenergic receptors. Reactivity of purified IgG from cardiomyopathy sera is inhibited by human cardiac myosin, and B1 or B2 adrenergic receptors but not by the alpha 2 adrenergic receptor or BSA.
Antibody-mediated PKA activation by sera and purified IgG from myocarditis and cardiomyopathies is blocked by cardiac myosin and beta receptors
To investigate the potential role of autoantibodies in myocarditis and cardiomyopathy, we tested the sera for antibody-induced PKA heart cell signaling in a rat heart cell line H9c2 in vitro. In the heart, cyclic AMP-dependent PKA signaling activated by stimulation of the beta-adrenergic receptor is a mechanism to enhance myocardial performance in response to stress and exercise. To determine if PKA signaling was activated by autoantibodies present in the sera of patients with myocarditis and cardiomyopathy, heart cells in culture were treated with the sera (1:100 dilution) or purified IgG (100μg/ml). Figure 5 illustrates the levels of PKA signaling in the heart cells. Myocarditis sera signaled PKA at a higher rate (range 45–125% above basal level) than the cardiomyopathy sera (range 30–80% above basal level) Normal basal levels were at the range of 0–25% above the basal PKA level in the heart cells.
Figure 5.
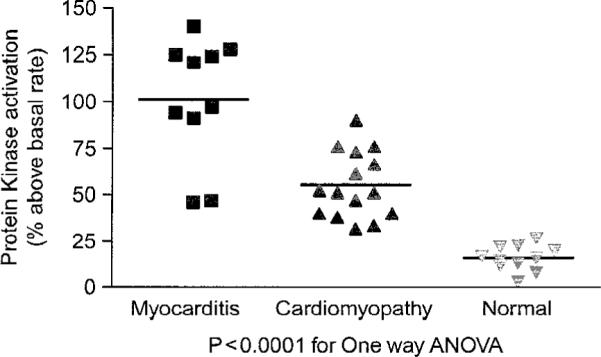
Antibody-mediated cell signaling by sera from myocarditis and cardiomyopathy. H9c2 heart cells in culture were treated with sera (1:100 dilution) or purified IgG (100 μg/ml) and illustrate the levels of PKA signaling in the heart cells.
To determine if the antibody-induced heart cell signaling was mediated by the anti-beta receptor antibodies in the sera, propranolol, a beta-adrenergic receptor antagonist, is shown in Figure 6 to effectively inhibit the PKA activity of the purified IgG from selected cardiomyopathy sera. Purified IgG from cardiomyopathy activated PKA significantly above basal PKA levels while purified normal IgG did not. To further prove that IgG was the basis of the PKA activation, anti-IgG beads absorbed the PKA activity of sera while BSA beads had no effect (Figure 7).
Figure 6.
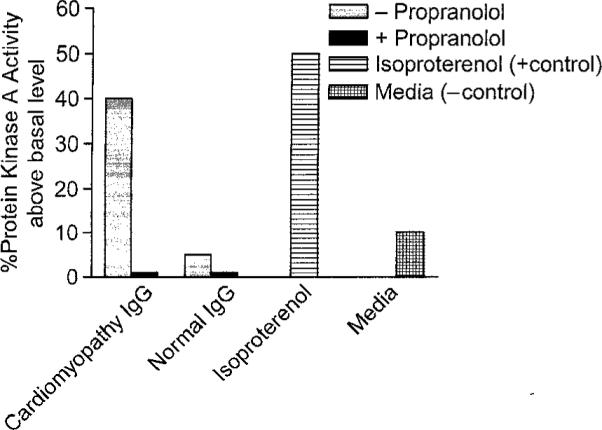
Propranolol, a beta-adrenergic receptor antagonist, is shown to effectively inhibit the PKA activity of the purified IgG from selected cardiomyopathy sera. Purified IgG from cardiomyopathy activated PKA significantly above basal PKA levels while purified normal IgG did not. H9c2 heart cells in culture were treated with purified IgG and PKA activity measured in counts per minute as described in materials and methods.
Figure 7.

