

Abstract
The evolution of actin gene families is characterized by independent expansions and contractions across the eukaryotic tree of life. Here, we assess diversity of actin gene sequences within three lineages of the genus Arcella, a free-living testate (shelled) amoeba in the Arcellinida. We established four clonal lines of two morphospecies, Arcella hemisphaerica and A. vulgaris, and assessed their phylogenetic relationship within the “Amoebozoa” using small subunit ribosomal DNA (SSU-rDNA) genealogy. We determined that the two lines of A. hemisphaerica are identical in SSU-rDNA, while the two A. vulgaris are independent genetic lineages. Furthermore, we characterized multiple actin gene copies from all lineages. Analyses of the resulting sequences reveal numerous diverse actin genes, which differ mostly by synonymous substitutions. We estimate that the actin gene family contains 40–50 paralogous members in each lineage. None of the three independent lineages share the same paralog with another, and divergence between actins reaches 29% in contrast to just 2% in SSU-rDNA. Analyses of effective number of codons (ENC), compositional bias, recombination signatures, and genetic diversity in the context of a gene tree indicate that there are two groups of actins evolving with distinct patterns of molecular evolution. Within these groups, there have been multiple independent expansions of actin genes within each lineage. Together, these data suggest that the two groups are located in different regions of the Arcella genome. Furthermore, we compare the Arcella actin gene family with the relatively well-described gene family in the slime mold Dictyostelium discoideum and other members of the Amoebozoa clade. Overall patterns of molecular evolution are similar in Arcella and Dictyostelium. However, the separation of genes in two distinct groups coupled with recent expansion is characteristic of Arcella and might reflect an unusual pattern of gene family evolution in the lobose testate amoebae. We provide a model to account for both the existence of two distinct groups and the pattern of recent independent expansion leading to a large number of actins in each lineage.
Keywords: Amoebozoa, actin gene family, codon usage, Arcella, purifying selection, gene duplication
Introduction
Though actin is one of the most abundant proteins in eukaryotic cells and has been the subject of many studies, much remains to be understood about the tempo and mode of evolution of this gene family (Reisler and Egelman 2007). Actin cytoskeletal functions are well characterized (Goodson and Hawse 2002), but actins and actin-related proteins are also implicated in nuclear processes (Chen and Shen 2007; Reisler and Egelman 2007; Pederson 2008).
There is a high level of structural and sequence conservation among actin proteins between disparate organisms and between eukaryotic and bacterial homologues, such as MreB (Hightower and Meagher 1986; van den Ent et al. 2001; Goodson and Hawse 2002; Erickson 2007). However, different lineages show different patterns of evolution in their sets of actin paralogs across the eukaryotic tree of life (Goodson and Hawse 2002; Wade et al. 2009).
In multicellular eukaryotes, appearance of distinct actin gene duplicates and subsequent innovation is associated with tissue differentiation. In the green algal lineage (including land plants), an increase of paralogs up to 18 in the soybean (McDowell et al. 1996) is associated with an increase in morphological complexity (Bhattacharya et al. 2000). In animals, the appearance of specific muscle and cytoplasmic actin types is ancient, and subsequent duplications within each type seem independent, yielding up to six actin genes in vertebrates and arthropods (Kusakabe et al. 1997; Hooper and Thuma 2005).
Our understanding of actin gene family evolution in microbial eukaryotes is incomplete. Across the estimated ∼70 lineages of microbial eukaryotes (Patterson 1999; Parfrey et al. 2006), the breadth of knowledge on actin diversity is largely limited to organisms with completed genomes (Reisler and Egelman 2007). In addition, diverse lineages such as dinoflagellates (Bachvaroff and Place 2008), Foraminifera (Flakowski et al. 2006), and red algae (Wu et al. 2009) have been shown to contain large collections of actin gene paralogs of up to 28, 7, and 10 genes, respectively. In these three cases, paralogs are divided into two groups with different evolutionary characteristics. In all three cases, there are significantly more synonymous substitutions than replacement ones, indicating purifying selection. In the dinoflagellate Amphidinium carterae (Bachvaroff and Place 2008), both actin groups contain introns and are tandemly organized. There is indication that the size of introns and expression differs between the two groups.
The Amoebozoa contain many familiar amoeboid organisms, including Amoeba proteus, Dictyostelium discoideum, and the testate lobose amoebae (Arcellinida), that are the subject of this study. Most knowledge in this group stems from studies in the model slime molds (Dictyostelium and Physarum) and some pathogenic lineages (Entamoeba and Acanthamoeba). The completed genome of D. discoideum (Eichinger et al. 2005) reveals a 41-member set of actin paralogs encompassing 17 paralogs that code for the exact same amino acid sequence (Act8-group), 7 potential pseudogenes, and 16 other paralogs ranging from canonical actins to very divergent proteins (Joseph et al. 2008). Identical paralogs for the most highly expressed type of actin (Act8-group) are spread across four chromosomes.
The remaining ∼14 major lineages in Amoebozoa remain largely unexplored with respect to gene family evolution (Pawlowski and Burki 2009). We have investigated the actin gene family in the lobose testate amoebae (Arcellinida). The Arcellinida are characterized by the presence of a test (shell), but despite a 750-Ma fossil record (Porter et al. 2003) and high abundance in numerous environments (Smith et al. 2008), the Arcellinida remain relatively understudied. We have isolated four clonal lines that represent two morphospecies, Arcella hemisphaerica and A. vulgaris. We established their relationship by analyzing small subunit ribosomal DNA (SSU-rDNA) genealogies and characterized their actin genes. Analyses of actin gene genealogies coupled with analysis of the ENC, genetic diversity indices, and recombination signatures reveal that actin genes in the Arcellinida are under an intriguing mode of evolution, which combines paralogy predating the divergence of these morphospecies with recent independent gene expansions.
Materials and Methods
Taxa Studied
We isolated and cultured four lineages of Arcella spp. for this study. Two lineages of A. hemisphaerica were isolated from commercial cultures, both marketed as A. vulgaris. The “Blue” lineage was purchased from Connecticut Valley Biological Supply Company, Southampton, MA, and is the same strain described in Tekle et al. (2008) (table 1; fig. 1a). The “Red” lineage was purchased from Carolina Biological, Burlington, NC (table 1; fig. 1b). The two A. vulgaris lineages were isolated from nature by sampling two geographically separated lake sediments in Massachusetts, USA (table 1). The “SC” lineage was isolated from Lyman Lake at the Smith College Campus, Northampton, MA (fig. 1d), and the WP lineage was isolated at Weeks Pond in Falmouth, MA (fig. 1c). After starting initial mixed cultures, individual organisms were picked and washed to start clonal cultures (from a single organism) by placing cells into autoclaved pond water and adding 0.05 volume cereal grass media (Fisher Scientific, Cat. No. NC9735391) as well as bacteria. Identification follows Lahr and Lopes (2009), briefly A. hemisphaerica are 60- to 80-μm wide (fig. 1a and b), with a markedly semicircular lateral profile; A. vulgaris are 100- to 120-μm wide (fig. 1c and d), with a slightly flattened lateral profile and the presence of a rim on the border where the abapertural and apertural surfaces meet.
