

Abstract
The acylphloroglucinol hyperforin (Hyp) from St. John's wort possesses anti-inflammatory and anti-carcinogenic properties which were ascribed among others to the inhibition of 5-lipoxygenase. Here, we investigated whether Hyp also interferes with prostanoid generation in biological systems, particularly with key enzymes participating in prostaglandin (PG)E2 biosynthesis, i.e., cyclooxygenases (COX)-1/2 and microsomal PGE2 synthase (mPGES)-1 which play key roles in inflammation and tumorigenesis. Similar to the mPGES-1 inhibitors MK-886 and MD-52, Hyp significantly suppressed PGE2 formation in whole blood assays starting at 0.03–1 μM, whereas the concomitant generation of COX-derived 12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid, thromboxane B2, and 6-keto PGF1α was not significantly suppressed up to 30 μM. In cell-free assays, Hyp efficiently blocked the conversion of PGH2 to PGE2 mediated by mPGES-1 (IC50 = 1 μM), and isolated COX enzymes were not (COX-2) or hardly (COX-1) suppressed. Intraperitoneal (i.p.) administration of Hyp (4 mg kg−1) to rats impaired exudate volume and leukocyte numbers in carrageenan-induced pleurisy associated with reduced PGE2 levels, and Hyp (given i.p.) inhibited carrageenan-induced mouse paw edema formation (ED50 = 1 mg kg−1) being superior over indomethacin (ED50 = 5 mg kg−1). We conclude that the suppression of PGE2 biosynthesis in vitro and in vivo by acting on mPGES-1 critically contributes to the anti-inflammatory efficiency of Hyp.
Keywords: hyperforin, St. John's wort, prostaglandin E2, cyclooxygenase, inflammation
Introduction
Hyperforin (Hyp), a polyprenylated acylphloroglucinol (Figure 1A), is assumed to be one of the main active components of St. John's wort, which is frequently used for the treatment of mild to moderate depressions (Muller, 2003). Besides its antidepressant activity, Hyp also exerts potent anti-inflammatory and anti-tumoral effects (Medina et al., 2006). Hyp was shown to reduce tumor cell growth (Schempp et al., 2002; Hostanska et al., 2003), cancer invasion, metastasis (Dona et al., 2004), angiogenesis (Martinez-Poveda et al., 2005), and lymphatic capillary growth (Rothley et al., 2009). On the cellular level, Hyp decreases proliferation rates and causes apoptosis of leukemic cells (Schempp et al., 2002; Quiney et al., 2006) and various cancer cell lines (Schempp et al., 2002; Hostanska et al., 2003; Dona et al., 2004), and also inhibits proliferation of non-transformed T lymphocytes (Schempp et al., 2000). In regard of its anti-inflammatory potential, Hyp blocks several pro-inflammatory functions of leukocytes in vitro such as chemotaxis and chemoinvasion (Dell'Aica et al., 2007; Lorusso et al., 2009), suppresses receptor-mediated Ca2+-mobilization and eicosanoid release in leukocytes (Albert et al., 2002; Feisst and Werz, 2004), and down-regulates effector functions of activated T lymphocytes (Cabrelle et al., 2008). In vivo, Hyp reduced croton-oil-induced ear edema in mice after topical application (Sosa et al., 2007), impaired acute neutrophil recruitment and enhanced resolution in a pulmonary bleomycin-induced inflammation model reducing consequent fibrosis (Dell'Aica et al., 2007), and suppressed carrageenan-induced rat pleurisy when given intraperitoneally (i.p.; Feisst et al., 2009). Also for humans, topical treatment of atopic dermatitis with a Hypericum cream, standardized to 1.5% Hyp, revealed significant therapeutic efficacy (Schempp et al., 2003). However, despite these well-recognized anti-carcinogenic and anti-inflammatory effects, the underlying molecular mechanisms and targets of Hyp are incompletely understood.
Figure 1.

Effects of hyperforin on prostanoid formation in LPS-stimulated human whole blood. (A) Chemical structure of Hyp. (B,C) Heparinized human whole blood, treated with 1 μM TX synthase inhibitor CV4151 and 50 μM aspirin, was pre-incubated with the indicated agents or vehicle (DMSO) for 5 min at room temperature, and then, prostanoid formation was induced by addition of 10 μg ml−1 LPS. After 5 h at 37°C, PGE2 (B) was extracted from plasma by RP-18 solid phase extraction, separated by RP-HPLC, and quantified by ELISA as described. 6-Keto PGF1α (C) and TXB2 (D) were directly determined in the plasma by ELISA. The 100% values correspond to prostanoid levels in the range of 14–27 ng ml−1 PGE2, 3–5 ng ml−1 6-keto PGF1α, and 47–64 ng ml−1 TXB2 in the individual experiments, respectively. MK-886 (30 μM), MD-52 (6 μM), indomethacin (Indo, 50 μM), and celecoxib (Cele, 20 μM) were used as controls. Data are given as mean ± SE, n = 3–4, *p < 0.05, ***p < 0.001 vs. vehicle (0.1% DMSO) control, ANOVA + Tukey HSD post hoc tests.
The lipid mediator prostaglandin (PG)E2 is a key player in inflammation, pain, fever, and cancer but is also known to regulate physiological functions in the gastrointestinal tract, in the kidney, and in the immune and nervous system (Sugimoto and Narumiya, 2007). PGE2 is formed from arachidonic acid (AA) by cyclooxygenase (COX)-catalyzed synthesis of PGH2 and further transformation by PGE2 synthases (Samuelsson et al., 2007). The microsomal PGE2 synthase (mPGES)-1 is induced by pro-inflammatory stimuli and is responsible for excessive PGE2 generation connected to pathologies (Jakobsson et al., 1999). Co-expression studies indicate a preferred functional coupling between mPGES-1 and COX-2 (Murakami and Kudo, 2006), and data from mPGES-1 knockout mice and animal studies with selective mPGES-1 inhibitors suggest key roles of mPGES-1 in inflammation, pain, fever, atherosclerosis, and tumorigenesis (Samuelsson et al., 2007; Friesen and Mancini, 2008; Koeberle and Werz, 2009).
We have previously shown that Hyp inhibits leukotriene biosynthesis in neutrophils (IC50 = 1–2 μM) by interference with the C2-like domain of 5-lipoxygenase (5-LO), and we observed an inhibitory effect of Hyp also on COX-1 activity in human platelets (IC50 = 3 μM; Albert et al., 2002; Feisst et al., 2009). However, COX-2 activity (measured as 6-keto PGF1α synthesis) in monocytic Mono Mac 6 cells was not affected up to 30 μM Hyp. This is surprising because Hammer et al. (2007) recently demonstrated that Hyp inhibits the release of PGE2 from lipopolysaccharide (LPS)-stimulated murine (RAW264.7) macrophages, a process coupled to COX-2 as well. Here, we identified mPGES-1 as functional target of Hyp and suggest that interference with mPGES-1 suppresses PGE2 biosynthesis in vivo and contributes to the anti-inflammatory effectiveness in related animal models.
