

Abstract
Modern biomedical research greatly benefits from large-scale genome-sequencing projects ranging from studies of viruses, bacteria, and yeast to multicellular organisms, like Caenorhabditis elegans. Comparative genomic studies offer a vast array of prospects for identification and functional annotation of human ortholog genes. We presented a novel comparative proteomic approach for assembling human gene contigs and assisting gene discovery. The C. elegans proteome was used as an alignment template to assist in novel human gene identification from human EST nucleotide databases. Among the available 18,452 C. elegans protein sequences, our results indicate that at least 83% (15,344 sequences) of C. elegans proteome has human homologous genes, with 7,954 records of C. elegans proteins matching known human gene transcripts. Only 11% or less of C. elegans proteome contains nematode-specific genes. We found that the remaining 7,390 sequences might lead to discoveries of novel human genes, and over 150 putative full-length human gene transcripts were assembled upon further database analyses.
[The sequence data described in this paper have been submitted to the GenBank data library under accession nos. AF132936–AF132973, AF151799–AF151909, and AF152097.]
Over the past decade, important advances in large-scale DNA sequencing have dramatically changed the scope of biologic research and computation biology. Complete genome sequences of viruses, bacteria, and yeast have been obtained (Fleischmann et al. 1995; Fraser et al. 1995; Goffeau et al. 1996), as has information about the composition of the entire genome, gene structure, gene regulation, and gene product functions of these model organisms (Dujon 1996). The recent completion of the C. elegans genome has marked another milestone as the first completed genome from a multicellular organism (The C. elegans Sequencing Consortium 1998). Many important findings will be generated by data mining through the whole 97 MB genome sequences of this nematode (Chervitz et al. 1998). Another extraordinary and crucial genome sequencing project is the Human Genome Project (HGP), formally proposed in 1993, and which started large-scale DNA sequencing in 1997 (Collins and Galas 1993; Olson 1993; Watson 1990). The goal of identifying all human genes could be achieved by the year 2003 or sooner (Collins et al. 1998). Biomedical investigators will soon face new challenges in the post-genome era once all human genes have been identified and characterized through the HGP. Genes affecting human diseases and biology will be easily identified and comprehensively studied (Rawlings and Searls 1997). In addition, comparative analysis between completed genomes from various organisms will provide a better overview regarding the origin of life, phylogenomics, and functional genomics (Tatusov et al. 1997). Critical information on important motifs and functions of protein or on more complex genetic circuitry will be revealed by such comparative genomics.
Expressed sequence tags (ESTs) are nucleotide sequences generated from single-pass cyclic deoxyribonucleic acid (cDNA) sequences of the ends of randomly selected clones from many different cDNA libraries (Adams et al. 1991). The global nucleic acid databases, such as GenBank, as of 1999 contained over one billion bases in more than three million sequence entries, including more than three million entries of ESTs in dbEST (Boguski et al. 1993). This is a rich resource for biomedical investigations involving gene identification and characterization (Aaronson et al. 1996; Miller et al. 1997). Bioinformation tools are being established to access this enormous amount of data and to extract functional and tissue-specific expression information (i.e., data mining) (Pietu et al. 1999). Molecular cloningin silico is becoming a primary procedure for many laboratories to assemble full-length mRNA sequences from dbEST following the identification of novel gene fragments (Marchese et al. 1999; Rossi et al. 1997; Tao et al. 1999). New human genes might also be identified by searching EST databases with query sequences of xenolog origins—we use the term “comparative gene identification” (CGI) for such an approach in this article. However, there are still limitations inherent from dbEST, which greatly reduce the effectiveness of such an approach. Sequencing errors can result from many of these methodologies due to automated-sequencing methods and difficult templates, limitations on readable lengths of sequence, alternatively spliced transcripts, and occasionally preprocessed mRNA or genomic DNA fragment contamination in cDNA libraries. Gene transcripts are often fragmented and not easily assembled into one continuous contig by using nucleotide sequences as the primary alignment basis, such as UniGene (Boguski and Schuler 1995; Schuler 1997) and HGI (Adams et al. 1995; White and Kerlavage 1996) databases. Therefore, a new strategy is necessary to eliminate nucleotide sequence errors.