Table 1.
Lineages of Arcella Used in this Study.
| Lineage | Source | Coordinates | Isolation | SSU GenBank |
| A. hemisphaerica Blue | CT V. Bio. Cat# L 1B | — | May 2005 | EU273445 |
| A. hemisphaerica Red | Carolina Cat# 131310 | — | March 2007 | — |
| A. vulgaris SC | Lyman Lake, MA | N42°19′07″; W72°38′24″ | April 2007 | HM853761 |
| A. vulgaris WP | Weeks Pond, MA | N41°33′21″; W70°36′52″ | November 2008 | HM853762 |
Note.—The lineages A. hemisphaerica Blue and A. vulgaris SC have been deposited at the American Type Culture Collection (ATCC). The other two lineages perished before preservation.
FIG. 1.

Representative images of Arcella lineages used in this study. (a) A. hemisphaerica Blue lineage. (b) A. hemisphaerica Red lineage. (c) A. vulgaris WP lineage. (d) A. vulgaris SC lineage: The image shows six individuals undergoing plasmogamy, where multiple individuals fuse their cytoplasm. It is unknown whether nuclear fusion also occurs. Images a, b and d are Hoffman interference contrast, image c is differential interference contrast. Scale bars are 50 um.
DNA Extraction and Amplification Experiments
For DNA extraction, 100–1,000 clonal individuals were harvested multiple times for each isolate. Individuals were either handpicked or harvested by spinning culture flasks into DNA extraction buffer (100 mM NaCl, 10 mM Tris, 25 mM EDTA, 0.5% sodium dodecyl sulfate, and 10 μg/ml proteinase K). DNA was extracted following a standard 2 phenol:1 chloroform extraction followed by cold ethanol precipitation. Amplification of target genes was achieved by polymerase chain reaction (PCR) in a PTC-200 Thermal Cycler (MJ Research, Waltham, MA) with Phusion HotStart polymerase (Cat. No. M0254; New England BioLabs), using concentrations of reagents per manufacturers recommendations (1× HF buffer, 1.5 mM MgCl2, 0.2 mM each deoxyribonucleotide triphosphate, and 0.01 U/μl polymerase), except for primers that were used at a final concentration 2- to 4-fold higher than recommended (1–2 μm for each) in a total reaction volume of 25 μl. Cycling conditions were 98° for 3 min; followed by 35–60 cycles at 98° for 15 s, 56° for 15 s, and 72° for 90 s; and then a final extension at 72° for 5 min. The number of cycles varied from 35 to 60 according to conditions inherent to different DNA extractions (see supplementary table 1, Supplementary Material online and Detection and Avoidance of Artifactual PCRRecombinants [chimeras] section for details). We performed single-celled PCRs by picking an individual, washing it three times in autoclaved water, and transferring directly into a PCR master mixture. Primers for SSU-rDNA genes are eukaryote specific from Medlin et al. (1988), and primers for actin genes are either eukaryote specific from Tekle et al. (2007) or Arcella specific as described in Lahr and Katz (2009) (see supplementary table 1, Supplementary Material online for details). The target amplicon for Arcella actins were either 669- or 795-bp long depending on the primer set used. Cloning experiments were performed for all amplified products using the Invitrogen TOPO cloning kit exactly as described in Lahr and Katz (2009). All plasmids containing inserts were purified using a PureLink kit (Invitrogen). The positive colonies were either amplified individually using a Big Dye Terminator kit (PerkinElmer) and run on an ABI 3100 automated sequencer at the Center for Molecular Biology (Smith College, Northampton, MA) or in a 96-well format at the Pennsylvania State University Nucleic Acid Facility (University Park, PA).
Detection and Avoidance of Artifactual PCR Recombinants (chimeras)
Avoiding Chimera Formation
PCR amplification yields artifactual recombinants (chimeras) when multiple closely related target sequences are present in the mixture (Judo et al. 1998). We chose to rely on an empirical strategy, performing extensive PCR-artifact formation experiments in our study system to determine chimera-reducing PCR conditions (Lahr and Katz 2009). We have found that PCR recombinants result of a combination of too many amplification cycles and too much starting DNA template. Both conditions were determined on a sample-by-sample basis, given differences in genomic DNA extractions (number of individuals in the culture for example), and thus differ from experiment to experiment (supplementary table 1, Supplementary Material online). Hence, most PCRs were performed across a gradient of DNA concentrations and amplification cycles to choose the most chimera-restricting amplification possible (lowest cycle number coupled with lowest initial DNA concentration). Only those PCR products were cloned and sequenced, the others were discarded. In addition, we have performed all PCRs using three times the recommended extension time and 2- to 4-fold more concentrated primers according to general guidelines for chimera reduction (Judo et al. 1998).
Determining a Data Set of “Real” Sequences
We chose a conservative approach to distinguish “real” and “chimeric” sequences based on observed experimental properties of chimera formation (Lahr and Katz 2009). The appearance of a specific gene sequence in two or more independent PCRs is almost certainly indicative of a “real” sequence. Hence, we consider all sequences in this condition to be “real” (a total of 41 sequences for A. hemisphaerica, 16 for A. vulgaris SC, and 3 for A. vulgaris WP). Additionally, when a particular gene sequence is found multiple times in the same PCR experiment (i.e., multiple clones), there is a reduced chance that this sequence is a recombinant, though this chance is larger than using the former criterion. This probability increases with sampling effort, and we consider that for A. hemisphaerica, with a total sampling effort of 440 clones and 30 PCRs, sequences that appeared three or more times can be considered “real” for a total of four additional sequences. For A. vulgaris SC, with a quarter of that effort, we also consider clones that appeared three times or more as “real,” an additional four gene sequences. For A. vulgaris WP, with only 43 clones sequenced, we consider that clones, which appeared two or more times to be “real,” yielding an additional five gene sequences. Using these two criteria, we come to a final data set that includes 45 actin genes for A. hemisphaerica, 20 genes for A. vulgaris SC, and 8 genes for A. vulgaris WP.
Total Number of Actin Genes Per Lineage
We estimated the total number of actin genes in each lineage using estimation tools most commonly used in ecological sampling freely available in the package EstimateS 8.0 (Colwell 2006). We have used only actin sequences that were considered nonchimeric for these estimations. We used two subpartitions of the data set to estimate diversity: 1) a conservative data set that considers each PCR a sample and uses only genes that were found in two or more independent PCRs as real paralogs and 2) a more liberal data set that considers a real paralog every gene that was found three or more times for A. hemisphaerica and two or more times for the A. vulgaris SC lineage as described above. In A. hemisphaerica, sampling was more intense, and we were able to use interpolation methods (Mao τ) to estimate the total diversity. For A. vulgaris SC, not enough samples were taken to plateau the accumulation curve. Hence, we used an extrapolation method (MMMeans) to estimate total diversity. Estimates were calculated using 1,000 randomizations and sampling without replacement. The remaining lineage, A. vulgaris WP, had too few samples to allow a consistent estimate of total diversity, but the pattern of discovery of new genes is similar to the other two lineages, and we expect results to apply to this lineage as well.