Materials and Methods
Reagents
Hyperforin dicyclohexylammonium salt and the Hypericum perforatum extract were obtained from Dr. Willmar Schwabe GmbH & Co (Karlsruhe, Germany), respectively. The compounds and the extract were dissolved in dimethyl sulfoxide (DMSO) and kept in the dark at −20°C, and freezing–thawing cycles were kept to a minimum. For animal studies, Hyp was dissolved in DMSO and diluted with saline achieving a final DMSO concentration of 2–4%. The thromboxane (TX) synthase inhibitor CV4151 (Kato et al., 1985) and the mPGES-1 inhibitor 2-(2-chlorophenyl)-1H-phenanthro[9,10-d]-imidazole (MD-52; Côté et al., 2007) were kindly provided by Dr. S. Laufer (University of Tübingen, Germany) and Dr. M. Schubert-Zsilavecz (University of Frankfurt, Germany), respectively. The antibody against human mPGES-1 was from Cayman Chemical (Ann Arbor, MI, USA), antibodies against COX-2 and β-actin were obtained from Sigma-Aldrich (Deisenhofen, Germany) and the antibody against cleaved poly(ADP-ribose)polymerase (PARP) was purchased from Cell Signaling Technology (Danvers, MA, USA). Materials used: DMEM high glucose (4.5 g l−1) medium, penicillin, streptomycin, trypsin–EDTA solution, sodium pyruvate, PAA (Coelbe, Germany); PGH2, Larodan (Malmö, Sweden); 11β-PGE2, MK-886, PGB1, human recombinant COX-2, ovine isolated COX-1, Cayman Chemical (Ann Arbor, MI, USA). λ-Carrageenan type IV isolated from Gigartina aciculaire and G. pistillata was purchased from Sigma-Aldrich (Milan, Italy). 3H-PGE2 was from PerkinElmer Life Sciences (Milan, Italy) and PGE2 antibody from Sigma-Aldrich (Milan, Italy). All other chemicals were obtained from Sigma-Aldrich (Deisenhofen, Germany) unless stated otherwise.
Cells and cell viability assay
Human lung carcinoma A549 cells were cultured in DMEM high glucose (4.5 g l−1) medium supplemented with heat-inactivated fetal calf serum (FCS, 10%, v v−1), penicillin (100 U ml−1), and streptomycin (100 μg ml−1) at 37°C in a 5% CO2 incubator. After 3 days, confluent cells were detached using 1 × trypsin–EDTA solution and reseeded at 2 × 106 cells in 20 ml medium in 175 cm2 flasks. Cell viability was assessed using the colorimetric thiazolyl blue tetrazolium bromide dye reduction assay (MTT assay). A549 cells (4 × 104 cells per 100 μl medium) were plated into a 96-well microplate and incubated at 37°C and 5% CO2 for 16 h. Then, Hyp (30 μM) was added, and the samples were incubated for another 5 h. Thiazolyl blue tetrazolium bromide (20 μl, 5 mg ml−1) was added and the incubations were continued for 4 h. The formazan product was solubilized with sodium dodecylsulfate (SDS, 10%, m v−1 in 20 mM HCl), and the absorbance was measured at 595 nm relative to the absorbance of vehicle (DMSO)-treated control cells using a multiwell scanning spectrophotometer (Victor3 plate reader, PerkinElmer, Rodgau-Juegesheim, Germany).
Animals
Male adult CD1 mice (25–35 g, Harlan, Milan, Italy) and Wistar Han rats (190–200 g, Harlan, Milan, Italy) were housed in a controlled environment and provided with standard rodent chow and water. Animal care complied with Italian regulations on protection of animals used for experimental and other scientific purpose (Ministerial Decree 116192) as well as with the European Economic Community regulations (Official Journal of E.C. L 358/1 12/18/1986).
Determination of PGE2, 6-keto PGF1α, and TXB2 formation in LPS-stimulated human whole blood
Peripheral blood from healthy adult volunteers, who had not received any medication for at least 2 weeks under informed consent, was obtained by venepuncture and collected in syringes containing heparin (20 U ml−1). For determination of PGE2 and 6-keto PGF1α, aliquots of whole blood (0.8 ml) were mixed with the TX synthase inhibitor CV4151 (1 μM) and with aspirin (50 μM) to establish experimental conditions where prostanoids are essentially produced via the COX-2 pathway (Koeberle et al., 2008). For the determination of TXB2, whole blood aliquots were not pretreated with aspirin and CV4151. A total volume of 1 ml was adjusted with sample buffer (10 mM potassium phosphate buffer pH 7.4, 3 mM KCl, 140 mM NaCl, and 6 mM D-glucose). After pre-incubation with the indicated compounds for 5 min at room temperature, the samples were stimulated with LPS (10 μg ml−1) for 5 h at 37°C. Prostanoid formation was stopped on ice, the samples were centrifuged (2300 × g, 10 min, 4°C), and 6-keto PGF1α and TXB2 were quantified in the supernatant using a 6-keto PGF1α or TXB2 high sensitivity EIA kit (Assay Designs, Ann Arbor, MI, USA) according to the manufacturer's protocol. PGE2 was determined after solid phase extraction and HPLC separation using a PGE2 high sensitivity EIA kit (Assay Designs, Ann Arbor, MI, USA) as described (Koeberle et al., 2008). The mPGES-1 inhibitors MK-886 (30 μM) and MD52 (6 μM), the COX-1/2 inhibitor indomethacin (50 μM) and the COX-2-selective inhibitor celecoxib (20 μM) were used as control. Care should be taken when interpreting the controls in respect to COX-1/2 isoenzyme specificity which was not further evaluated for our experimental settings.
Determination of prostanoid formation from exogenous AA in human whole blood
Heparinized human blood, supplemented with penicillin (100 U ml−1) and streptomycin (100 μg ml−1), was treated with 10 μg ml−1 LPS for 16 h at 37°C and 5% CO2. Then, CV4151 (1 μM) was added, and after pre-incubation with the indicated compounds for 10 min at 37°C, prostanoid formation was initiated by addition of 20 μM AA. In contrast to the LPS-stimulated whole blood assay described above, whole blood aliquots were not pretreated with aspirin (inhibition of COX-1 product formation) because both COX-1 and -2 are described to contribute to mPGES-1-dependent PGE2 synthesis at high AA concentrations (Murakami et al., 2000). PGE2 or 6-keto PGF1α formation within 10 min was determined as described for LPS-stimulated whole blood. Calculated prostanoid levels were corrected by the amount of PGE2 formed during pre-stimulation with LPS.
For determination of the COX product 12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid (12-HHT), freshly drawn human blood (2 ml) was pre-incubated with the indicated compounds at 37°C for 10 min, and formation of 12-HHT was initiated by addition of 30 μM Ca2+-ionophore A23187 plus 100 μM AA. After 10 min at 37°C, formed 12-HHT was extracted from plasma and analyzed by HPLC using PGB1 as internal standard as described (Capdevila et al., 1995).
Activity assays of isolated COX-1 and COX-2
Inhibition of the activities of isolated ovine COX-1 and human COX-2 was performed as described (Koeberle et al., 2008). Briefly, purified COX-1 (ovine, 50 units) or COX-2 (human recombinant, 20 units) were diluted in 1 ml reaction mixture containing 100 mM Tris buffer pH 8, 5 mM glutathione, 5 μM hemoglobin, and 100 μM EDTA at 4°C and pre-incubated with the test compounds for 5 min. Samples were pre-warmed for 60 s at 37°C, and AA (5 μM for COX-1, 2 μM for COX-2) was added to start the reaction. After 5 min at 37°C, the COX product 12-HHT was extracted and then analyzed by HPLC as described (Albert et al., 2002).