During evolution, protein primary sequences and structures are often better conserved across species than are nucleotide sequences (Eisen 1998). This is largely due to the degeneracy in the genetic codon usage and confinement on protein functional domains in primary sequences and tertiary frameworks (Gish and States 1993). Ortholog genes from distantly related species have often been discovered by protein sequence alignments, but not by nucleotide sequences (Tatusov et al. 1997; Yuan et al. 1998). Therefore, our CGI approach is beneficial, because it provides a better alignment scaffold with protein sequence queries. In this study, we propose to use the completed C. elegans proteome as a template to assist human gene identification and further EST dataset reductions. We use the available C. elegans protein sequences (proteome) identified through the C. elegans genome project. With the available 18,452 protein sequences analyzed, 7,954 sequences matched known human genes; ∼3,000 sequences were less informative; and the remaining 7,390 sequences might lead to identification of novel human genes. Indeed, further analysis of these 7,390 sequences has thus far identified 150 novel human full-length gene transcripts, verifying the effectiveness of this comparative gene identification approach.
Results
Dataset Reductions and Error-corrections by CGI
Identification of ortholog genes provides important information about the functional and structural conservation within these orthologs throughout evolution. The concept of comparative gene identification (CGI) has been previously used by many laboratories to search for orthologous genes once a particular gene of interest has been identified in another species. Instead of evaluating one gene at a time, we used the available C. elegans genome protein sequences (proteome) as a scaffold for identifying novel human genes. A similar approach was previously used to identify Drosophila-related human ESTs with functional significance, although the number of genes examined was smaller than entire proteome of C. elegans in this study (Banfi et al. 1996, 1997).
We created a Java-based computer program to automatically perform TBLASTN (BLAST version 2.0.4) searches against HGI datasets (release 3.3) and for analysis of these data. The TBLASTN algorithm was originally designed for comparing protein sequence (C. elegans proteome in this study) queries against translated nucleotide databases (dbEST; HGI; UniGene-human). The main feature of our approach utilizes protein sequences as scaffolds to align and correct dbEST entries. It is well known that dbEST entries often contain errors from many sources (such as errors associated with autosequencing methods). These mistakes prevent the proper joining of two EST entries into a complete contig by nucleotide-based assembly programs. In addition, frame-shifting insertion or deletion of sequences often generates incorrectly translated products and thus greatly affects gene discovery and functional annotations of ESTs through database interrogations. Our CGI approach can be used in an endeavor to correct some of these problems.
One example of the error correction potential of the CGI approach is illustrated in Fig. 1a. A C. elegans protein sequence of 264 amino acids (GenBank Accession number AF022982) was used as an initial query to search the HGI database obtained from The Institute for Genome Research (TIGR). This protein sequence matched one HGI entry—THC195430. In the original blast search report, two different reading frames matched this C. elegans query with a high degree of similarity (62% and 67%). One reading frame ended at position 502 of THC195430 and matched amino acid residue 159 of the C. elegans query protein (Fig. 1b). On the other hand, a second reading frame started at position 502 and matched amino acid residue 160 of the nematode protein (Fig. 1b). Therefore, this approach identified a possible error at position 502 of THC195430, consistent with a possible deletion or insertion of one or two bases at position 502. To verify this, the region around position 502 was evaluated and compared with the human dbEST database. One EST entry (X84715) was indicated among several possible matches. This match was used to correct the reading frame by inserting a carboxy nucleotide at position 502 (Fig. 1c), creating a continuous translatable reading frame, which more effectively matched the C. elegans query protein.
Figure 1.

Illustration of CGI gene identification by error correction and gap closure. (a and b), A C. elegans protein sequence of 264 amino acids (AF022982) was used in an initial search against the HGI database. Two different reading frames (frame-1 and frame-2) in THC195430 matched the C. elegans query. Positions of amino acid residues on AF022982 are listed in bold, and positions of nucleotide sequences of THC195430 are underlined. Please note the overlapping nucleotide sequence at position 502 at both reading frames. (b) The TBLASTN results around position 502. A high-level of similarity was observed at both reading frames. (c) Upon further sequence analysis on dbEST, one dbEST entry (X84715) was used to correct the reading frame by inserting a carboxy nucleotide at position 502 as indicated by the arrowhead. A continuous translatable reading frame was generated following the correction. (d) CGI gene identification by a gap closure procedure. In these cases, two separated THC entries were linked by a C. elegans protein scaffold and the gap sequences were determined by performing RT-PCR experiments with primers designed from each THC entry.
In several cases, two separated THC entries were linked by a C. elegans protein scaffold and the gap sequences were determined by performing reverse-transcription-polymerase chain reaction (RT-PCR) experiments with primers designed from each THC entry (Fig. 1d). Nevertheless, it is invaluable to have the possible reading frame identified, which is essential for further error correction attempts. Our CGI approach offers a powerful solution to assemble fragmented ESTs by providing putative translation reading frames. It is possible to discover sequencing errors even in a single EST entry as illustrated in Fig. 1a. This represents a novel approach in comparing and correcting dbEST errors and in performing gene identification on the proteome scale. Therefore, gene identification is accelerated by further EST database reduction. In addition, functional annotation of the gene product is accelerated,because the CGI approach only focuses on the coding region by using protein sequences as initial queries. This gene identification effort should prove to be beneficial to the HGP.