Activation of PKA by IgG in myocarditis serum. Anti-IgG beads remove the antibody-mediated PKA activity while BSA coated beads do not. Therefore, autoantibodies which mediate cell signaling in cardiomyocytes can be blocked by anti-IgG.
In addition, antibody-mediated cell signaling of PKA was blocked by antigen-specific inhibition with cardiac myosin or the beta-adrenergic receptor but not by the alpha-adrenergic receptor or BSA (Figure 8). Therefore, autoantibodies in the sera act as an agonist of the beta-adrenergic receptor activating PKA levels in heart cells. The results suggest that in human myocarditis and cardiomyopathy sera, antibodies can mediate cell signaling in cardiomyocytes which can be blocked by anti-IgG, or by the specific antigens human cardiac myosin and the beta-adrenergic receptor, and can be inhibited as well by the beta-blocker propranolol.
Figure 8.
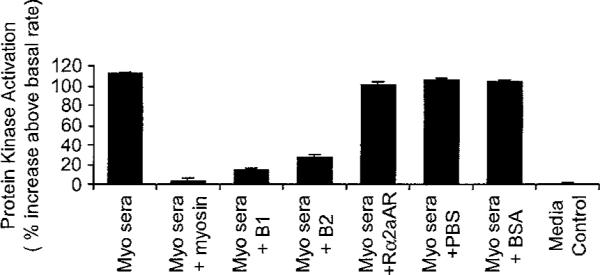
Autoantibodies which can mediate PKA cell signaling in cardiomyocytes can be blocked by the specific antigens human cardiac myosin and beta-adrenergic receptors. Antibodies do not react with the Rα2aAR or BSA. Antigen-specific inhibition pattern was similar to cardiomyopathy sera compared to myocarditis sera as shown in PKA assay.
Discussion
Cardiac myosin is well established in animal models as a prominent autoantigen in the induction of myocarditis [39,50–56] and has been associated with rheumatic carditis [57–59] and cardiomyopathy [1]. Anti-cardiac myosin antibodies have been associated with both bacterial or viral infections [8,59,60] where autoimmunity develops against the heart. In our study, we define the human cardiac myosin epitopes in human myocarditis and cardiomyopathies and establish a mechanism to explain how anti-cardiac myosin autoantibodies may contribute to heart disease. In our study we show that cardiac myosin epitopes recognized in cardiomyopathy and myocarditis were found primarily in the S2 region of the cardiac myosin rod. Compared to myocarditis, the number of cardiac myosin epitopes recognized by antibodies in cardiomyopathies were increased.
In human myocarditis, autoantibodies have been observed in myocardium [4,5], but mechanisms by which autoantibody against cardiac myosin may contribute to heart disease have not been established. Our data supports the hypothesis that anti-cardiac myosin antibodies may play a role in the pathogenesis of myocarditis and cardiomyopathy because they crossreact with mimicking antigens present on the surface of heart cells, such as the beta-adrenergic receptor, which may have pathophysiological effects on the heart. In our most recent studies in the Lewis rat model of cardiac myosin-induced myocarditis, anti-cardiac myosin antibodies reacted with the cell surface of cardiomyocytes and crossreacted with the beta-adrenergic receptor [16]. The reaction with the beta-adrenergic receptor induced cell signaling and PKA induction which eventually may lead to apoptosis of heart cells [16]. Using a more defined antigen, cardiac myosin pathogenic peptide S2-16, anti-cardiac myosin antibodies were induced by immunization which targeted the beta-adrenergic receptor on the heart cell surface and induced cAMP-dependent PKA activity in heart cells. In addition and further proof of mimicry, anti-cardiac myosin mAbs from myocarditis in the Lewis rat reacted in Western blots with the 67 kDa beta-adrenergic receptor and the 200 kDa human cardiac myosin molecule [16]. Thus, we wanted to extrapolate our findings from the Lewis rat into human myocarditis and determine if antibodies against cardiac myosin and the beta-adrenergic receptor were present in human disease and also could function as signaling antibodies.