Preparation of crude mPGES-1 in microsomes of A549 cells and determination of PGE2 synthase activity
Preparation of A549 cells and determination of mPGES-1 activity was performed as described previously (Koeberle et al., 2008). In brief, cells were incubated for 16 h at 37°C and 5% CO2, the medium was replaced, 1 ng ml−1 interleukin (IL)-1β was added, and cells were incubated for another 48 h. After sonication, the homogenate was subjected to differential centrifugation at 10,000 × g for 10 min and 174,000 × g for 1 h at 4°C. The pellet (microsomal fraction) was resuspended in 1 ml homogenization buffer (0.1 M potassium phosphate buffer pH 7.4, 1 mM phenylmethanesulfonyl fluoride, 60 μg ml−1 soybean trypsin inhibitor, 1 μg ml−1 leupeptin, 2.5 mM glutathione, and 250 mM sucrose), and the total protein concentration was determined. Microsomal membranes were diluted in potassium phosphate buffer (0.1 M, pH 7.4) containing 2.5 mM glutathione to a final protein concentration of approx. 70 μg ml−1. Test compounds or vehicle were added, and after 15 min at 4°C, the reaction (100 μl total volume) was initiated by addition of PGH2 (20 μM, final concentration). After 1 min at 4°C, the reaction was terminated using stop solution (100 μl; 40 mM FeCl2, 80 mM citric acid, and 10 μM of 11β-PGE2 as internal standard). PGE2 was separated by solid phase extraction and analyzed by HPLC as described (Koeberle et al., 2008).
SDS-page and western blot
Cells (4 × 106 cells) were resuspended in 50 μl PBS buffer pH 7.2, mixed with the same volume of 2 × SDS/PAGE sample loading buffer (20 mM Tris–HCl, pH 8, 2 mM EDTA, 5% (m v−1) SDS, and 10% (v v−1) β-mercaptoethanol), and boiled for 5 min at 95°C. Aliquots (20 μl) corresponding to equivalents of 0.8 × 106 cells were mixed with 4 μl glycerol/0.1% bromophenol blue (1:1, v v−1), and proteins were separated by SDS-PAGE. After electroblotting to nitrocellulose membrane (GE Healthcare, Munich, Germany) and blocking with 5% BSA for 1 h at room temperature, membranes were washed and incubated with primary antibodies overnight at 4°C. The membranes were washed and incubated with a 1:1000 dilution of alkaline phosphatase-conjugated immunoglobulin G for 3 h at room temperature. After washing, proteins were visualized with nitro blue tetrazolium and 5-bromo-4-chloro-3-indolylphosphate.
Carrageenan-induced paw edema in mice
Mice were divided into groups (n = 10 for each group) and lightly anesthetized with enflurane 4% mixed with O2, 0.5 l min−1, N2O 0.5 l min−1. Each group of animals received subplantar administration of saline (0.05 ml) or of λ-carrageenan type IV 1% (w v−1, 0.05 ml) in saline. The paw was marked in order to immerge it always at the same extent in the measurement chamber. The volume was measured by using a hydroplethismometer, specially modified for small volumes (Ugo Basile, Milan, Italy), immediately before subplantar injection and 2, 4, and 6 h thereafter (Posadas et al., 2004). The assessment of paw volume was performed always in double blind and by the same operator. The increase in paw volume was calculated by subtracting the initial paw volume (basal) to the paw volume measured at each time point.
Carrageenan-induced pleurisy in rats
Rats were anesthetized with enflurane 4% mixed with O2, 0.5 l min−1, N2O 0.5 l min−1 and submitted to a skin incision at the level of the left sixth intercostal space. The underlying muscle was dissected, and saline (0.2 ml) or λ-carrageenan type IV 1% (w v−1, 0.2 ml) was injected into the pleural cavity. The skin incision was closed with a suture, and the animals were allowed to recover. At 4 h after the injection of carrageenan, the animals were killed by inhalation of CO2. The chest was carefully opened, and the pleural cavity was rinsed with 2 ml saline solution containing heparin (5 U ml−1). The exudate and washing solution were removed by aspiration, and the total volume was measured. Any exudate that was contaminated with blood was discarded. The amount of exudate was calculated by subtracting the volume injected (2 ml) from the total volume recovered. Leukocytes in the exudate were resuspended in phosphate buffered saline (PBS) and counted with an optical light microscope in a Burker's chamber after vital trypan blue staining.
The amount of PGE2 in the supernatant of centrifuged exudate (800 × g for 10 min) was assayed by radioimmunoassay according to manufacturer's protocol. The results are expressed as nanograms per rat and represent the mean ± SE of 10 rats.
Statistics
Data are expressed as mean ± SE. IC50 values were calculated by nonlinear regression using SigmaPlot 9.0 (Systat Software Inc., San Jose, USA) one site binding competition. The program Graphpad Instat (Graphpad Software Inc., San Diego, CA, USA) was used for statistical comparisons. Statistical evaluation of the data was performed by one-way or two-way ANOVAs for independent or correlated samples followed by Tukey HSD post hoc tests. A P value < 0.05 (*) was considered significant.
Results
Effects of hyperforin on prostanoid formation in human whole blood
Hyperforin was recently shown to inhibit the production of PGE2 in LPS-stimulated RAW264.7 mouse macrophages (Hammer et al., 2007). Here, we investigated the effects of Hyp on prostanoid formation in human whole blood pre-incubated with Hyp for 5 min and stimulated with LPS for 5 h. Hyp concentration-dependently suppressed PGE2 formation with an apparent IC50 of approx. 3 μM reaching significant inhibition at 1 μM (25 ± 3% inhibition; Figure 1B). Acute (5 h) cytotoxic effects of Hyp (up to 30 μM) can be excluded as determined by an A549 cell viability assay measuring mitochondrial dehydrogenase activity (see Materials and Methods, data not shown). The selective mPGES-1 inhibitor MD-52 (Côté et al., 2007) and the combined 5-LO-activating protein (FLAP), COX-1 and mPGES-1 inhibitor MK-886 (Claveau et al., 2003; Koeberle et al., 2009b) were used as reference (see also Koeberle et al., 2008). PGE2 levels were neither completely suppressed by the mPGES-1 inhibitors nor by Hyp, even not at high Hyp concentrations (30 μM) or prolonged incubation times (24 h, data not shown). In contrast, the COX-1/2 inhibitor indomethacin (50 μM) and the COX-2 selective celecoxib (20 μM) effectively reduced PGE2 (Figure 1B) almost to the level of the unstimulated control (PGE2: 14.2 ± 8.4%). Celecoxib and/or indomethacin also inhibited the formation of 6-keto PGF1α (the stable metabolite of PGI2, Figure 1C) and TXB2 (Figure 1D) as expected, whereas Hyp (up to 30 μM), MD-52, and MK-886 failed to suppress the formation of 6-keto PGF1α (Figure 1C) and TXB2 (Figure 1D) under the same experimental conditions. Together, unlike COX inhibitors but as like mPGES-1 inhibitors, Hyp selectively suppresses PGE2 formation but not the synthesis of other prostanoids such as 6-keto PGF1α or TXB2.
Hyperforin may suppress PGE2 formation by interference with LPS-signaling, mPGES-1 expression or liberation of AA as substrate for COX enzymes. Thus, heparinized human whole blood was first incubated with LPS for 16 h to induce the expression of COX-2 and mPGES-1 (in contrast to constitutively expressed COX-1 and cPGES). Then, the blood was pre-incubated with Hyp for 10 min, and prostanoid formation was initiated by addition of 20 μM AA. After 10 min at 37°C, generation of PGE2 and 6-keto PGF1α was determined in plasma as described above. PGE2 formation was potently inhibited by Hyp in a concentration-dependent manner with an IC50 of 0.25 μM reaching significance at 30 nM (Figure 2A), whereas formation of 6-keto PGF1α was not significantly affected up to 30 μM (Figure 2B), again indicating selective suppression of PGE2 biosynthesis by Hyp. Note that Hyp (3–30 μM) was more efficient in suppressing PGE2 synthesis after long-term pre-stimulation with LPS (induction of COX-2 and mPGES-1, Figure 2A) than during short term stimulation (Figure 1B) supporting that Hyp interferes with the inducible COX-2/mPGES-1 pathway.