Comparative Gene Identification with C. elegans Proteome
We used 18,452 C. elegans protein sequences of the Wormpep dataset (blast server version) as the initial query set to search against TIGR/HGI database containing 234,460 entries. TBLASTN program was selected to compare protein queries against DNA databases and defaulted BLAST parameters were used to obtain high-stringency search results. All search data were stored in a specially designed FileMaker Pro database, referred to as IBMS (Integrated Bioinformatics Management System) as shown in Fig. 2. This database was designed for better organization and easy manipulation of gene sequences and integration of various biological functional information. There were different layouts designed to store basic information (Fig. 2), nucleotide and translated protein information, the original C. elegans query protein information, and final translated product BLASTP search results. The final contig assembly information of each CGI gene was recorded in the IBMS-CR layout. The complete IBMS database for CGI genes can be obtained by request or via anonymous ftp at 140.109.41.19. To analyze BLAST results, we used simplified criteria to classify C. elegans proteome genes into several categories based on the first-matched (often best-matched) THC entry. There were 7,954 records of C. elegans proteome that matched known human gene sequences. Those records (1,038) with very short peptide lengths (≤100 a.a. residues) were rejected and not used as search queries due to sensitivity limitations of the BLAST program (Oliver et al. 1992). Although short open reading frames and their gene products do exist in the Escherichia coli and yeast genomes (Andrade et al. 1997; Wasinger and Humphery-Smith 1998), one would need to use different BLAST parameters for their identification. Our main focus in this study was to generate full-length continuous contigs by linking fragmented ESTs. There were 2,070 records that did not produce informative matches against the HGI database; some of these proteins could represent genes specific to C. elegans. There were 7,390 records matched to THC entries without genotype assignment; these might represent C. elegans orthologs of novel human genes. Among these records, 3,456 genes had two or more uncharacterized THC entries or long BLAST match areas in the first matched uncharacterized THC entry. These genes were considered to be unique and novel gene transcripts in the HGI database. The remaining 3,934 records might be assigned to possible new genes or members of gene families, which contained homologous matches to known human genes.
Figure 2.

Illustration of IBMS CGI gene database. Following the determination of CGI genes, more comprehensive analysis was performed to obtain the possible full-length nucleotide sequences by dbEST searches; to determine the optimum open-reading frame; to search the UniGene-human database for chromosome localization, tissue distribution, and EST matches; and a final BLAST analysis to confirm its novelty and protein family annotation. All information was then stored in a customized FileMaker Pro-based IBMS CGI gene database. Only the basic layout is shown here. There are different easy-viewing layouts designed to store nucleotide and translated protein information, the original C. elegans query protein information (possible ortholog gene), the full-length contig assembly information and the final BLASTP search results.
In total, 15,344 out of 18,452 proteins were found to have matched human THC entries. This indicates that at least 83% of C. elegans proteome potentially have human orthologs. This number is much higher than the reported 36% matches when only 4,979 human sequences were used for comparison (The C. elegans Sequencing Consortium 1998). Our results were attributed to the much larger human gene transcript dataset used in our analysis. On the other hand, only 11% or less of the C. elegans proteome contains nematode-specific genes (2,070 out of 18,452 sequences).
These results were similar to our analyses performed with the mouse EST data set (MGI). There were 4,151 records of C. elegans proteome matched to known mouse genes; 11,407 records possible matched novel genes; and only 1,856 of 18,452 (10%) genes that might be nematode-specific when comparing the mouse database. There were 15,558 out of 18,452 proteins found to have matched mouse THC entries. These results indicate that at least 84% of C. elegans proteome potentially have mouse orthologs. The numbers were remarkably similar to those presented for HGI searches. Accurate numbers comparing C. elegans and human proteomes will be obtained following the completion of the HGP in the near future. Nonetheless, our results indicated that most of the C. elegans proteome were conserved throughout its evolution to human and mouse.