SSU-rDNA Analysis
Sequences of representative organisms in the Amoebozoa were retrieved from GenBank (fig. 2 lists all accession numbers). Taxon sampling reflects an effort to include representatives of all major lineages in the Amoebozoa (Pawlowski 2008; Tekle et al. 2008; Pawlowski and Burki 2009). One Arcella sequence used in previous reconstructions of the Arcellinida (A. artocrea AY848969) is likely a contaminant (Mitchell EAD, personal communication) and was not included in this study. Alignments were constructed in SeaView (Galtier et al. 1996; Gouy et al. 2010) with alignment algorithm MAFFT (Katoh et al. 2009) using the L-INS-I setting and adjusted manually in MacClade (Maddison WP and Maddison DR 2005). We have masked the alignment to exclude regions that had over 50% missing data as well as ambiguously aligned sites identified by ALISCORE, using default settings (Misof B and Misof K 2009). The resulting alignment is 1,587 sites (available as supplementary material, alignment 1, Supplementary Material online). We generated a second more conservative alignment, by manually excluding ambiguous regions, to a total of 1,357 sites. Phylogenetic reconstruction was made using RAxML-HPC 7.0.4 (Stamatakis et al. 2008) through the online server CIPRES Portal 2.0 (Miller et al. 2009), using the GTRGAMMA model of nucleotide substitution and running 200 automatic rapid bootstraps followed by a slow search for the best-scoring ML tree. The GTRGAMMA model was selected as the most appropriate model for our data set through a ModelTest analysis performed on HyPhy (Pond et al. 2005).
FIG. 2.

Most likely SSU-rDNA gene tree of the Amoebozoa. For this reconstruction, we used an alignment consisting of 55 taxa and 1,587 characters and performed a maximum likelihood analysis on RaxML using the GTR model of evolution and 200 bootstrap replicates (−lnL = 29300.662). The position of two new isolates of A. vulgaris and A. hemisphaerica is indicated in bold. Thicker branches represent nodes that have >75% BS. Branches are drawn to scale. Dashed lines represent paraphyletic groupings.
Actin Genealogical Analysis
Actins from other Amoebozoa were obtained from GenBank accessions, curated Genome databases, and expressed sequence tag (EST) databases. From genome databases, we included every available actin paralog, except four variant putative actin genes (>30% divergence at nucleotide level and >30% divergence at amino acid level) in D. discoideum (Joseph et al. 2008). To collect actins from ESTs, we first performed a basic local alignment search tool search with one described actin against the EST database of the same organism. We then constructed contigs using SeqMan, and with a cutoff point of 1%, we established putative actin paralogs. The data set, including 73 Arcella actins and another 103 Amoebozoa actins (alignment and accession numbers are available as supplementary material, alignment 2, Supplementary Material online), was aligned on SeaView (Galtier et al. 1996; Gouy et al. 2010) using the alignment algorithm MAFFT (Katoh et al. 2009) set to L-INS-I optimization. Proper codon alignment was confirmed visually. The final total alignment consists of 179 sequences, 1,134 nucleotide sites, or 378 amino acid sites, which we designate “Actin Alignment A” (supplementary material, alignment 2, Supplementary Material online, also see supplementary fig. 1, Supplementary Material online). Phylogenetic analyses were performed at the nucleotide level considering all sites as well as third positions excluded with RAxML-HPC 7.0.4 in the CIPRES Portal 2.0, using the GTRGAMMA model of nucleotide substitution and running automatic bootstrapping followed by a slow ML search. An additional analysis limiting the data set to only the 795 homologous sites that were amplified in Arcella, which we designate “Actin Alignment B,” was performed (see supplementary fig. 1, Supplementary Material online). The translated data sets consisting of 378 total sites and 265 homologous sites were also analyzed on RAxML using the JTT model of amino acid substitution, chosen through a ProtTest analysis performed on HyPhy (Pond et al. 2005).
Codon Usage and Compositional Bias
We determined the codon usage and compositional bias for collections of actin paralogs in lineages that are represented by three or more actin sequences, totaling 149 actin sequences that do not have internal stop codons or frameshift deletions in “Actin Alignment B” (which comprises only the 795 base pair region that contains the largest Arcella amplicons, see supplementary fig. 1, Supplementary Material online) using the algorithm CodonW (Peden 1999) as implemented in the online server MOBYLE (Neron et al. 2009). We calculated the ENC, total GC content and GC content at 4-fold degenerate sites.
Genetic Diversity Indices
We have used tools available for nucleotide diversity calculation at the online server DPDB (Casillas et al. 2005) to calculate average pairwise distance in the group (k = measured by averaging all pairwise uncorrect P distances), the average number of nucleotide differences per site (π), the number of segregating sites (S), and respective variances. The alignment used for this analysis is the most restrictive in terms of length because we only used the 669 homologous base pair region that is available for our shortest Arcella amplicons (“Actin Alignment C” in supplementary fig. 1, Supplementary Material online). Each group of actin sequences representing an Amoebozoa taxon was analyzed separately, and the Arcella data set was divided into Groups 1 and 2 from the phylogenetic reconstruction.
Recombination Detection
We used the algorithm GARD (Pond et al. 2006) implemented in the online server Datamonkey (Pond and Frost 2005) to infer historical recombination between actins in each Arcella lineage as well as other collections of sequences in each Amoebozoa lineage separately. Analyses were run by first calculating the most appropriate model of substitution for each case, then running the genetic algorithm estimating site-to-site variation with beta–gamma distribution and four rate classes. The output is given in terms of most likely points for recombination in the data set, and those points are further submitted to the Kishino–Hasegawa test because initial detection of points could be due to rate heterogeneity. We then used the statistically significant recombination points to divide the data set into partitions and independently reconstructed ML trees using RAxML. Despite incomplete sampling in the A. vulgaris lineages, we can infer which genes have recombined by analyzing their relative position in different partitions. We cannot infer, in most cases, exactly which other gene they recombined with.
Comparison of Actin Gene Copies Across Eukaryotes
We have performed a comparison of average pairwise distances between chosen eukaryotes with large numbers of actin paralogs in their genomes. We aligned GenBank deposits for actin genes in the Amoebozoa Entamoeba histolytica (seven actins) and D. discoideum (29 actins) and the dinoflagellate Am. carterae (28 actins); the red algae Flintiella sanguinaria (10 actins) and Glaucosphaera vaculolata (7 actins); the Metazoa Drosophila melanogaster and Homo sapiens (6 actins for each); and the plants Glycine max (18 actins), Zea mays (12 actins), and Arabidopsis thaliana (10 actins). We calculated uncorrected pairwise distances both at the nucleotide and at the amino acid levels using Paup 4.0 beta 10 (Swofford 2003) and averaged the distances for each taxon.