Figure 2.

Effects of hyperforin on arachidonic acid-induced prostanoid formation in human whole blood. (A,B) Heparinized human whole blood was treated with 10 μg ml−1 LPS for 16 h at 37°C and 5% CO2, supplemented with TX synthase inhibitor CV4151 (1 μM), and pre-incubated with Hyp or vehicle (DMSO) for 10 min at 37°C. (A) Then, PGE2 formation was initiated with 20 μM AA, and PGE2 formed within 10 min was separated by RP-HPLC and quantified by ELISA. The 100% value corresponds to PGE2 levels in the range of 18–31 ng ml−1 in the individual experiments, respectively. (B) 6-keto PGF1α was directly determined in the plasma by ELISA. The 100% value corresponds to 6-keto PGF1α levels in the range of 4–7 ng ml−1. Indomethacin (Indo, 50 μM) and celecoxib (Cele, 20 μM) were used as controls. (C) 12-HHT formation in whole blood. Heparinized whole blood was pre-incubated with Hyp or vehicle (DMSO) for 10 min, and AA (100 μM) and Ca2+-ionophore (30 μM) were added to induce 12-HHT product formation. After 10 min at 37°C, 12-HHT was extracted form the plasma by RP-18 solid phase extraction and analyzed by RP-HPLC as described. The 100% value corresponds to 1.5–2.4 μg ml−1 12-HHT. Indomethacin (Indo, 20 μM) and aspirin (ASA, 30 μM) were used as controls. Data are given as mean ± SE, n = 3–5, *p < 0.05, **p < 0.01 or ***p < 0.001 vs. vehicle (0.1% DMSO) control, ANOVA + Tukey HSD post hoc tests.
In order to assess the effects of Hyp on the activity of COX-1 in whole blood (constitutively expressed in platelets), freshly withdrawn human blood (no incubation with LPS) was pre-incubated with Hyp for 10 min at 37°C, treated with Ca2+-ionophore and 100 μM AA for another 10 min, and the formation of 12-HHT (mainly derived from platelet COX-1) was determined. Hyp (up to 30 μM) failed to suppress 12-HHT formation under these assay conditions, whereas indomethacin and aspirin almost completely blocked it (Figure 2C). In conclusion, the data suggest that Hyp potently and selectively blocks PGE2 formation in whole blood without markedly affecting COX-1/2.
Effects of hyperforin on the activity of isolated COX-1 and COX-2
We determined the direct interference of Hyp with the catalytic activity of isolated COX enzymes. Purified ovine COX-1 and human recombinant COX-2 were pre-incubated with Hyp for 5 min prior to addition of AA (5 μM for COX-1, 2 μM for COX-2). Formation of 12-HHT, the major COX-1/2-derived product under these experimental conditions (Capdevila et al., 1995), was analyzed by RP-HPLC. Hyp concentration-dependently reduced 12-HHT formation by COX-1 with an IC50 = 12 μM (Figure 3A), whereas COX-2 was not significantly inhibited up to 30 μM (Figure 3B). The reference compounds indomethacin (10 μM) and celecoxib (5 μM) potently inhibited 12-HHT formation, as expected.
Figure 3.

Effects of hyperforin on the activity of isolated COX-1 and -2. Purified ovine COX-1 [(A) 50 units] or human recombinant COX-2 [(B) 20 units] were added to a COX reaction mix containing 5 mM glutathione. The COX enzymes were pre-incubated with the test compounds for 5 min, and then, the reaction was started with 5 μM (COX-1) or 2 μM (COX-2) AA. After 5 min at 37°C, the formation of 12-HHT was determined by RP-HPLC as described. The 100% values in individual experiments are in the range of 110–140 ng ml−1 and 90–120 ng ml−1 12-HHT for COX-1 and COX-2 assays, respectively. Indomethacin (Indo, 10 μM) and celecoxib (Cele, 5 μM) were used as controls. Data are given as mean ± SE, n = 3, *p < 0.05, ***p < 0.001 vs. vehicle (0.1% DMSO) control, ANOVA + Tukey HSD post hoc tests.
Inhibition of mPGES-1 activity by hyperforin
Because Hyp potently inhibited PGE2 formation in whole blood from AA but failed to block COX-2, inhibition of PGE2 synthesis downstream of COX-2 (i.e., interference with PGE2 synthases) appeared reasonable. mPGES-1 is the major PGE2 synthase under pathological conditions related to inflammation and cancer (Samuelsson et al., 2007), and its expression is strongly increased in blood upon LPS treatment, primarily contributing to PGE2 formation in blood (Mosca et al., 2007). To assess the effects of Hyp on mPGES-1 activity, microsomal preparations of IL-1β-stimulated A549 cells, a rich source of mPGES-1 (Jakobsson et al., 1999), were used. Microsomes were pre-incubated with Hyp for 15 min, and then, PGE2 formation was initiated by addition of 20 μM PGH2 as substrate for mPGES-1. MK-886, used as reference compound, concentration-dependently inhibited PGE2 formation with an IC50 of 2.1 μM (data not shown), which is in agreement with the literature (Koeberle et al., 2008). As shown in Figure 4A, Hyp concentration-dependently suppressed PGE2 formation with an IC50 of 1 μM, and at 10 μM, 85 ± 2% inhibition was evident. Decreasing the PGH2 concentration to 1 μM did not significantly alter the potency of Hyp (data not shown). Interestingly, octahydro-hyperforin was equally effective, whereas the acylphloroglucinol core or the closely related polyprenylated acylphloroglucinol humulone (Figure 4B) failed to significantly inhibit PGE2 formation up to 10 μM (Figure 4A). These data suggest that defined structural arrangements are required for Hyp's inhibitory effect on mPGES-1. Finally, also an ethanolic extract (60% ethanol, v v−1) of St. John's wort proved to be efficient in suppressing PGE2 formation with an ED50 = 4 μg ml−1 (Figure 4C).
Figure 4.

Effects of hyperforin on the activity of mPGES-1. (A) Concentration–response curves for Hyp, octahydro-hyperforin, the acylphloroglucinol core, and humulone. (B) Chemical structures of octahydro-hyperforin, the acylphloroglucinol core, and humulone. (C) Concentration–response curves for a St. John's wort extract. (A,C) For studies of mPGES-1 inhibition, microsomal preparations of IL-1β-stimulated A549 cells were pre-incubated with vehicle (DMSO) or the test compounds at the indicated concentrations for 15 min at 4°C, and the reaction was started with 20 μM PGH2. After 1 min at 4°C, the reaction was terminated using a stop solution containing FeCl2 and 11β-PGE2 (1 nmol) as internal standard. The 100% values in the individual experiments are in the range of 3–4 μg ml−1 PGE2. Data are given as mean ± SE, n = 3–4. (D) Reversibility of mPGES-1 inhibition by Hyp. Microsomal preparations of IL-1β-stimulated A549 cells were pre-incubated with 3 μM inhibitor for 15 min at 4°C. An aliquot was diluted 10-fold to obtain an inhibitor concentration of 0.3 μM. For comparison, microsomal preparations were pre-incubated for 15 min with 0.3 μM Hyp or with vehicle (DMSO), and then, 20 μM PGH2 was added (no dilution). Then, all samples were incubated for 1 min on ice, and PGE2 formation was analyzed as described by RP-HPLC. Data are given as mean ± SE, n = 3–4, **p < 0.01 vs. vehicle (0.1% DMSO) control, ANOVA + Tukey HSD post hoc tests.