In our study, crossreactive anti-cardiac myosin/anti-beta receptor antibodies were clearly present in myocarditis or cardiomyopathy. The presence of low levels of antibodies in normal healthy subjects suggested that some level is present in normal healthy subjects where these antibodies are not strongly reactive with cardiac myosin or its epitopes and may lack high specificity or avidity for antigen. In myocarditis or cardiomyopathy, functional signaling by antibody would require high enough affinity to signal the beta-adrenergic receptor. In other diseases where we have found signaling to be a potential cause of disease, only mAbs with high enough avidity can trigger the receptor [16,61] or in certain cases become cytotoxic [35].
It is well known that binding of the beta-adrenergic receptor triggers the major signaling pathway controlling heart rate and contractility [62]. It has also been shown that excessive stimulation of the beta receptor with isoproterenol (beta receptor-agonist) causes cardiomyocyte toxicity, myocardial scarring, and congestive heart failure [63,64]. The effect of anti-cardiac myosin antibody on the beta receptor suggests that cardiac myosin antibodies can modulate myocardial function and severity of cardiomyopathy and heart failure. In our most recent study, monoclonal antibodies specific for cardiac myosin confirmed the cross-reactive mimicry between cardiac myosin and the beta receptors and passively transferred rat anti-cardiac myosin/anti-beta-adrenergic receptor IgG produced dilated cardiomyopathy and apoptosis in Lewis rat [16]. In addition, immunization of animals with peptides of the beta-adrenergic receptor also led to a cardiomyopathic phenotype [65].
Although we do not know the exact cause of the presence of anti-cardiac myosin/anti-beta-adrenergic receptor antibodies in diabetic cardiomyopathy, it is possible that in diabetes the cardiac myosin molecule is exposed to the immune system and the increased recognition of cardiac myosin leads to recognition of more of the cardiac myosin peptides in both S2 and LMM-fragments. Alternatively, the increased susceptibility to repeated infections in diabetics could also lead to elevated levels of anti-cardiac myosin antibodies. Whatever the cause of the anti-cardiac myosin antibodies, the anti-human cardiac myosin autoanti-bodies in diabetic cardiomyopathy behave similarly to those in dilated cardiomyopathy patients and crossreact with the beta-adrenergic receptor and signal PKA. Without longitudinal or prospective studies, it is not clear if the autoantibodies against myosin are a risk factor in disease or if epitope spreading can occur in cardiomyopathies. Autoantibodies have been reported to occur months to years prior to clinical onset of symptoms in diseases such as systemic lupus erythe-matosus [66]. Therefore, the presence of autoantibody may be a risk factor in development of heart disease associated with cardiomyopathies.
Although we have not identified the regions of crossreactivity between the beta-adrenergic receptors and human cardiac myosin, studies are in progress with a human mAbs which recognizes both antigens and will be used to identify common structural regions of the molecules. The crossreactivity and mimicry between cardiac myosin and the beta-adrenergic receptor may contribute to the development of antibodies against the beta-adrenergic receptors during the progression of disease.
Acknowledgements