Results
SSU-rDNA Analysis
Maximum likelihood analysis of the SSU-rDNA gene (fig. 2) including a total of 55 taxa and 1,587 characters is largely concordant with other recently published reconstructions of the Amoebozoa (Smirnov et al. 2005; Nikolaev et al. 2006; Tekle et al. 2008; Pawlowski and Burki 2009; Parfrey et al. 2010). Most major lineages are recovered with high bootstrap supports (BS): The Amoebidae, Leptomyxidae, Echinamoebidae, Archamoebae, Dictyosteliida, and Acanthamoebidae are all monophyletic and receive full support (BS = 100). The Flabellinea and the Hartmannellidae are recovered as paraphyletic. This analysis uses an alignment with liberal masking. When analysis is repeated with more stringent masking, yielding an alignment with 1,357 sites similar to Tekle et al. (2008), the relationships remain the same, and BS increase slightly (data not shown). The Arcellinida are recovered with low support (BS < 50%), and the structure of the Arcellinida subtree agrees with more focused reconstructions (Nikolaev et al. 2005; Lara et al. 2007, 2008), with the exception that the genus Argynnia appears closely related to Arcella in our reconstruction as opposed to Heleopera sphagni in Lara et al. (2008).
The Arcella lineages are monophyletic within the Arcellinida (BS = 100). The SSU-rDNA sequences for both lineages of A. hemisphaerica (Red and Blue) are identical, which may not be surprising as these were both obtained from biological supply companies. Based on this and the sharing of numerous identical actin sequences, we consider these to be two populations of the same species. However, the two A. vulgaris SSU-rDNAs differ by 1.7% and do not share any actin sequences. This indicates that these two lineages are independently evolving units. Furthermore, the phylogenetic reconstruction places A. vulgaris WP isolate as a sister group to A. hemisphaerica (BS = 98) to the exclusion of the A. vulgaris SC isolate.
Actin Genes Identified
We identified a total of 166 distinct actin sequences from one lineage of A. hemisphaerica (two populations that we interpret as the same genetical lineage) and two lineages of A. vulgaris. Given the possibility of chimera formation and taking into account the results from a previous experimental approach to PCR in this system (Lahr and Katz 2009), we use the sole criterion of redundancy to exclude chimeric sequences. We include 45 sequences of A. hemisphaerica that were found in at least two separate PCRs or were represented by three or more clones in a single PCR experiment. For both lineages of A. vulgaris, we analyze 28 sequences (20 from the SC lineage and 8 from the WP lineage) that were found in at least two PCRs or were represented by two or more clones. These 73 sequences, along with representatives from other Amoebozoa lineages, were used in subsequent phylogenetic, recombination, diversity, and codon usage analyses.
After chimera exclusion, we have estimated the total number of actins likely to be present in each lineage, using tools for estimating total species richness commonly used by ecologists (table 3). For the A. hemisphaerica data set, the most appropriate statistic is the species accumulation curve calculated by the Mao Tau parameter. The estimate for this lineage is 45 ± 1 actin genes. For A. vulgaris, only the SC lineage was sampled sufficiently enough to enable estimation of total number of actin genes. In this case, there are fewer samples than in A. hemisphaerica. Hence, it is more appropriate to use an extrapolation method instead of a species accumulation curve. The estimate for this lineage lies within 25–50 actin genes, consistent with the estimate for A. hemisphaerica. We conclude that each lineage has around 50 actin paralogs in their genome (table 3).
Table 3.
Estimates of Total Number of Actin Genes for Lineages of Arcella.
|
A. hemisphaerica |
A. vulgaris SC |
|||
| ≥2 PCRs (n = 365) | ≥3 Clones (n = 381) | ≥2 PCRs (n = 85) | ≥2 Clones (n = 111) | |
| Sobs (Mao τ) | 41 ± 0 | 45 ± 1 | 16 ± 0 | 27 ± 2 |
| MMMeans | 55 | 61 | 25.1 | 50 |
Note.—Sobs = expected total number of sequences sample-based statistic, using the Mao Tau calculation; MMMeans = expected total number of actin sequences by functional extrapolation, based on the Michaelis–Menten richness estimator, computed analytically; n = number of actin sequences used to calculate the statistic. We did not perform estimates of diversity in the lineage A. vulgaris WP due to low sampling effort. The most appropriate statistic for each data set is in bold. Only actin sequences that were deemed nonchimeric were used for estimation.
Unique gene sequence discovery varied with intensity of sampling effort. Sampling efforts were greater in Ar. hemisphaerica, where we obtained a total of 41 nonchimeric gene sequences (table 2). The two populations (Blue and Red) share 30 of 45 gene sequences. These 45 sequences (HM853688–HM853732) are 1–29% divergent from each other at the nucleotide level. The large majority of polymorphisms are synonymous substitutions; thus, the amino acid sequences are identical for 36 actin genes. Ahem_act19 shows major modifications (20 amino acid substitutions), Ahem_act45 shows 9 substitutions, and seven other sequences show 1–3 amino acid substitutions. Three sequences show deletions: Ahem_act33 has a frameshifting deletion of 26 nucleotides and no amino acid modifications, Ahem_act38 shows two in-frame deletions and one frameshifting and three amino acid modifications if made to be in-frame, and Ahem_act41 has a one-nucleotide frameshifting deletion as well as a six-nucleotide in-frame deletion, and two amino acid substitutions if made to be in-frame (fig. 3).
Table 2.
Summary of Actin PCR Experiments on Arcella Lineages.
| Lineage | Pop PCRs | SC PCRs | Clones | Genes | Nonchimera |
| A. hemisphaerica Blue | 15 | 3 | 246 | 69 | 41 |
| A. hemisphaerica Red | 6 | 6 | 194 | 58 | 33 |
| A. vulgaris SC | 11 | NA | 132 | 48 | 20 |
| A. vulgaris WP | 8 | NA | 43 | 20 | 8 |
NOTE.—Pop PCR = PCR performed on DNA extracted from a clonal culture; SC PCR = PCR performed on a single cell. A complete table is available as supplementary table 1 (Supplementary Material online); clones = number of clones sequenced; genes = number of distinct actin gene sequences obtained; nonchimera = number of distinct actin sequences determined to be nonchimeric.
FIG. 3.

Recent frameshifting deletions in Arcella actin genes. The amino acid alignment compares three actin sequences to the most common actin found (Ahem_act01). Identities are shown as dots, and substitutions are indicated with respective amino acid symbol. Dashes show in-frame deletions, and gray areas show frameshifting deletions with number of nucleotides deleted. Note that although all three sequences show at least one frameshifting deletion, the amino acid sequence remains largely unchanged, suggesting that these deletion events are recent.
For both isolates identified as the morphospecies A. vulgaris, sampling was less intense than in A. hemisphaerica. We have obtained 20 distinct sequences for the SC isolate (HM853733–HM853752) and 8 for the WP isolate (HM853753–HM853760). The levels of divergence are similar to those found in A. hemisphaerica with up to 27% nucleotide divergence in pairwise comparisons, and most sequences (16 in the SC lineage and 7 in the WP lineage) code for the same amino acid sequence. The most divergent sequence found is AvulSC_act09 (ten amino acid substitutions, including a stop codon); other four show two to three amino acid substitutions (AvulSC_act20, AvulSC_act17, AvulSC_act18, and AvulWP_act02). No nucleotide sequences are shared between isolates, but the most common coding sequence is the same across all four Arcella analyzed (59 of 73 sequences).