To investigate whether Hyp inhibits PGE2 synthesis in a reversible manner, microsomal preparations of A549 cells were pre-incubated with Hyp and subjected to wash-out experiments. Hyp at 0.3 μM failed to efficiently block PGE2 synthesis, whereas PGE2 formation was efficiently blocked at 3 μM (Figure 4D). mPGES-1 activity was restored by 10-fold dilution of the sample containing 3 μM Hyp (Figure 4D) implying a reversible mode of inhibition.
Effects of hyperforin on the expression of COX-2 and mPGES-1 and induction of apoptotic cell death
We investigated whether Hyp might suppress PGE2 formation by interfering with COX-2 or mPGES-1 expression during prolonged incubation times (24 h). In fact, Hyp concentration-dependently inhibited the induction of COX-2 and mPGES-1 with an EC50 of 1–3 μM in IL-1β-stimulated A549 cells (a model lung carcinoma cell line to study mPGES-1 functions), and also β-actin levels were slightly reduced at 10 μM (Figure 5). Since previous studies showed that Hyp causes apoptosis of various cancer cell lines (Schempp et al., 2002; Hostanska et al., 2003; Dona et al., 2004), we reasoned that reduced expression of COX-2 and mPGES-1 in A549 cells by Hyp may be related to induction of apoptotic cell death. In fact, the decrease of COX-2 and mPGES-1 protein correlated with the induction of apoptosis as determined by the cleavage of PARP (Figure 5), and the cell viability was strongly reduced after 24 h (EC50 ≈ 3 μM, not shown).
Figure 5.
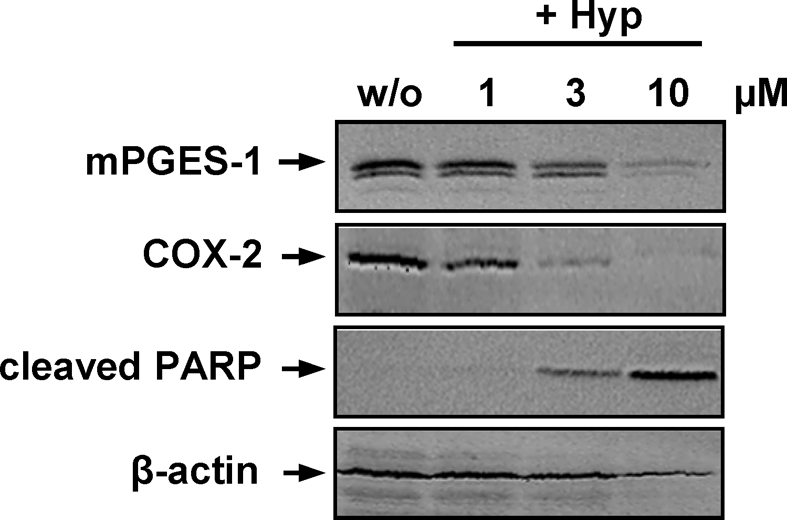
Effects of hyperforin on apoptosis and the expression of COX-2 and mPGES-1. A549 cells, 60% confluent, were incubated with 2 ng ml−1 interleukin-1β together with vehicle (DMSO) or Hyp in cell culture medium containing 2% (v v−1) fetal calf serum. Cells were harvested after 24 h, total cell lysates were prepared and analyzed for the induction of COX-2 (72 kDa), mPGES-1 (16 kDa) and cleaved PARP (89 kDa) using SDS-PAGE and Western blotting. β-Actin (45 kDa) was used as loading control. Data are representatives of two to three independent experiments.
Hyperforin suppresses carrageenan-induced mouse paw edema
The anti-inflammatory properties of Hyp were investigated in carrageenan-induced mouse paw edema in which PGE2 (and also other mediators) critically contributes to the inflammatory response (Guay et al., 2004). Hyp (0.25, 1, 4 mg kg−1) was administered i.p. 30 min prior to injection of carrageenan into the mouse paw. The increase in paw volume was time-dependently assessed and reached a maximum at 4-h post-carrageenan treatment (Figure 6A). In fact, in mice treated with 0.25, 1, and 4 mg kg−1 of Hyp, the peak of the response to carrageenan at 4 h was reduced by 35, 63, and 78%, respectively, whereas indomethacin (5 mg kg−1), used as reference, caused 57% inhibition. Moreover, comparison of the areas under the curves of each group, between 2 and 6 h after carrageenan injection, yielded similar degrees of inhibition confirming a more potent effect of Hyp (ED50 = 1 mg kg−1) over indomethacin (ED50 = 5 mg kg−1; Figure 6B).
Figure 6.

Effects of hyperforin on carrageenan-induced mouse paw edema. (A) Animals (n = 10 for each experimental group) were treated i.p. with 0.25, 1, and 4 mg kg−1 Hyp, 5 mg kg−1 indomethacin, or vehicle (2% DMSO) 30 min before carrageenan subplantar injection. (B) Inhibition of edema formation caused by the treatment with the indicated compounds as percentage of vehicle control calculated from the relative areas under the curves. Data are given as mean ± SE. n = 10, *p < 0.05, **p < 0.01, ***p < 0.001 vs. vehicle (2% DMSO) control, ANOVA + Tukey HSD post hoc tests.
Hyperforin suppresses carrageenan-induced pleurisy in rats
Hyperforin has previously been shown to suppress carrageenan-induced pleurisy in rats by decreasing pleural LTB4 levels (Feisst et al., 2009). Here, we further assessed the formation of PGE2 during pleurisy in order to get some information about its ability to inhibit PGE2 synthesis in vivo. Injection of carrageenan into the pleural cavity of rats (DMSO 4% group) elicited an acute inflammatory response characterized by the accumulation of fluid that contained large numbers of inflammatory cells (Table 1). As observed in carrageenan-induced paw edema, Hyp (4 mg kg−1 i.p., 30 min before carrageenan) significantly inhibited the inflammatory response 4 h after carrageenan injection as demonstrated by the significant attenuation of exudate formation (64%) and cell infiltration (50%). Indomethacin (5 mg kg−1) also reduced exudate formation and cell infiltration (75 and 65%, respectively), and its anti-inflammatory action is not significantly different from Hyp (Table 1). In comparison with the corresponding exudates from DMSO-treated rats, exudates of Hyp-treated animals exhibited significantly decreased PGE2 levels, and indomethacin almost completely suppressed PGE2 formation as expected (Table 1).
Table 1.
Effect of Hyp on rat carrageenan-induced pleurisy 4 h after i.p. injection.
| Treatment | Exudate volume (ml) | Inflammatory cells (×106) | PGE2 (ng/rat) |
|---|---|---|---|
| Vehicle (DMSO) | 0.44 ± 0.04 | 103.0 ± 15.4 | 1.81 ± 0.16 |
| Hyperforin (4 mg kg−1) | 0.16 ± 0.05*** | 51.3 ± 7.2** | 0.97 ± 0.15*** |
| Indomethacin (5 mg kg−1) | 0.11 ± 0.04*** | 35.8 ± 4.2*** | 0.19 ± 0.05*** |
Data are given as mean ± SE, n = 10, **p < 0.01, ***p < 0.001 vs. vehicle (DMSO 4%), ANOVA + Tukey HSD post hoc tests.