We thank Dr Mark Hemric for preparation of human cardiac myosin, Mona Cantu, R. N., N. P. for recruiting study subjects at the University of Colorado Health Sciences Center, Dr Nadia Mertens-Ellis, Dr Ya Li and Dr Carol Cox for helpful discussions, Dr Ken Jackson and the Molecular Biology Resource Facility at the University of Oklahoma Heatlh Sciences Center for synthesis of the human cardiac myosin peptides. We thank the many human subjects whose participation made our study possible. Our work was supported by grant HL56257 to MWC from the National Heart Lung and Blood Institute (NHLBI). MWC is the recipient of a NHLBI Award HL35282.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- [1].Caforio ALP, Goldman JH, Haven AJ, Baig KM, McKenna WJ. Evidence for autoimmunity to myosin and other heart-specific autoantigens in patients with dilated cardiomyopathy and their relatives. Int J Cardiol. 1996;54:157–163. doi: 10.1016/0167-5273(96)02593-4. [DOI] [PubMed] [Google Scholar]
- [2].Brown CA, O'Connell JB. Myocarditis and idiopathic dilated cardiomyopathy. Am J Med. 1995;99:309–314. doi: 10.1016/S0002-9343(99)80164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Felker GM, Hu W, Hare JM, Hruban RH, Baughman KI, Kasper EK. The spectrum of dilated cardiomyopathy. The Johns Hopkins experience with 1278 patients. Medicine. 1999;78:270–283. doi: 10.1097/00005792-199907000-00005. [DOI] [PubMed] [Google Scholar]
- [4].Aretz HT. Myocarditis: The Dallas criteria. Hum Pathol. 1987;18:619–624. doi: 10.1016/s0046-8177(87)80363-5. [DOI] [PubMed] [Google Scholar]
- [5].Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ, Jr, et al. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol. 1987;1:3–14. [PubMed] [Google Scholar]
- [6].Maisch B, Deeg P, Liebau G, Kochsiek K. Diagnostic relevance of humoral and cytotoxic immune reactions in primary and secondary dilated cardiomyopathy. Am J Cardiol. 1983;52:1072–1078. doi: 10.1016/0002-9149(83)90535-0. [DOI] [PubMed] [Google Scholar]
- [7].Maisch B, Trostel-Soeder R, Stechemesser E, Berg PA, Kochsiek K. Diagnostic relevance of humoral and cell-mediated immune reactions in patients with acute viral myocarditis. Clin Exp Immunol. 1982;48:533–545. [PMC free article] [PubMed] [Google Scholar]
- [8].Woodruff JF. Viral myocarditis—a review. Am J Pathol. 1980;101:425–466. [PMC free article] [PubMed] [Google Scholar]
- [9].Towbin JA, Bowles KR, Bowles NE. Etiologies of cardiomyo-pathy and heart failure. Nat Med. 1999;5:266–267. doi: 10.1038/6474. [DOI] [PubMed] [Google Scholar]
- [10].Huber SA, Job LP, Woodruff JF. Lysis of infected myofibers by coxsackievirus B3 immune lymphocytes. Am J Pathol. 1980;98:681–694. [PMC free article] [PubMed] [Google Scholar]
- [11].Cooper LT., Jr. Giant cell myocarditis: Diagnosis and treatment. Herz. 2000;25:291–298. doi: 10.1007/s000590050023. [DOI] [PubMed] [Google Scholar]
- [12].Cooper LT, Jr., Berry GJ, Shabetai R. Idiopathic giant-cell myocarditis—natural history and treatment. Multicenter Giant Cell Myocarditis Study Group Investigators. N Engl J Med. 1997;336:1860–1866. doi: 10.1056/NEJM199706263362603. [DOI] [PubMed] [Google Scholar]
- [13].Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Gatewood SJ, et al. Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the pronbrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am J Pathol. 2004;165:1883–1894. doi: 10.1016/s0002-9440(10)63241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Steele RA, et al. IL-12 protects against coxsackievirus B3-induced myocarditis by increasing IFN-gamma and macrophage and neutrophil populations in the heart. J Immunol. 2005;174:261–269. doi: 10.4049/jimmunol.174.1.261. [DOI] [PubMed] [Google Scholar]
- [15].Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, et al. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–2019. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li Y, Heuser JS, Cunningham LC, Kosanke SD, Cunningham MW. Mimicry and antibody-mediated cell signaling in autoimmune myocarditis. J Immunol. 2006;177:8234–8240. doi: 10.4049/jimmunol.177.11.8234. [DOI] [PubMed] [Google Scholar]