Actins in the Arcellinida
Maximum likelihood analysis at the nucleotide level of the 73 actin genes described in the present study reveals that instead of yielding monophyletic clades, the genes interdigitate between the three lineages (the two populations of A. hemisphaerica that we interpret as the same genetical lineage as well as the independent lineages A. vulgaris SC and A. vulgaris WP; fig. 4). These analyses were performed both including and excluding third positions. There is no correspondence between actin genealogy and morphospecies or SSU-rDNA relationship for the Arcella. Instead, the gene copies fall into two groups, with a well-supported split (BS = 96): Group 1 is paraphyletic, whereas Group 2 is monophyletic and falls within Group 1 (fig. 4). A phylogenetic analysis excluding third positions to minimize the effect of saturation reveals the same pattern (fig. 5a). The interdigitation indicates that gene duplication predated the divergence of these strains. Yet there is also evidence of independent gene copy expansion within the three lineages as evidenced by the shallow clustering of paralogs from within a lineage in the actin topology.
Fig. 4.

Most likely genealogy of the actin gene family in the genus Arcella inferred from maximum likelihood. For this reconstruction, we performed a RaxML analysis using the GTR model of evolution with 300 bootstrap replicates on the data set “Actin Alignment B,” which consists of 795 nucleotides; third positions are included. Genes are colored according to lineage. Most genes (59) code the exact same amino acid sequence. A minority of genes (14) encode divergent amino acid sequences; these are indicated by the number of AA substitutions. Sequences with an asterisk (*) represent putative pseudogenes. Thicker branches represent nodes that have >75% BS. All branches are drawn to scale. Dashed lines represent paraphyletic groupings.
FIG. 5.

Genealogy of actin gene families across the Amoebozoa, showing multiple independent expansion in different lineages. (A) nucleotide tree without third positions inferred from the “Actin Alignment A,” which consists of 179 sequences and 756 (after excluding third positions from a total of 1,134 sites) using maximum likelihood with the GTR model of substitution and running 650 bootstrap replicates (−lnL = 5991.952). (B) amino acid tree inferred from translated “Actin Alignment A,” which consists of 179 sequences and 378 amino acid sites, using maximum likelihood with the JTT model of substitution and running 1,000 bootstrap replicates (−lnL = 4392.092). Thick branches represent >75% BS for the backbone of the tree. All branches are drawn to scale. Dashed lines represent paraphyletic groupings.
Actins in the Amoebozoa
The maximum likelihood reconstruction of the actin gene tree recovers most major lineages with moderate/high support (fig. 5a): The Amoebidae, Dictyosteliida, Entamoebida, and Arcellinida are all monophyletic (BS > 75). The reconstruction based on amino acid sequences is topologically similar regarding the placement of Arcella actin paralogs with the reconstruction at the nucleotide level (fig. 5b). Collections of paralogs within each lineage appear to have expanded independently in each species in the Amoebozoa for which sufficient data exist (e.g., intense PCR study, EST analyses). In almost all cases, actin paralogs of a given species group together to the exclusion of paralogs in another species’ gene family. There are two exceptions: The D. purpureum set of actin genes is a monophyletic group that falls within the D. discoideum family, and in Arcella, the three lineages interdigitate, that is, no one isolate is monophyletic to the exclusion of others.
Codon Usage and Base Composition
We compared codon usage and base composition for sets of actin genes in lineages with three or more representative sequences (fig. 6). In the genus Arcella, there are two separate groups of genes based on codon usage, and these correspond to the Groups 1 and 2 recovered in the phylogenetic reconstruction (fig. 4): Group 1 is moderately biased with average ENC of 34.6, and Group 2 is less constrained with average ENC of 42.3 (fig. 6). Group 1 also has higher GC content in 4-fold degenerate sites, with an average of 65% compared with 44% in Group 2.
Fig. 6.

ENC versus GC content in 4-fold degenerate sites for actin gene families in the Amoebozoa. This analysis is based on a subset of “Actin Alignment B” and comprises 149 sequences and 795 base pairs. The three Arcella species are depicted in non-filled symbols. The slime molds represented by two Dictyostelium species and Physarum polycephalum are depicted in crosses and dashes. The Archamoebae, represented by two parasitic Entamoeba species, and the free-living Mastigamoeba balamuthi are depicted in grey-filled symbols. The parasitic Acanthamoeba castellani is depicted in grey-filled triangles. Most gene families are restricted to an area of low ENC as well as highly biased GC content, consistent with actins being highly expressed genes. The Arcella on the other hand are able to explore a more relaxed space regarding both ENC and GC content to the middle-upper area of the graph.
In contrast, sets of actin paralogs in the other Amoebozoa lineages analyzed are restricted to a range of codon usage that is biased and low, with ENC generally less than 30 (fig. 6). Base compositions for the actin gene are highly variable in the Amoebozoa: Mastigamoeba balamuthi has an average GC composition of 65% and E. dispar has 35%. These organisms have biased codon usage for the actin gene, probably reflecting GC bias in the genome.
Recombination among Actin Genes
We have searched for recombination among gene sequences in each Arcella lineage, using the online server GARD (table 4). For A. hemisphaerica, two putative points of recombination were detected (Kishino–Hasegawa test, P < 0.01). Analyzing trees for each partition allows determination of sequences that have likely recombined (table 4). Strikingly, sequences within Group 1 only recombine among themselves; the same is true for sequences in Group 2 with one exception: Ahem_act17 is a member of Group 2 and appears to have recombined with members of Group 1 for the first third of the sequence. For A. vulgaris WP, the one break point inferred by GARD is not statistically significant. For A. vulgaris SC, no inferred recombination break points were statistically significant. We are confident that these are historical and not artifactual recombination events because there are further point mutations in recombined segments. When using the same methodology for other sets of actin genes in Amoebozoa lineages, only the genus Dictyostelium shows statistically significant recombination between paralogs (table 4).
Table 4.
Number of Recombinants for Each Actin Gene Family in the Amoebozoa.
| # BP | #BP P < 0.05a | #Sequences | #Recombinants | |
| A. hemisphaerica | 2 | 2 | 15 (G1) | 4 |
| 30 (G2) | 4 | |||
| A. vulgaris WP | 1 | 0 | 1 (G1) | 0 |
| 7 (G2) | 2 | |||
| A. vulgaris SC | 0 | 0 | 1 (G1) | 0 |
| 19 (G2) | 0 | |||
| D. discoideum | 2 | 2 | 25 | 2 |
| D. purpureum | 1 | 1 | 11 | 3 |
| M. balamuthi | 0 | 0 | 12 | 0 |
| E. histolytica | 1 | 0 | 7 | 0 |
| E. dispar | 1 | 0 | 7 | 0 |
| Aca. castellani | 2 | 0 | 6 | 0 |
Note.—BP = number of inferred recombination points, G1 = Arcella Group 1 actins, G2 = Arcella Group 2 actins.
P values are calculated by the Kishino–Hasegawa test after break point (BP) inference by GARD.
Genetic Diversity Indices
We calculated genetic diversity indices to elucidate general patterns of molecular evolution (table 5). Arcella sequences were analyzed separately according to the two phylogenetic Groups 1 and 2 (fig. 4). Both Arcella groups show a high propensity for substitution, revealed by a high number of segregating sites per site (Group 1 S = 0.23; Group 2 S = 0.37). Additionally, Group 2 shows higher average nucleotide differences per site (π = 0.11) than Group 1 (π = 0.07). D. discoideum is the only other Amoebozoa that shows a comparable average pairwise distance, intermediate between Group 1 and Group 2 (π = 0.08).