Discussion
Systematic investigations of Hyp's pharmacological action within the PGE2 biosynthetic pathway revealed mPGES-1 as a putative molecular target of Hyp. Thus, Hyp potently and concentration-dependently inhibited the enzymatic conversion of PGH2 to PGE2 catalyzed by mPGES-1 in a defined cell-free assay, diminished the formation of PGE2 in human whole blood without affecting the synthesis of other COX-derived prostanoids (i.e., 6-keto PGF1α, TXB2, and 12-HHT), and reduced PGE2 levels in the pleurisy model in vivo. COX-2, phospholipases, or LPS-signaling components as possible point of attack were excluded based on cell-free and cellular assays of COX-2 activity and by experiments applying exogenous AA as substrate for COX enzymes. Discrepancies were observed in the potency of Hyp to inhibit COX-1 activity between whole blood (Figure 2C) and cell-free assays (Figure 3A) or platelet COX-1 activity assays (Feisst and Werz, 2004) which apparently do not solely depend on plasma protein binding and are not readily understood. The pharmacological relevance of the inhibition of mPGES-1 and, as previously reported, 5-LO (Feisst and Werz, 2004) is supported by the beneficial effect of Hyp during eicosanoid (mainly PGE2)-driven acute inflammation in mouse paw edema and rat pleurisy. We suggest that the dual interference with mPGES-1 and 5-LO forms the basis of the anti-inflammatory effectiveness of Hyp and St. John's wort observed in vitro and in vivo (Schempp et al., 2003; Medina et al., 2006; Hammer et al., 2007; Sosa et al., 2007).
St. John's wort is frequently used for the treatment of mild and moderate depressions but also to intervene with inflammatory disorders, peptic ulcers, and skin wounds (Medina et al., 2006). Although the anti-depressive, anti-inflammatory, and anti-carcinogenic effects of St. John's wort extracts could be ascribed to Hyp (Muller, 2003; Medina et al., 2006), the underlying molecular mechanisms and molecular targets of Hyp are still elusive. Postulated anti-inflammatory mechanisms include down-regulation of the expression of the chemokine receptor CXCR3 on activated T cells (Cabrelle et al., 2008), down-modulation of matrix metalloproteinase 9, seemingly due to inhibition of leukocyte elastase, and reduced expression of the adhesion molecule CD11b in neutrophils (Dell'Aica et al., 2007), as well as suppression of Ca2+ mobilization, leukocyte elastase release, and formation of reactive oxygen species in neutrophils (Feisst and Werz, 2004). In addition, we recently identified Hyp to inhibit 5-LO and COX-1 in pro-inflammatory eicosanoid biosynthesis in vitro (Albert et al., 2002) thereby exhibiting a unique molecular interference of 5-LO at its C2-like domain (Feisst et al., 2009). Nevertheless, most of these postulated anti-inflammatory mechanisms have been essentially identified and characterized based on cell-free assays and assays based on isolated cells, and thus, the pharmacological relevance of these postulates is unclear. These assays lack the influence of a physiologically relevant environment as compared to studies using whole blood or tissues, and important factors (plasma protein binding, cell–cell interactions, etc.) that may potentially modulate the efficacy are therefore neglected. Moreover, the effective concentrations of Hyp for intervention with certain targets or mechanisms are often substantially higher (>1 μM) than Hyp levels reached in vivo after oral administration of St. John's wort preparations. Thus, daily oral intake of 3 × 300 mg hypericum extract by human volunteers led to Hyp steady-state concentrations of 0.28 μM (Biber et al., 1998). In light of these facts, suppression of PGE2 formation by Hyp was evident in human whole blood with significant suppressive effects at 0.03–1 μM, congruent with the ability of Hyp to directly interfere with mPGES-1 activity in the cell-free assay (IC50 = 1 μM).
Hyperforin almost completely blocked mPGES-1 activity in the cell-free assay, but PGE2 formation in LPS-stimulated whole blood was only partially inhibited (residual PGE2 formation approx. 50% of vehicle control) as observed for mPGES-1 inhibitors such as MK-886 (this study and Koeberle et al., 2008), MD-52 (this study and Koeberle et al., 2009a), and others (Koeberle and Werz, 2009). In contrast, COX inhibitors (i.e., indomethacin or celecoxib) were markedly more effective to suppress PGE2 formation in whole blood. Possibly, constitutively expressed homeostatic PGE synthases contribute to PGE2 synthesis under these conditions and are not inhibited by Hyp. In fact, when whole blood was first pre-incubated with LPS to induce COX-2- and mPGES-1-expression (in contrast to the expression of constitutively expressed COX-1 or PGE2 synthases like cPGES and mPGES-2, Murakami and Kudo, 2006), Hyp showed an increased efficiency to reduce PGE2 formation. The contribution of PGE2 synthases others than mPGES-1 may also explain the incomplete suppression of PGE2 formation in the pleurisy model by Hyp as compared to indomethacin. Future studies will address the dissection of the different enzymatic pathways leading to PGE2 biosynthesis in cellular assays and focus on the respective interference with Hyp.
The anti-inflammatory efficiency of Hyp in vivo was confirmed in carrageenan-induced rat pleurisy and further demonstrated for carrageenan-induced mouse paw edema. During paw edema formation, PGE2 levels were significantly elevated (Harada et al., 1982; Guay et al., 2004), and COX inhibitors were shown to prevent the inflammatory response (Gemmell et al., 1979). Also in the pleurisy model, PGE2 essentially contributes to the inflammatory response following carrageenan stimulation (Harada et al., 1996; Kawamura et al., 2000). Hyp efficiently suppressed paw edema and was even more potent in this respect than indomethacin. Similarly, Hyp impaired PGE2 levels in the exudates of rats during pleurisy and markedly attenuated the concomitant exudate formation and inflammatory cell infiltration. However, our data cannot exclude that inhibition of 5-LO (as previously reported in cell-free and cellular studies and for carrageenan-induced rat pleurisy; Albert et al., 2002; Feisst et al., 2009) contributes to the suppression of PGE2 formation by reducing leukocyte recruitment. Taken together, both mPGES-1 and 5-LO seem to be relevant targets underlying the therapeutic efficiency of Hyp in eicosanoid-related disorders.
Besides inflammation, excessive PGE2 formation is also associated with tumorigenesis, particularly of colon, breast, prostate, and lung carcinoma. Pharmacological intervention with PGE2 formation (COX-2 selective inhibitors) has shown a chemopreventive potential (Rao and Reddy, 2004), and in vitro studies suggest induction of apoptosis as critical step (Jana, 2008). mPGES-1 is overexpressed in various cancers such as non-small cell lung cancer, invasive breast cancer, colorectal cancer, and gastric cancer (Samuelsson et al., 2007). Hyp induced apoptosis of various cancer cells such as mammary carcinoma, squamous cell carcinoma, malignant melanoma, and lymphoma cells (Schempp et al., 2002; Hostanska et al., 2003; Dona et al., 2004; Quiney et al., 2006), and we could demonstrate that also human A549 lung carcinoma cells, constitutively expressing mPGES-1 (Jakobsson et al., 1999), undergo apoptosis in response to Hyp (EC50 ≈ 3 μM) as monitored by the cleavage of PARP. It is tempting to speculate whether a functional correlation between the inhibition of PGE2 formation and the induction of apoptosis by Hyp exists. Interestingly, also major depression represents an inflammatory process (Leonard, 2007), where PGE2 levels are significantly elevated, and suppression of COX-2 by celecoxib showed clinical efficacy (Muller et al., 2006). A recent meta-analysis suggests significant effectiveness of St. John's wort in major depression (Linde et al., 2008) but whether inhibition of mPGES-1 contributes to the antidepressant activity of Hyp cannot be answered so far.