150 Potential Full-length Novel Human Genes Identified through CGI
One significant advantage of using this CGI approach is in determining the starting position of a gene transcript with high-level conservation between some orthologs. Following the establishment of the IBMS C. elegans database, we first examined the 7,390 C. elegans Wormpep records for novel full-length human genes. As expected, most THCs for matching required further extension of their 5′-ends to complete the coding region. However, some genes were found to have nonoverlapping THC entries and lacked a middle segment. These represented excellent opportunities for validating our CGI approach. If these nonoverlapping THC entries were derived from a unique gene transcript, as suggested by our C. elegans protein scaffold, we should have been able to close the gap sequence by an RT-PCR approach as illustrated in Fig. 1d.
We therefore performed gap-closure experiments to identify gap sequences by designing specific primers from each end of the gap using THC sequence information. We selected twelve genes containing possible 5′-end initiation ATG sites and only one gap for assembling full-length transcripts. Eleven out of twelve genes were successfully amplified with this approach— CGI-1, CGI-2, CGI-5, CGI-7, CGI-13, CGI-17, CGI-19, CGI-27, CGI-40, CGI-41, CGI-42 (some of these are listed in Fig. 3). YJ12405 was the only gene that did not yield a successful product in the gap-closure experiment with several attempts; this might be due to low or nonexpression of this gene in the human gastric cancer cell line used. Among these genes, CGI-42 was recently reported as the human protein serine/threonine phosphatase 4 regulatory subunit (Greller and Tobin 1999). The CGI-42 gene contains 933 amino acids and completely matches the reported sequences, further validating our approach for identifying novel human genes. More human genes might be identified by this gap-closure procedure, as well as by the RACE method to complete their 5′-end sequences.
Figure 3.

Simplified output list of CGI genes used for tissue blot analysis by IBMS database. They are CGI-1, CGI-2, CGI-7, CGI-13, CGI-17, CGI-19, CGI-27. The full list (CGI-1 to CGI-151) can be obtained as IBMS database for CGI genes by request or via anonymous ftp at 140.109.41.19.
Following the examination of 7,390 records for full-length human genes, more than 150 candidate genes were identified: CGI-1 to CGI-151. All CGI gene information was stored in a custom-built database—the IBMS CGI database. Complete information for each CGI gene was stored, including identification and GenBank accession number, EST expression pattern, chromosome localization obtained from the UniGene-human database, nucleotide and protein sequences, C. elegans ortholog sequence, BLASTP results, the putative protein family annotation, and the final contig assembly information. Potential functional matches were noted from BLASTP search results. There are many CGI proteins matched to yeast or C. elegans gene products. Some of these are related to prokaryotic genes, such as Mycobacterirum dehydrogenase for CGI-93. An example of a simple output is illustrated in Fig. 3.
The identification of 150 full-length human genes with bioinformatic resources in a relatively short period of time could serve as a contribution to the HGP. It was not possible to verify each nucleotide of every CGI gene identified due to limited laboratory resources, and there may be some discrepancies in nucleotide sequences due to inherited errors from EST entries or single nucleotide polymorphisms. For example, CGI-8 and CGI-71 were discovered later to be the same transcript with only three different base insertions in different regions; this was confirmed by a new EST entry. It is also possible that human CGI genes contain much longer 5′-end sequences compared with C. elegans genes, and we might not have the complete coding region for CGI genes at that point in our study. In spite of these reservations, we are confident that most CGI genes indeed contain their full coding regions. Additional comparisons were performed with CGI-1 to CGI-151, excluding CGI-71 as stated above.
The degree of conservation between CGI genes and their C. elegans orthologs surpasses our original expectations. The average protein length of human CGI genes is 304 amino acids; their C. elegans counterparts have an average length of 312 residues. The largest protein from CGI analysis has 933 amino acids and the shortest has 106 residues. The protein-length distribution of CGI-1 to CGI-151 is illustrated in Fig. 4a. The only protein length that lies outside the figure is CGI-142, which contains only 203 amino acids, whereas the original C. elegans ortholog is 1,095 amino acids. However, the CGI-142 sequence matched to that of the amino-terminal sequence of the C. elegans protein. We consider this a full-length gene because of its BLASTP matches to human and mouse hepatoma-derived growth factors, which are 240 and 237 amino acid residues in length. These known growth factors also shared 76% similarity with CGI-142 throughout the protein sequence with a higher degree of similarity at their amino-terminal (not shown). Perhaps CGI-142 should be considered a parallel of human hepatoma-derived growth factor. Another possibility is that the large 1,095 amino acid C. elegans protein may be mispredicted or may be generated as a result of gene restructuring that occurred during evolution.
Figure 4.