Table 5.
Genetic Diversity Indices for Actin Gene Families Across the Amoebozoa.
| S | π Nucleotide (SD) | N | Source | |
| Arcella Group 2 | 0.37 | 0.11 (0.015) | 52 | PCR |
| D. discoideum | 0.27 | 0.08 (0.011) | 22 | Genome |
| Arcella Group 1 | 0.23 | 0.07 (0.011) | 17 | PCR |
| D. purpureum | 0.05 | 0.02 (0.004) | 11 | EST |
| Aca. castellani | 0.04 | 0.02 (0.005) | 6 | EST |
| M. balamuthi | 0.04 | 0.01 (0.004) | 13 | EST |
| E. dispar | 0.01 | 0.01 (0.003) | 6 | Genome |
| E. histolytica | 0.01 | 0.01 (0.003) | 7 | Genome |
Note.—S = number of segregating sites per site, π = average number of nucleotide differences per site, SD = standard deviation assuming free recombination, N = number of genes used to calculate indices, source = indicates whether sequences were obtained from Whole Genome Projects, EST Projects, or PCR-based experiments.
Discussion
The two main observations for the actin gene family in the genus Arcella are as follows: 1) The gene family is organized in two distinct groups whose members share similar patterns of molecular evolution and 2) there have been recent independent expansions within each group. To establish the pattern of molecular evolution, we first assessed the phylogenetic position of two isolates from each of the morphospecies, A. hemisphaerica and A. vulgaris. Analysis of both SSU-rDNA and sharing of identical actin gene sequences indicate that the two A. hemisphaerica isolates represent the same genetical lineage, whereas the two A. vulgaris isolates are independently evolving. The genus Arcella forms a monophyletic clade in maximum likelihood genealogies of the SSU-rDNA and actin genes (figs. 2 and 4). However, the two A. vulgaris morphospecies are not monophyletic, indicating that there might be more genetic divergence than seen at the phenotypical level. Though taxon sampling is limited here, these data also provide a framework for additional phylogenetic hypotheses. For example, the genus Argynnia previously assigned to Hyalospheniidae (Lara et al. 2008) is recovered here as a sister group to Arcella with moderate support (BS = 67). This might be indicative of an unpredicted relationship between some testate amoebae with chitinous shells (Arcella) and others with biomineralized siliceous plates (Argynnia).
The collection of actin gene copies in the genus Arcella is organized in two distinct genomic groups based on multiple lines of evidence. Phylogenetic analysis reveals a well-supported split in a paraphyletic Group 1 and a nested monophyletic Group 2 (fig. 4). Group 1 has lower codon usage and higher GC content (fig. 6) as well as lower substitution rates (table 5) than Group 2. Recombination inference indicates that members in each group recombine mostly among themselves (table 4). Although most of the classic literature lists testate amoebae as asexual organisms, evidence for meiosis (Mignot and Raikov 1992) validates our recombination inferences. Because recombination via unequal crossing-over is more likely within physically close segments, this pattern is consistent with two or more groups of tandemly arrayed actin paralogs in separate parts of the genome (different chromosomes or chromosomal regions).
The two groups of actin paralogs experience strong purifying selection as they maintain a common coding sequence. The majority of Arcella genes (59 of 73) encode exactly the same amino acid sequence, even though uncorrected nucleotide divergence reaches 29%. The slime mold D. discoideum presents a similar scenario: The core group of 17 highly expressed actins (Act8-group) have the same coding sequence and are separated in groups across four chromosomes (Joseph et al. 2008). However, there are two significant differences in D. discoideum compared with the pattern of actin evolution observed for Arcella spp. First, D. discoideum does not show a separation in two distinct groups of actins with respect to codon usage and compositional bias (fig. 6). Second, there is no evidence of maintenance of ancient paralogous groups within different species as D. discoideum sequences form a single clade. In contrast, the two distinct groups of Arcella sp. sequences are interdigitated showing that they predate the divergence of the three lines: A. hemisphaerica, A. vulgaris SC, and A. vulgaris WP.
The second main observation of this study is the evidence for recent and rapid duplications within each lineage and most likely within each group. Within Group 2, there are multiple closely related copies for each of the three lineages of Arcella studied: A. hemisphaerica, A. vulgaris SC, and A. vulgaris WP (fig. 4). Within Group 1, there are multiple closely related actin copies for the A. hemisphaerica, but only one copy for each of the two other lineages, which might reflect either incomplete sampling or really a large reduction in this group of actins for the two lineages. The recency of these gene family expansions is evidenced by the presence of frameshifting deletions in paralogs that have no additional amino acid substitutions (fig. 3). We consider these as an indication of recent recombination because a locus that is no longer useful should quickly accumulate mutations.
We propose a model consistent with our main observations: Arcella has a large collection of actin genes encoded in two distinct regions of the genome that evolve under strong purifying selection and yet are also expanding (fig. 7). The two groups are subject to different evolutionary pressures as evidenced by differing levels of codon usage. These two groups may be evolving under distinct regulatory constraints or the mutational background differs between different areas of the genome or both. At the same time, there are multiple independent expansions at the tips of the actin tree, especially within Group 2 where we see higher rates of recombination (fig. 3; table 4).
FIG. 7.

A hypothetical model for actin gene family evolution among species in the genus Arcella. The branching order for species is obtained from the SSU-rDNA reconstruction, and the branching order for actin paralogs is exactly as in figure 4. The first event depicted is the separation of actins in two genomic groups (Grey and Black), which predates the divergence of lineages. Following separation, each group is under distinct regulatory constraints. Perhaps, actins located in different areas are activated/deactivated following the life cycle, thus may be subject to different evolutionary pressures. Furthermore, speciation happens, with maintenance of the two actin groups in all three lineages. Within each lineage, there is a high level of independent duplications, the mechanism for which might be either a recombinational hotspot or a DRGR.
There are at least two mechanisms to explain the pattern of recent expansion: 1) Group 2 actins are in a recombination hotspot or 2) actins are the target of developmentally regulated genome rearrangements (DRGR). If Group 2 actins are in a recombination hotspot, we should expect the appearance and elimination of genes at a higher than usual rate. Somatic events that alter genomes of either specific cells or at specific life cycle stages are referred to as DRGR (Zufall et al. 2005). Arcella might show the kind of DRGR Zufall et al. (2005) classified as genome-wide rearrangements. The actins in this scenario would be amplified many times, as in a ciliate macronucleus, and might even reside on extrachromosomal pieces of DNA. Under this scenario, the expansion pattern observed in the tips of our tree (fig. 4) really depicts one genomic copy and many “extra” copies. Other Amoebozoa are known to have extrachromosomal rDNA (D. discoideum and E. histolytica). Additionally, Amo. proteus, which is more closely related to Arcella, has been shown to exhibit DNA synthesis outside of cell division (see Parfrey et al. 2008 for a review).