In conclusion, Hyp potently inhibits cellular and cell-free PGE2 formation via interference with mPGES-1 at physiologically achievable plasma concentrations and shows anti-inflammatory effectiveness in vivo. Hyp also suppresses leukotriene formation by inhibition of 5-LO (Albert et al., 2002), which is also involved in inflammation and carcinogenesis (Werz and Steinhilber, 2006) like mPGES-1 (Murakami and Kudo, 2006). Dual inhibition of mPGES-1 and 5-LO might therefore provide a molecular basis for Hyp's anti-inflammatory and anti-carcinogenic properties. Such a pharmacological profile would be advantageous, particularly in view of the emerging link between chronic inflammation and cancer, where mPGES-1 and 5-LO may act as potential players.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Albert D., Zundorf I., Dingermann T., Muller W. E., Steinhilber D., Werz O. (2002). Hyperforin is a dual inhibitor of cyclooxygenase-1 and 5-lipoxygenase. Biochem. Pharmacol. 64, 1767–1775 [DOI] [PubMed] [Google Scholar]
- Biber A., Fischer H., Romer A., Chatterjee S. S. (1998). Oral bioavailability of hyperforin from hypericum extracts in rats and human volunteers. Pharmacopsychiatry 31(Suppl. 1), 36–43 [DOI] [PubMed] [Google Scholar]
- Cabrelle A., Dell'Aica I., Melchiori L., Carraro S., Brunetta E., Niero R., Scquizzato E., D'Intino G., Calza L., Garbisa S., Agostini C. (2008). Hyperforin down-regulates effector function of activated T lymphocytes and shows efficacy against Th1-triggered CNS inflammatory-demyelinating disease. J. Leukoc. Biol. 83, 212–219 [DOI] [PubMed] [Google Scholar]
- Capdevila J. H., Morrow J. D., Belosludtsev Y. Y., Beauchamp D. R., DuBois R. N., Falck J. R. (1995). The catalytic outcomes of the constitutive and the mitogen inducible isoforms of prostaglandin H2 synthase are markedly affected by glutathione and glutathione peroxidase(s). Biochemistry 34, 3325–3337 10.1021/bi00010a023 [DOI] [PubMed] [Google Scholar]
- Claveau D., Sirinyan M., Guay J., Gordon R., Chan C. C., Bureau Y., Riendeau D., Mancini J. A. (2003). Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J. Immunol. 170, 4738–4744 [DOI] [PubMed] [Google Scholar]
- Côté B., Boulet L., Brideau C., Claveau D., Ethier D., Frenette R., Gagnon M., Giroux A., Guay J., Guiral S., Mancini J., Martins E., Masse F., Methot N., Riendeau D., Rubin J., Xu D., Yu H., Ducharme Y., Friesen R. W. (2007). Substituted phenanthrene imidazoles as potent, selective, and orally active mPGES-1 inhibitors. Bioorg. Med. Chem. Lett. 17, 6816–6820 [DOI] [PubMed] [Google Scholar]
- Dell'Aica I., Niero R., Piazza F., Cabrelle A., Sartor L., Colalto C., Brunetta E., Lorusso G., Benelli R., Albini A., Calabrese F., Agostini C., Garbisa S. (2007). Hyperforin blocks neutrophil activation of matrix metalloproteinase-9, motility and recruitment, and restrains inflammation-triggered angiogenesis and lung fibrosis. J. Pharmacol. Exp. Ther. 321, 492–500 [DOI] [PubMed] [Google Scholar]
- Dona M., Dell'Aica I., Pezzato E., Sartor L., Calabrese F., Della Barbera M., Donella-Deana A., Appendino G., Borsarini A., Caniato R., Garbisa S. (2004). Hyperforin inhibits cancer invasion and metastasis. Cancer Res. 64, 6225–6232 10.1158/0008-5472.CAN-04-0280 [DOI] [PubMed] [Google Scholar]
- Feisst C., Pergola C., Rakonjac M., Rossi A., Koeberle A., Dodt G., Hoffmann M., Hoernig C., Fischer L., Steinhilber D., Franke L., Schneider G., Radmark O., Sautebin L., Werz O. (2009). Hyperforin is a novel type of 5-lipoxygenase inhibitor with high efficacy in vivo. Cell. Mol. Life Sci. 66, 2759–2771 10.1007/s00018-009-0078-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feisst C., Werz O. (2004). Suppression of receptor-mediated Ca2+ mobilization and functional leukocyte responses by hyperforin. Biochem. Pharmacol. 67, 1531–1539 [DOI] [PubMed] [Google Scholar]
- Friesen R. W., Mancini J. A. (2008). Microsomal prostaglandin E2 synthase-1 (mPGES-1): a novel anti-inflammatory therapeutic target. J. Med. Chem. 51, 4059–4067 [DOI] [PubMed] [Google Scholar]
- Gemmell D. K., Cottney J., Lewis A. J. (1979). Comparative effects of drugs on four paw oedema models in the rat. Agents Actions 9, 107–116 10.1007/BF02024141 [DOI] [PubMed] [Google Scholar]
- Guay J., Bateman K., Gordon R., Mancini J., Riendeau D. (2004). Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J. Biol. Chem. 279, 24866–24872 [DOI] [PubMed] [Google Scholar]
- Hammer K. D., Hillwig M. L., Solco A. K., Dixon P. M., Delate K., Murphy P. A., Wurtele E. S., Birt D. F. (2007). Inhibition of prostaglandin E(2) production by anti-inflammatory Hypericum perforatum extracts and constituents in RAW264.7 mouse macrophage cells. J. Agric. Food Chem. 55, 7323–7331 10.1021/jf0710074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada Y., Hatanaka K., Kawamura M., Saito M., Ogino M., Majima M., Ohno T., Ogino K., Yamamoto K., Taketani Y., Yamamoto S., Katori M. (1996). Role of prostaglandin H synthase-2 in prostaglandin E2 formation in rat carrageenin-induced pleurisy. Prostaglandins 51, 19–33 10.1016/0090-6980(95)00168-9 [DOI] [PubMed] [Google Scholar]
- Harada Y., Tanaka K., Uchida Y., Ueno A., Oh-Ishi S., Yamashita K., Ishibashi M., Miyazaki H., Katori M. (1982). Changes in the levels of prostaglandins and thromboxane and their roles in the accumulation of exudate in rat carrageenin-induced pleurisy – a profile analysis using gas chromatography-mass spectrometry. Prostaglandins 23, 881–895 10.1016/0090-6980(82)90131-9 [DOI] [PubMed] [Google Scholar]
- Hostanska K., Reichling J., Bommer S., Weber M., Saller R. (2003). Hyperforin a constituent of St John's wort (Hypericum perforatum L.) extract induces apoptosis by triggering activation of caspases and with hypericin synergistically exerts cytotoxicity towards human malignant cell lines. Eur. J. Pharm. Biopharm. 56, 121–132 10.1016/S0939-6411(03)00046-8 [DOI] [PubMed] [Google Scholar]
- Jakobsson P. J., Thoren S., Morgenstern R., Samuelsson B. (1999). Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. U.S.A. 96, 7220–7225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana N. R. (2008). NSAIDs and apoptosis. Cell. Mol. Life Sci. 65, 1295–1301 10.1007/s00018-008-7511-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K., Ohkawa S., Terao S., Terashita Z., Nishikawa K. (1985). Thromboxane synthetase inhibitors (TXSI). Design, synthesis, and evaluation of a novel series of omega-pyridylalkenoic acids. J. Med. Chem. 28, 287–294 [DOI] [PubMed] [Google Scholar]