Analyses of human CGI proteins and their original C. elegans protein queries. (a) Protein length. (b) Matched areas of sequence length. (c) Similarity percentage. Analyses were performed for protein length and BLAST results of CGI-1 to CGI-151 (excluding CGI-71, which is almost identical to CGI-8 with three bases inserted). The average length of human CGI genes is 304 amino acids (106∼ 933). C. elegans proteins have an average length of 312 residues (107∼1160). Matched areas from BLAST analysis results averaged 255 residues (85∼692). The average homology percentage is 41% (20%∼71%) and the average similarity percentage is 59% (34%∼87%).
Following BLASTP analysis under default conditions (but with the filter option off), the average matched-residue number is 255, with the range from 85 to 692 residues (Fig. 4b). This indicates that close to 84% of the primary protein framework of individual CGI proteins (255/304 a.a.) is similar to that of C. elegans orthologs. The homology between human CGI genes and C. elegans genes is approximately 41% (20% to 71% in range) and similarity is even more striking at 59% (34% to 87%), as shown in Fig. 4c. This high degree of conservation is another validation of our CGI approach. It is likely that the high-stringency default BLAST parameters used here would lead to identifying CGI genes with higher similarities. It will be interesting to use various BLAST settings in our program for future CGI gene identifications and to further refine these comparative evaluations.
Tissue Expression Profiles on Selected CGI Genes
To demonstrate expression of CGI genes, hybridization experiments were performed with a human tissue Master Blot from CLONTECH (Palo Alto, CA). In Fig. 5, gap-closure fragments from CGI-7, CGI-17, and CGI-27 genes (listed in Fig. 3) were used as probes for hybridization against human tissue blots. All three genes were expressed in most tissues. CGI-27 is more abundant in many tissues compared to CGI-7 and CGI-17, correlating with its dbEST matches (48 matches vs. 20 matches for CGI-17). CGI-7 codes for a 503 amino acid protein similar to a yeast nonsense-mediated mRNA decay protein. It is expressed more in fetal heart, fetal kidney, fetal liver, fetal lung, placenta, liver, salivary gland, pituitary gland, pancreas, stomach, and heart (Fig. 5a). CGI-17 encodes a 385-residue protein similar to Drosophila pelota protein. It is expressed more abundantly in fetal kidney, fetal spleen, fetal lung, placenta, lung, spleen, kidney, and adrenal gland (Fig. 5b). CGI-27 codes for a 297 protein similar to a hypothetical yeast protein. It is most expressed in testis, thymus, skeletal muscle, and placenta (Fig. 5c). In summary, these genes seemed to be ubiquitously expressed. Tissue distribution prediction based on UniGene information (as shown in Fig. 3) did not always match well with the hybridization results. It is likely that coverage of cDNA libraries in dbEST-human is not comprehensive enough, although there were more than a hundred libraries. Many cDNA libraries were derived from tumor tissues and cell lines, which may have altered gene expression patterns. For low abundance transcripts, they might not be properly represented in the EST libraries with fewer clones sequenced. Therefore, predicting tissue expression patterns with UniGene information could be used for reference only. On the contrary, the numbers of EST entries in dbEST (as listed in Fig. 3) seemed to reflect better the general expression level of the identified CGI genes (Fig. 5).