- [17].Morad M, Davies NW, Ulrich G, Schultheiss HP. Antibodies against ADP-ATP carrier enhance Ca2+ current in isolated cardiac myocytes. Am JPhysiol. 1988;255:H960–H964. doi: 10.1152/ajpheart.1988.255.4.H960. [DOI] [PubMed] [Google Scholar]
- [18].Schultheiss HP, Schulze K, Schauer R, Witzenbichler B, Strauer BE. Antibody-mediated imbalance of myocardial energy metabolism. A causal factor of cardiac failure? Circ Res. 1995;76:64–72. doi: 10.1161/01.res.76.1.64. [DOI] [PubMed] [Google Scholar]
- [19].Galvin JE, Hemric ME, Kosanke SD, Factor SM, Quinn A, Cunningham MW. Induction of myocarditis and valvulitis in Lewis rat by different epitopes of cardiac myosin and its implications in rheumatic carditis. Am J Pathol. 2002;160:297–306. doi: 10.1016/S0002-9440(10)64373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kodama M, Matsumoto U, Fujiwara M, Masani F, Izumi T, Shibata A. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol. 1990;57:250–262. doi: 10.1016/0090-1229(90)90039-s. [DOI] [PubMed] [Google Scholar]
- [21].Li Y, Heuser JS, Kosanke SD, Hemric ME, Cunningham MW. Cryptic epitope identified in rat and human cardiac myosin S2 region induces myocarditis in the Lewis rat. J Immunol. 2004;172:3225–3234. doi: 10.4049/jimmunol.172.5.3225. [DOI] [PubMed] [Google Scholar]
- [22].Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987;139:3630–3636. [PubMed] [Google Scholar]
- [23].Kuan AP, Chamberlain W, Malkeil S, Lieu HD, Factor SM, Diamond B, et al. Genetic control of autoimmune myocarditis mediated by myosin-specific antibodies. Immunogenetics. 1999;49:595–596. doi: 10.1007/s002510050466. [DOI] [PubMed] [Google Scholar]
- [24].Kuan AP, Zuckier L, Liao L, Factor SM, Diamond B. Immunoglobulin isotype determines pathogenicity in antibody-mediated myocarditis in naive mice. Circ Res. 2000;86:281–285. doi: 10.1161/01.res.86.3.281. [DOI] [PubMed] [Google Scholar]
- [25].Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med. 1995;181:1123–1131. doi: 10.1084/jem.181.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Neumann DA, Lane JR, Wulff SM, Allen GS, LaFond-Walker A, Herskowitz A, et al. In vivo deposition of myosin-specific autoantibodies in the hearts of mice with experimental autoimmune myocarditis. J Immunol. 1992;148:3806–3813. [PubMed] [Google Scholar]
- [27].Caforio AL, Mahon NJ, McKenna WJ. Cardiac autoantibodies to myosin and other heart-specific autoantigens in myocarditis and dilated cardiomyopathy. Autoimmunity. 2001;34:199–204. doi: 10.3109/08916930109007385. [DOI] [PubMed] [Google Scholar]
- [28].Lauer B, Schannwell M, Kuhl U, Strauer BE, Schultheiss HP. Antimyosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol. 2001;37:1106–1110. doi: 10.1016/s0735-1097(99)00485-4. [DOI] [PubMed] [Google Scholar]
- [29].Maisch B, Deeg P, Liebau G, Kochsiek K. Diagnostic relevance of humoral and cytotoxic immune reactions in primary and secondary dilated cardiomyopathy. Am J Cardiol. 1983;52:1072–1078. doi: 10.1016/0002-9149(83)90535-0. [DOI] [PubMed] [Google Scholar]
- [30].Neumann DA, Burek CL, Baughman KL, Rose NR, Herskowitz A. Circulating heart-reactive antibodies in patients with myocarditis or cardiomyopathy. J Am Coll Cardiol. 1990;16:839–846. doi: 10.1016/s0735-1097(10)80331-6. [DOI] [PubMed] [Google Scholar]
- [31].Felix SB, Staudt A, Friedrich GB. Improvement of cardiac function after immunoadsorption in patients with dilated cardiomyopathy. Autoimmunity. 2001;34:211–215. doi: 10.3109/08916930109007387. [DOI] [PubMed] [Google Scholar]
- [32].Rose NR. The significance of autoimmunity in myocarditis. Ernst Schering Res Found Workshop. 2006;55:141–154. doi: 10.1007/3-540-30822-9_9. [DOI] [PubMed] [Google Scholar]