- Kawamura M., Hatanaka K., Saito M., Ogino M., Ono T., Ogino K., Matsuo S., Harada Y. (2000). Are the anti-inflammatory effects of dexamethasone responsible for inhibition of the induction of enzymes involved in prostanoid formation in rat carrageenin-induced pleurisy? Eur. J. Pharmacol. 400, 127–135 [DOI] [PubMed] [Google Scholar]
- Koeberle A., Siemoneit U., Buhring U., Northoff H., Laufer S., Albrecht W., Werz O. (2008). Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J. Pharmacol. Exp. Ther. 326, 975–982 [DOI] [PubMed] [Google Scholar]
- Koeberle A., Northoff H., Werz O. (2009a). Curcumin blocks prostaglandin E2 biosynthesis through direct inhibition of the microsomal prostaglandin E2 synthase-1. Mol. Cancer Ther. 8, 2348–2355 10.1158/1535-7163.MCT-09-0290 [DOI] [PubMed] [Google Scholar]
- Koeberle A., Siemoneit U., Northoff H., Hofmann B., Schneider G., Werz O. (2009b). MK-886, an inhibitor of the 5-lipoxygenase-activating protein, inhibits cyclooxygenase-1 activity and suppresses platelet aggregation. Eur. J. Pharmacol. 608, 84–90 10.1016/j.ejphar.2009.02.023 [DOI] [PubMed] [Google Scholar]
- Koeberle A., Werz O. (2009). Inhibitors of the microsomal prostaglandin E(2) synthase-1 as alternative to non steroidal anti-inflammatory drugs (NSAIDs) – a critical review. Curr. Med. Chem. 16, 4274–4296 [DOI] [PubMed] [Google Scholar]
- Leonard B. E. (2007). Inflammation, depression and dementia: are they connected? Neurochem. Res. 32, 1749–1756 [DOI] [PubMed] [Google Scholar]
- Linde K., Berner M. M., Kriston L. (2008). St John's wort for major depression. Cochrane Database Syst. Rev. CD000448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorusso G., Vannini N., Sogno I., Generoso L., Garbisa S., Noonan D. M., Albini A. (2009). Mechanisms of hyperforin as an anti-angiogenic angioprevention agent. Eur. J. Cancer 45, 1474–1484 [DOI] [PubMed] [Google Scholar]
- Martinez-Poveda B., Quesada A. R., Medina M. A. (2005). Hyperforin, a bio-active compound of St. John's Wort, is a new inhibitor of angiogenesis targeting several key steps of the process. Int. J. Cancer 117, 775–780 [DOI] [PubMed] [Google Scholar]
- Medina M. A., Martinez-Poveda B., Amores-Sanchez M. I., Quesada A. R. (2006). Hyperforin: more than an antidepressant bioactive compound? Life Sci. 79, 105–111 [DOI] [PubMed] [Google Scholar]
- Mosca M., Polentarutti N., Mangano G., Apicella C., Doni A., Mancini F., De Bortoli M., Coletta I., Polenzani L., Santoni G., Sironi M., Vecchi A., Mantovani A. (2007). Regulation of the microsomal prostaglandin E synthase-1 in polarized mononuclear phagocytes and its constitutive expression in neutrophils. J. Leukoc. Biol. 82, 320–326 [DOI] [PubMed] [Google Scholar]
- Muller N., Schwarz M. J., Dehning S., Douhe A., Cerovecki A., Goldstein-Muller B., Spellmann I., Hetzel G., Maino K., Kleindienst N., Moller H. J., Arolt V., Riedel M. (2006). The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatry 11, 680–684 [DOI] [PubMed] [Google Scholar]
- Muller W. E. (2003). Current St John's wort research from mode of action to clinical efficacy. Pharmacol. Res. 47, 101–109 [DOI] [PubMed] [Google Scholar]
- Murakami M., Kudo I. (2006). Prostaglandin E synthase: a novel drug target for inflammation and cancer. Curr. Pharm. Des. 12, 943–954 [DOI] [PubMed] [Google Scholar]
- Murakami M., Naraba H., Tanioka T., Semmyo N., Nakatani Y., Kojima F., Ikeda T., Fueki M., Ueno A., Oh S., Kudo I. (2000). Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J. Biol. Chem. 275, 32783–32792 [DOI] [PubMed] [Google Scholar]
- Posadas I., Bucci M., Roviezzo F., Rossi A., Parente L., Sautebin L., Cirino G. (2004). Carrageenan-induced mouse paw oedema is biphasic, age-weight dependent and displays differential nitric oxide cyclooxygenase-2 expression. Br. J. Pharmacol. 142, 331–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiney C., Billard C., Faussat A. M., Salanoubat C., Ensaf A., Nait-Si Y., Fourneron J. D., Kolb J. P. (2006). Pro-apoptotic properties of hyperforin in leukemic cells from patients with B-cell chronic lymphocytic leukemia. Leukemia 20, 491–497 10.1038/sj.leu.2404098 [DOI] [PubMed] [Google Scholar]
- Rao C. V., Reddy B. S. (2004). NSAIDs and chemoprevention. Curr. Cancer Drug Targets 4, 29–42 10.2174/1568009043481632 [DOI] [PubMed] [Google Scholar]
- Rothley M., Schmid A., Thiele W., Schacht V., Plaumann D., Gartner M., Yektaoglu A., Bruyere F., Noel A., Giannis A., Sleeman J. P. (2009). Hyperforin and aristoforin inhibit lymphatic endothelial cell proliferation in vitro and suppress tumor-induced lymphangiogenesis in vivo. Int. J. Cancer 125, 34–42 [DOI] [PubMed] [Google Scholar]
- Samuelsson B., Morgenstern R., Jakobsson P. J. (2007). Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol. Rev. 59, 207–224 [DOI] [PubMed] [Google Scholar]
- Schempp C. M., Kirkin V., Simon-Haarhaus B., Kersten A., Kiss J., Termeer C. C., Gilb B., Kaufmann T., Borner C., Sleeman J. P., Simon J. C. (2002). Inhibition of tumour cell growth by hyperforin, a novel anticancer drug from St. John's wort that acts by induction of apoptosis. Oncogene 21, 1242–1250 10.1038/sj.onc.1205190 [DOI] [PubMed] [Google Scholar]
- Schempp C. M., Windeck T., Hezel S., Simon J. C. (2003). Topical treatment of atopic dermatitis with St. John's wort cream--a randomized, placebo controlled, double blind half-side comparison. Phytomedicine 10(Suppl. 4), 31–37 [DOI] [PubMed] [Google Scholar]
- Schempp C. M., Winghofer B., Ludtke R., Simon-Haarhaus B., Schopf E., Simon J. C. (2000). Topical application of St John's wort (Hypericum perforatum L.) and of its metabolite hyperforin inhibits the allostimulatory capacity of epidermal cells. Br. J. Dermatol. 142, 979–984 10.1046/j.1365-2133.2000.03482.x [DOI] [PubMed] [Google Scholar]
- Sosa S., Pace R., Bornancin A., Morazzoni P., Riva A., Tubaro A., Della Loggia R. (2007). Topical anti-inflammatory activity of extracts and compounds from Hypericum perforatum L. J. Pharm. Pharmacol. 59, 703–709 10.1211/jpp.59.5.0011 [DOI] [PubMed] [Google Scholar]
- Sugimoto Y., Narumiya S. (2007). Prostaglandin E receptors. J. Biol. Chem. 282, 11613–11617 [DOI] [PubMed] [Google Scholar]
- Werz O., Steinhilber D. (2006). Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol. Ther. 112, 701–718 [DOI] [PubMed] [Google Scholar]