Figure 5.
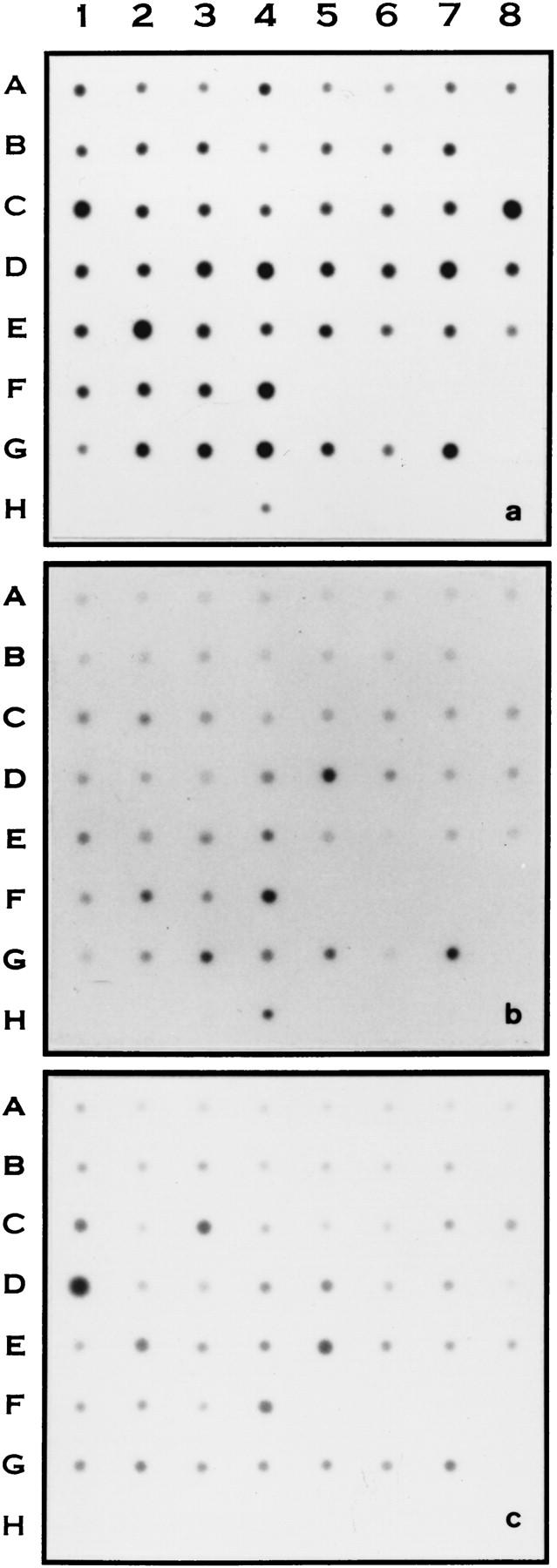
RNA master blot analysis of CGI genes in human tissues. Master tissue blots were hybridized with cloned RT-PCR-amplified fragments of human CGI genes as indicated. (a) CGI-7. (b) CGI-17. (c) CGI-27. The exposure time for each blot was 3 days for A, 7 days for B and 20 hours for C. The tissue distribution on the blot from left to right in order (1–8) was: A, whole brain; amygdala; caudate nucleus; cerebellum; cerebral cortex; frontal lobe; hippocampus; medulla oblongata. B, occipital pole; putamen; substantia nigra; temporal lobe; thalamus; subthalamic; nucleus; spinal cord. C, heart; aorta; skeletal muscle; colon; bladder; uterus; prostate; stomach. D, testis; ovary; pancreas; pituitary gland; adrenal gland; thyroid gland; salivary gland; mammary gland. E, kidney; liver; small intestine; spleen; thymus; peripheral leukocyte; lymph node; bone marrow. F, appendix, lung, trachea, placenta. G, fetal brain; fetal heart; fetal kidney; fetal liver; fetal spleen; fetal thymus; fetal lung. H, yeast total RNA; yeast tRNA; E. coli rRNA; E. coli DNA; Poly r(A); human C0t DNA; human DNA; human DNA.
To validate the actual message transcript sizes, we selected four more gap-closure fragments—CGI-1, CGI-2, CGI-13, CGI-19 from Fig. 3–for human tissue northern blot analysis, as shown in Fig. 6. CGI-2 encodes a 692 amino acid protein similar to E. coli glucose-inhibited division protein A. It is expressed primarily in heart, skeletal muscle, kidney, and liver (Fig. 6a). CGI-19 is expressed mostly in kidney, liver, and placenta (Fig. 6b); it is related to the human UDP-galactose transporter-related isozymes. CGI-1, related to bovine endothelin-converting enzyme-2, codes a 642 amino acid protein. It is expressed in testis, liver, heart, and thyroid (Fig. 6c). CGI-13 (455 amino acids) is similar only to a C. elegans protein. It is expressed in testis, kidney, pancreas, liver, heart, thyroid, spinal cord, and adrenal gland (Fig. 6d). Overall, our results confirm the tissue-restrictive expression of these genes. In addition, the molecular weights of mRNA transcripts also validate the full-length nature of these CGI genes. The nucleotide sequence lengths (without polyA tails) of CGI-1 is ∼2.3kb; CGI-2 2.5 kb; CGI-13 2.0 Kb; and CGI-19 1.3 Kb. Only CGI-19 is smaller than the expected value from northern blots, whereas the others matched the sizes predicted from northern blots. An alternative mRNA was noted for CGI-1 (Fig. 6c, upper band), for which the precise mechanism of generation has yet to be determined. However, we did observe alternatively transcribed mRNAs in the gap regions of CGI-5, CGI-40, and CGI-41 genes (not shown).
Figure 6.