The pattern of actin evolution revealed for Arcella is unusual among eukaryotes (fig. 8). Other unicellular eukaryotes show either a limited number of actins encoding the same amino acid sequence (e.g., Entamoeba spp.) or a large number of actin gene copies with detectable positive selection for some members (e.g., Am. carterae). Animals and plants have multiple actin copies, all with divergent amino acid sequences attributed to adaptive evolution concerning tissue differentiation. Arcella has a large collection of genes that generally maintain the same coding sequence. Yet, actins within these amoebae appear to be evolving under varying tempos of gene duplication.
FIG. 8.
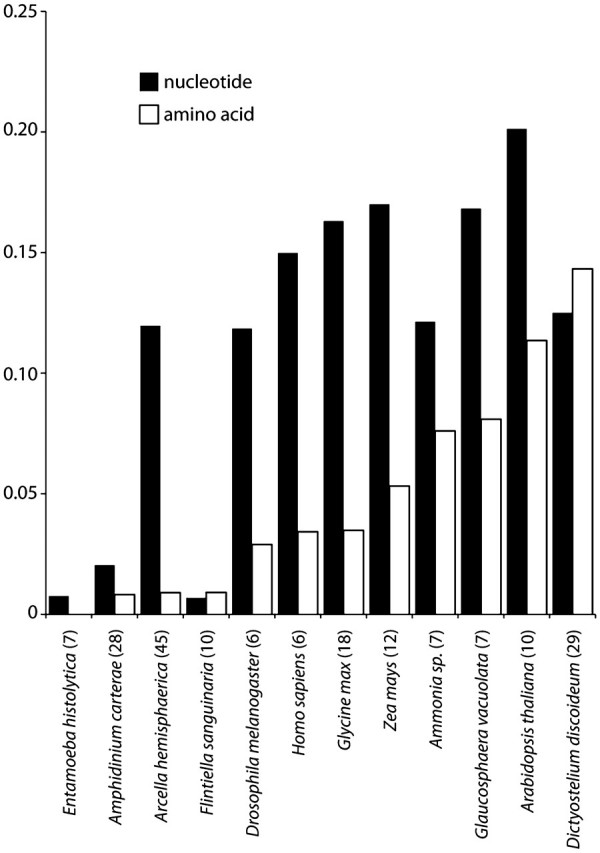
Average pairwise distances within actin gene paralogs for different eukaryotic taxa. The number of members in each family is indicated in parenthesis after the organism name on the x axis. The distances were calculated as uncorrected pairwise distances and then averaged over the number of actins in the taxon.
Supplementary Materials
Supplementary fig. 1, table 1, and alignments 1 and 2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We would like to thank Laura W. Parfrey, Micah Dunthorn, Yonas I. Tekle, Ben Normark, Michael Hood, and O. Roger Anderson for insightful discussions during this project. This work was supported by the National Institutes of Health (AREA award 1R15GM081865-01) and the National Science Foundation (OCE-0648713 and DEB 043115) to L.A.K. and the Conselho Nacional de Pesquisa e Desenvolvimento, Brazil (GDE200853/2007-4), to D.J.G.L.
References
- Bachvaroff TR, Place AR. From stop to start: tandem gene arrangement, copy number and trans-splicing sites in the dinoflagellate Amphidinium carterae. PLoS ONE. 2008;3:e2929. doi: 10.1371/journal.pone.0002929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya D, Aubry J, Twait EC, Jurk S. Actin gene duplication and the evolution of morphological complexity in land plants. J Phycol. 2000;36:813–820. [Google Scholar]
- Casillas S, Petit N, Barbadilla A. DPDB: a database for the storage, representation and analysis of polymorphism in the Drosophila genus. Bioinformatics. 2005;21:26–30. doi: 10.1093/bioinformatics/bti1103. [DOI] [PubMed] [Google Scholar]
- Chen MM, Shen XT. Nuclear actin and actin-related proteins in chromatin dynamics. Curr Opin Cell Biol. 2007;19:326–330. doi: 10.1016/j.ceb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Colwell RK. Estimates: Statistical estimation of species richness and shared species from samples. Version 8. Distributed by the author. Storrs (CT): Department of Ecology and Evolutionary Biology, University of Connecticut. 2006. [Google Scholar]
- Eichinger L, Pachebat JA, Glockner G, et al. (97 co-authors) The genome of the social amoeba Dictyostelium discoideum. Nature. 2005;435:43–57. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson HP. Evolution of the cytoskeleton. Bioessays. 2007;29:668–677. doi: 10.1002/bies.20601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flakowski J, Bolivar I, Fahrni J, Pawlowski J. Tempo and mode of spliceosomal intron evolution in actin of foraminifera. J Mol Evol. 2006;63:30–41. doi: 10.1007/s00239-005-0061-z. [DOI] [PubMed] [Google Scholar]
- Galtier N, Gouy M, Gautier C. Seaview and phylo_win: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- Goodson HV, Hawse WF. Molecular evolution of the actin family. J Cell Sci. 2002;115:2619–2622. doi: 10.1242/jcs.115.13.2619. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. Seaview version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- Hightower RC, Meagher RB. The molecular evolution of actin. Genetics. 1986;114:315–332. doi: 10.1093/genetics/114.1.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper SL, Thuma JB. Invertebrate muscles: muscle specific genes and proteins. Physiol Rev. 2005;85:1001–1060. doi: 10.1152/physrev.00019.2004. [DOI] [PubMed] [Google Scholar]
- Joseph JM, Fey P, Ramalingam N, Liu XI, Rohlfs M, Noegel AA, Möller-Taubenberger A, Glöckner G, Schleicher M. The actinome of Dictyostelium discoideum in comparison to actins and actin-related proteins from other organisms. PLoS ONE. 2008;3:e2654. doi: 10.1371/journal.pone.0002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judo MSB, Wedel AB, Wilson C. Stimulation and suppression of PCR-mediated recombination. Nucleic Acids Res. 1998;26:1819–1825. doi: 10.1093/nar/26.7.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Asimenos G, Toh H. Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol. 2009;537:39–64. doi: 10.1007/978-1-59745-251-9_3. [DOI] [PubMed] [Google Scholar]
- Kusakabe T, Araki I, Satoh N, Jeffery WR. Evolution of chordate actin genes: evidence from genomic organization and amino acid sequences. J Mol Evol. 1997;44:289–298. doi: 10.1007/pl00006146. [DOI] [PubMed] [Google Scholar]
- Lahr DJG, Katz LA. Reducing the impact of PCR-mediated recombination in molecular evolution and environmental studies using a new-generation high-fidelity DNA polymerase. Biotechniques. 2009;47:857–866. doi: 10.2144/000113219. [DOI] [PubMed] [Google Scholar]
- Lahr DJG, Lopes SGBC. Evaluating the taxonomic identity in 4 species of the lobose testate amoebae genus Arcella Ehrenberg, 1832. Acta Protozool. 2009;48:127–142. [Google Scholar]
- Lara E, Heger TJ, Ekelund F, Lamentowicz M, Mitchell EAD. Ribosomal RNA genes challenge the monophyly of the Hyalospheniidae (Amoebozoa: Arcellinida) Protist. 2008;159:165–176. doi: 10.1016/j.protis.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Lara E, Heger TJ, Mitchell EAD, Meisterfeld R, Ekelund F. SSU-rRNA reveals a sequential increase in shell complexity among the Euglyphid testate amoebae (Rhizaria: Euglyphida) Protist. 2007;158:229–237. doi: 10.1016/j.protis.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Maddison WP, Maddison DR. Macclade. v. 4.0.8. Sunderland (MA): Sinauer Associates; 2005. [Google Scholar]