- [33].Chow LH, Yuling Y, Linder J, McManus BM. Phenotypic analysis of infiltrating cells in human myocarditis. Arch Pathol Lab Med. 1989;113:1357–1362. [PubMed] [Google Scholar]
- [34].Cunningham MW, Antone SM, Gulizia JM, McManus BA, Gauntt CJ. Alpha-helical coiled-coil molecules: A role in autoimmunity against the heart. Clin Immunol Immunopathol. 1993;68:129–134. doi: 10.1006/clin.1993.1106. [DOI] [PubMed] [Google Scholar]
- [35].Cunningham MW, Antone SM, Gulizia JM, McManus BM, Fischetti VA, Gauntt CJ. Cytotoxic and viral neutralizing antibodies crossreact with streptococcal M protein, enteroviruses, and human cardiac myosin. Proc Natl Acad Sci USA. 1992;89:1320–1324. doi: 10.1073/pnas.89.4.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liao L, Sindhwani R, Rojkind M, Factor S, Leinwand L, Diamond B. Antibody-mediated autoimmune myocarditis depends on genetically determined target organ sensitivity. J Exp Med. 1995;187:1123–1131. doi: 10.1084/jem.181.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Antone SM, Adderson EE, Mertens NMJ, Cunningham MW. Molecular analysis of V gene sequences encoding cytotoxic anti-streptococcal/anti-myosin monoclonal antibody 36.2.2 that recognizes the heart cell surface protein laminin. J Immunol. 1997;159:5422–5430. [PubMed] [Google Scholar]
- [38].Tobacman LS, Adelstein RS. Enzymatic comparisons between light chain isozymes of human cardiac myosin subfragment-1. J Biol Chem. 1984;259:11226–11230. [PubMed] [Google Scholar]
- [39].Galvin JE, Hemric ME, Ward K, Cunningham MW. Cytotoxic monoclonal antibody from rheumatic carditis reacts with human endothelium: Implications in rheumatic heart disease. J Clin Invest. 2000;106:217–224. doi: 10.1172/JCI7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jaenicke T, Diederich KW, Haas W, Scheich J, Lichter P, Pfordt M, et al. Complete sequence of human b myosin. Genomics. 1990;8:194–207. doi: 10.1016/0888-7543(90)90272-v. [DOI] [PubMed] [Google Scholar]
- [41].Cunningham MW, Meissner HC, Heuser J, Leung DYM. Anti-human cardiac myosin autoantibodies in Kawasaki syndrome. J Immunol. 1999;163:1060–1065. [PubMed] [Google Scholar]
- [42].Cunningham MW, Swerlick RA. Polyspecificity of antistrep-tococcal murine monoclonal antibodies and their implications in autoimmunity. J Exp Med. 1986;164:998–1012. doi: 10.1084/jem.164.4.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Laemmli UK. Cleavage of structural proteins during ihe assembly of the head of bacteriophage T4. Nature. 1970;277:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- [44].Caforio AL, Goldman JH, Baig MK, Haven AJ, Dalla Libera L, Keeling PJ, et al. Cardiac autoantibodies in dilated cardiomyopathy become undetectable with disease progression. Heart. 1997;77:62–67. doi: 10.1136/hrt.77.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Caforio AL, Goldman JH, Haven AJ, Baig KM, Libera LD, McKenna WJ. Circulating cardiac-specific auloantibodies as markers of autoimmunity in clinical and biopsy-proven myocarditis. The myocarditis treatment trial investigators. Eur Heart J. 1997;18:270–275. doi: 10.1093/oxfordjournals.eurheartj.a015230. [DOI] [PubMed] [Google Scholar]
- [46].Caforio AL, Grazzini M, Mann JM, Keeling PJ, Bottazzo GF, McKenna WJ, et al. Identification of alpha- and beta-cardiac myosin heavy chain isoforms as major autoantigens in dilated cardiomyopathy. Circulation. 1992;85:1734–1742. doi: 10.1161/01.cir.85.5.1734. [DOI] [PubMed] [Google Scholar]
- [47].Caforio AL, Mahon NG, McKenna WJ. Clinical implications of anti-cardiac immunity in dilated cardiomyopathy. Ernst Schering Res Found Workshop. 2006:169–193. doi: 10.1007/3-540-30822-9_11. [DOI] [PubMed] [Google Scholar]
- [48].Caforio AL, Mahon NJ, McKenna WJ. Cardiac autoantibodies to myosin and other heart-specific autoantigens in myocarditis and dilated cardiomyopathy. Autoimmunity. 2001;34:199–204. doi: 10.3109/08916930109007385. [DOI] [PubMed] [Google Scholar]