Northern blot analysis of CGI genes in human tissues. Multiple tissue blots were hybridized with cloned RT-PCR-amplified fragments of human CGI genes as indicated. (a) CGI-2. (b) CGI-19. (c) CGI-1. (d) CGI-13. A and C were exposed for 4 days, whereas B and D were exposed for 10 days. Approximately 2 μg poly(A) RNA from these tissues was loaded in each lane. Tissues are indicated above each lane (BR: Brain, HE: Heart, S.Mu: Skeletal Muscle, CO: Colon, TH or THYM: Thymus, SP: Spleen, KI: Kidney, LI: Liver, S.IN: Small Intestine, PL: Placenta, LU: Lung, PBL: Peripheral Blood Leukocyte, PA: Pancreas, PR: Prostate, TE: Testis, OV: Ovary, ST: Stomach, THYR: Thyroid, S.CO: Spinal Cord, LY.N: Lymph Node, TR: Trachea, AD.G: Adrenal Gland, B.MA: Bone Marrow) and the marker sizes are 9.5 kb, 7.5 kb, 4.4 kb, 2.4 kb and 1.35 kb.
In conclusion, our studies have successfully identified 150 new human genes through a bioinformation-based approach to mining the current human EST databases. This CGI approach provides a new and powerful way for assembling human gene contigs without de novo sequencing by offering a protein primary sequence based scaffold from xenologous proteomes.
Discussion
With completion of the C. elegans genome project in 1998, biologists now have a powerful model organism for studying functions, regulations, and interactions of genes in an entire genome of higher eukaryotics. Following detailed bioinformational analysis, enormous amounts of valuable information on complete gene sets in constituting multicellular organisms will be revealed and thus will change our perspective on biomedical research (The C. elegans Sequencing Consortium 1998). Entire gene families [such as G proteins (Jansen et al. 1999) and nuclear receptors (Sluder et al. 1999)] can be rapidly identified and examined in greater detail in C. elegans and offer a better overview on gene functions and regulations during development. With the completion of the HGP in sight, we will soon be able to perform similar functional genomic studies with human gene families. However, we first have to identify all expressed human genes from genomic sequences, which is estimated to be about 5% of the entire genome. This task will be significant and essential in the next few years, and a substantial amount of work is now devoted to gene prediction (reviewed in Claverie 1997). Among more than a dozen gene-prediction bioinformation programs, none of them claims to be completely accurate. Wrongly predicted genes were reported in the nuclear receptor gene family in C. elegans (Sluder et al. 1999). A combination of several programs has been proposed to improve the prediction efficiency and reliability (Lin et al. 1999; Murakami and Takagi 1998). Nonetheless, experimental approaches will continue to be desirable for final verification of expressed transcripts.
In addition, predictions of alternatively transcribed messages expressed at various developmental stages remain impossible hurdles to the existing gene prediction programs. At this point, ESTs represent an alternative and wealthy resource in assisting gene identification and annotation in genomic sequences (Bailey et al. 1998). EST-based gene-indexing projects are extremely useful in functional genomic studies by providing early gene discovery and expression profiles (Burke et al. 1998; Marra et al. 1999). However, nucleotide-based clustering methods are greatly limited by errors generated in the autosequencing process and others. Our proteome-based CGI approach offers alternative scaffolds for identification of possible reading frames and sequence corrections of ESTs. The most significant contribution of the present study is the linking of fragmented ESTs by providing gap closure scaffolds. Another advantage of this approach is the identification of possible full-length gene transcripts, which is an important and challenging task in bioinformation-based in silico cloning projects. Although it is necessary to verify the translation initiation sites by experiments, our method provided a simple, fast, and economical technique to predict the start site of a translated open reading frame. In addition to the 150 genes identified here, more full-length genes could be identified with more EST information and with 5′-RACE experiments. We assumed that most highly conserved orthologous genes maintain their protein frameworks, including the approximate length of amino acid residues. It is possible that some genes acquire or lose functional domains during evolution (orthologs) and expansion (paralogs), and consequently the overall size of the encoded protein is severely changed. Alternatively, CGI could misjudge the initiation ATG site and the entire coding region of a transcript. One approach to improve prediction accuracy is to investigate the framework of a particular protein by adopting phylogenic analysis on conserved orthologs among completed genomes. This would also assist functional annotations on the identified human genes (Wu et al. 1998).
By utilizing UniGene-human and HGI databases, we reduced much of our efforts with already clustered consensus sequences. However, authentic nucleotide sequences of expressed transcripts in cells require validation by bench work due to existing errors or polymorphisms in ESTs. Nucleotide sequences of CGI genes might not be accurate at a single base level, because we used mostly ESTs from various libraries. Such single nucleotide polymorphisms have received much attention lately and most discoveries regarding gene diversity has been based upon EST information (Buetow et al. 1999; Cargill et al. 1999; Picoult-Newberg et al. 1999). In the present study, we tried to establish consensus nucleotide sequences out of possible useful ESTs first, which potentially incorporated different polymorphic sites into one gene transcript in some cases. This issue needs to be resolved by cloning and sequencing experiments; our main purpose here is to discover new human genes and obtain authentic nucleotide sequences.