- McDowell JM, Huang SR, McKinney EC, An YQ, Meagher RB. Structure and evolution of the actin gene family in Arabidopsis thaliana. Genetics. 1996;142:587–602. doi: 10.1093/genetics/142.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medlin L, Elwood HJ, Stickel S, Sogin ML. The characterization of enzymatically amplified eukaryotic 16s-like rRNA-coding regions. Gene. 1988;71:491–499. doi: 10.1016/0378-1119(88)90066-2. [DOI] [PubMed] [Google Scholar]
- Mignot J-P, Raikov IB. Evidence for meiosis in the testate amoeba Arcella. J Eukaryot Microbiol. 1992;39:287–289. [Google Scholar]
- Miller MA, Holder MT, Vos R, Midford PE, Liebowitz T, Chan L, Hoover P, Warnow T. The CIPRES portals. 2009. Cyberinfrastructure for Phylogenetic Research (CIPRES). [cited 2009 Aug 04]. Available from: http://www.phylo.org/sub_sections/portal. [Google Scholar]
- Misof B, Misof K. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst Biol. 2009;58:21–34. doi: 10.1093/sysbio/syp006. [DOI] [PubMed] [Google Scholar]
- Neron B, Menager H, Maufrais C, Joly N, Maupetit J, Letort S, Carrere S, Tuffery P, Letondal C. Mobyle: a new full web bioinformatics framework. Bioinformatics. 2009;25:3005–3011. doi: 10.1093/bioinformatics/btp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev SI, Berney C, Petrov NB, Mylnikov AP, Fahrni JF, Pawlowski J. Phylogenetic position of Multicilia marina and the evolution of Amoebozoa. Int J Syst Evol Microbiol. 2006;56:1449–1458. doi: 10.1099/ijs.0.63763-0. [DOI] [PubMed] [Google Scholar]
- Nikolaev SI, Mitchell EA, Petrov NB, Berney C, Fahrni J, Pawlowski J. The testate lobose amoebae (Order Arcellinida Kent, 1880) finally find their home within Amoebozoa. Protist. 2005;156:191–202. doi: 10.1016/j.protis.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Parfrey LW, Barbero E, Lasser E, Dunthorn M, Bhattacharya D, Patterson DJ, Katz LA. Evaluating support for the current classification of eukaryotic diversity. PLoS Genetics. 2006;2:2062–2073. doi: 10.1371/journal.pgen.0020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Grant J, Tekle YI, Lasek-Nesselquist E, Morrison HG, Sogin ML, Patterson DJ, Katz LA. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst Biol. 2010 doi: 10.1093/sysbio/syq037. Advance Access published July 28, 2010, doi:10.1093/sysbio/syq037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Lahr DJG, Katz LA. The dynamic nature of eukaryotic genomes. Mol Biol Evol. 2008;25:787–794. doi: 10.1093/molbev/msn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson DJ. The diversity of eukaryotes. Am Nat. 1999;154:S96–S124. doi: 10.1086/303287. [DOI] [PubMed] [Google Scholar]
- Pawlowski J. The twilight of Sarcodina: a molecular perspective on the polyphyletic origin of amoeboid protists. Protistology. 2008;5:22. [Google Scholar]
- Pawlowski J, Burki F. Untangling the phylogeny of amoeboid protists. J Eukaryot Microbiol. 2009;56:16–25. doi: 10.1111/j.1550-7408.2008.00379.x. [DOI] [PubMed] [Google Scholar]
- Peden JF. Analysis of codon usage. [thesis]. [United Kingdom]: University of Nottingham; 1999. [Google Scholar]
- Pederson T. As functional nuclear actin comes into view, is it globular, filamentous, or both? J Cell Biol. 2008;180:1061–1064. doi: 10.1083/jcb.200709082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21:2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Posada D, Gravenor MB, Woelk CH, Frost SDW. Automated phylogenetic detection of recombination using a genetic algorithm. Mol Biol Evol. 2006;23:1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- Porter SM, Meisterfeld R, Knoll AH. Vase-shaped microfossils from the neoproterozoic chuar group, grand canyon: a classification guided by modern testate amoebae. J Paleontol. 2003;77:409–429. [Google Scholar]
- Reisler E, Egelman EH. Actin structure and function: what we still do not understand. J Biol Chem. 2007;282:36133–36137. doi: 10.1074/jbc.R700030200. [DOI] [PubMed] [Google Scholar]
- Smirnov A, Nassonova E, Berney C, Fahrni J, Bolivar I, Pawlowski J. Molecular phylogeny and classification of the lobose amoebae. Protist. 2005;156:129. doi: 10.1016/j.protis.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Smith HG, Bobrov A, Lara E. Diversity and biogeography of testate amoebae. Biodivers Conserv. 2008;17:329–343. [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RaxML web servers. Syst Biol. 2008;57:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland (MA): Sinauer Associates; 2003. [Google Scholar]
- Tekle YI, Grant J, Anderson OR, Nerad TA, Cole JC, Patterson DJ, Katz LA. Phylogenetic placement of diverse amoebae inferred from multigene analyses and assessment of clade stability within 'Amoebozoa' upon removal of varying rate classes of SSU-rDNA. Mol Phylogenet Evol. 2008;47:339–352. doi: 10.1016/j.ympev.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Tekle YI, Grant J, Cole JC, Nerad TA, Anderson OR, Patterson DJ, Katz LA. A multigene analysis of Corallomyxa tenera sp. nov. suggests its membership in a clade that includes Gromia, Haplosporidia and Foraminifera. Protist. 2007;158:457–472. doi: 10.1016/j.protis.2007.05.002. [DOI] [PubMed] [Google Scholar]
- van den Ent F, Amos LA, Lowe J. Prokaryotic origin of the actin cytoskeleton. Nature. 2001;413:39–44. doi: 10.1038/35092500. [DOI] [PubMed] [Google Scholar]
- Wade RH, Garcia-Saez I, Kozielski F. Structural variations in protein superfamilies: actin and tubulin. Mol Biotechnol. 2009;42:49–60. doi: 10.1007/s12033-008-9128-6. [DOI] [PubMed] [Google Scholar]
- Wu M, Comeron JM, Yoon HS, Bhattacharya D. Unexpected dynamic gene family evolution in algal actins. Mol Biol Evol. 2009;26:249–253. doi: 10.1093/molbev/msn263. [DOI] [PubMed] [Google Scholar]
- Zufall RA, Robinson T, Katz LA. Evolution of developmentally regulated genome rearrangements in eukaryotes. J Exp Zool B Mol Dev Evol. 2005;304B:448–455. doi: 10.1002/jez.b.21056. [DOI] [PubMed] [Google Scholar]