- [49].Caforio AL, Mahon NJ, Tona F, McKenna WJ. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: Pathogenetic and clinical significance. Eur J Heart Fail. 2002;4:411–417. doi: 10.1016/s1388-9842(02)00010-7. [DOI] [PubMed] [Google Scholar]
- [50].Kodama M, Hanawa H, Zhang S, Saeki M, Koyama S, Hosono H, et al. FK506 therapy of experimental autoimmune myocarditis after onset of the disease. Am Heart J. 1993;126:1385–1392. doi: 10.1016/0002-8703(93)90538-k. [DOI] [PubMed] [Google Scholar]
- [51].Kodama M, Hanawa H, Saeki M, Hosono H, Inomata T, Suzuki K, et al. Rat dilated cardiomyopathy after autoimmune giant cell myocarditis. Circ Res. 1994;75:278–284. doi: 10.1161/01.res.75.2.278. [DOI] [PubMed] [Google Scholar]
- [52].Kodama M, Matsumoto Y, Fujiwara M, Massani M, Izumi T, Shibota A. A novel experimental model of giant cell myocarditis induced in rats by immunization with cardiac myosin fraction. Clin Immunol Immunopathol. 1991;57:250–262. doi: 10.1016/0090-1229(90)90039-s. [DOI] [PubMed] [Google Scholar]
- [53].Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987;139:3630–3636. [PubMed] [Google Scholar]
- [54].Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell mediated disease. J Immunol. 1991;147:2141–2147. [PubMed] [Google Scholar]
- [55].Galvin JE, Hemric ME, Kosanke SD, Factor SM, Quinn A, Cunningham MW. Induction of myocarditis and valvulitis in Lewis rats by different epitopes of cardiac myosin and its implications in rheumatic carditis. Am J Pathol. 2001;160:297–306. doi: 10.1016/S0002-9440(10)64373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liao L, Sindhwani R, Leinwand L, Diamond B, Factor S. Cardiac alpha-myosin heavy chains differ in their induction of myocarditis: Identification of pathogenic epitopes. J Clin Invest. 1993;92:2877. doi: 10.1172/JCI116909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cunningham MW, McCormack JM, Fenderson PG, Ho MK, Beachey EH, Dale JB. Human and murine antibodies cross-reactive with streptococcal M protein and myosin recognize the sequence GLN-LYS-SER-LYS-GLN in M protein. J Immunol. 1989;143:2677–2683. [PubMed] [Google Scholar]
- [58].Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Cunningham MW, McCormack JM, Talaber LR, Harley JB, Ayoub EM, Muneer RS, et al. Human monoclonal antibodies reactive with antigens of the group A streptococcus and human heart. J Immunol. 1988;141:2760–2766. [PubMed] [Google Scholar]
- [60].Rose NR. The role of infection in the pathogenesis of autoimmune disease. Semin Immunol. 1998;10:5–13. doi: 10.1006/smim.1997.0100. [DOI] [PubMed] [Google Scholar]
- [61].Kirvan CA, Swedo SE, Heuser S, Cunningham MW. Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat Med. 2003;9:914–920. doi: 10.1038/nm892. [DOI] [PubMed] [Google Scholar]
- [62].Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmem-brane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- [63].Freedman NJ, Lefkowitz RJ. Anti-beta-1-adrenergic receptor antibodies and heart failure: Causation, not just correlation. J Clin Invest. 2004;113:1379–1382. doi: 10.1172/JCI21748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jahns R, Boivin V, Hein L, Triebel S, Angermann CE, Ertl G, et al. Direct evidence for a beta 1-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J Clin Invest. 2004;113:1419–1429. doi: 10.1172/JCI20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jahns R, Boivin V, Hein L, Triebel S, Angermann CE, Ertl G, et al. Direct evidence for a beta 1-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J Clin Invest. 2004;113:1419–1429. doi: 10.1172/JCI20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]