Our approach identifies the possible initiation site of a coding region but does not pinpoint the mRNA starting position in one transcript. This is extremely difficult to do for most genes and requires extensive investigation to confirm the transcription starting sites. With the completion of HGP, this situation will be improved. Currently, it is more valuable to identify the coding regions and elucidate the functions of the genes. As gene discovery and annotation will be central to the human genome project in the next few years, our comparative proteomic CGI approach should serve as a critical and complementary method for identifying novel human genes through EST databases.
Methods
Query Sequences, Databases, and Blast Searches
The initial query sequence was obtained from the special Wormpep release for the C. elegans blast server at Sanger Center (Hinxton Hall, U.K.); it contained 18,452 protein sequences. The HGI data set (release 3.3) and MGI data set (release 1.0), containing 234,460 and 75,094 entries, respectively, were obtained from TIGR (Rockville, MD). These sets were formatted for our local blast server. BLAST server program (version 2.0.4) was obtained from NCBI and established locally on our SGI origin 200 server. Default TBLASTN parameters were used for analysis (e=10, v=50, b=50, and w=0). Java-written computer programs, executed on a Sun ultra-1 compatible clone, were designed to extract protein sequences from the original Wormpep data set; perform TBLASTN searches against the HGI database on our local server; and extract information from blast reports for subsequent analyses. We initially designed the program with simple rules to uncover two different THC entries linked by a single C. elegans protein. Later we modified the program to include long open-reading frames within one THC entry matched to the C. elegans protein as stated below. The programming rules are briefly summarized here. Because of the limitation on comparing short sequences, our program rejected C. elegans protein sequences with less than 100 amino acid residues. The program then selected the remaining protein sequences for TBLASTN search against HGI database. If the first significant BLAST match contained a gene description, we classified this query to have a known human ortholog. If the first BLAST match did not have a description and showed more than 50 matched amino acid residues, we considered this query to represent a potential novel ortholog gene. The remaining results were considered as not informative, because they contained no significant BLAST matches or only short matched areas.
In Silico Cloning
New human genes were manually inspected and full-length candidates were identified by assembling THC entries and new dbEST matches with the aid of MacVector software (Oxford Molecular Ltd., Oxford, U.K.). Error corrections manually within THC entries were necessary for most genes identified to establish full-length contigs. Automation of this process is now in progress. Since the C. elegans protein sequence was utilized as a scaffold in our search, we could identify the correct reading frame and used other EST matches to verify the THC sequences. Corrections were made to produce the matching translation sequences according to the C. elegans proteins. In some cases, human EST matches did not provide the necessary information and therefore mouse EST entries, if available, were used as the secondary scaffold. Mouse ESTs were also used as cross-references in searching for ATG initiation sites in human CGI genes. Additional database searches against the UniGene-human database provided information about possible tissue expression patterns and chromosome localizations. Functional annotations were made by BLASTP searches against GenBank protein data sets with final full-length CGI protein sequences. All information was stored in a FileMaker database program and made available as compact discs upon requests or through an anonymous ftp server at 140.109.41.19. The nucleotide sequence reported in this paper has been deposited in the GenBank database under accession no. AF132936 to AF132973, AF151799 to AF151909 and AF152097.
RT-PCR and Primers for Gap Closure Experiments
Many new genes contained gaps between THC entries or EST records. Instead of waiting for new EST entries, we used experimental approaches to identify the sequences within the gap. Gap closure experiments were performed by a RT-PCR method to link these separated THC entries together. We selected twelve genes containing only one gap, as well as possible initiation ATG start sites. Primer pairs were designed from each end of the gap sequences. The sequences of each primer are listed below:
CGI-01F: CATCTCCCTGGCTCAGGCTCAC
CGI-01R: CAACCTCCTGAAGCCAGCACTG
CGI-02F: CTTCCTTTGGTGGCATCGGAAAG
CGI-02R: GAGTCAAGTAACATGGCAGCTG
CGI-05F: GCCTCCATTCTAGAGGAAGTG
CGI-05R: GGCAGGTGGATCTGTTTACAG
CGI-07F: TCAACCACCAGGAACTTGG
CGI-07R: CTGGAACTCTATCTGAGTTC
CGI-13F: GGCAGGAGCCTTCCTTACA
CGI-13R: GAAGGAAATGTGTTGGGAC
CGI-17F: CATGAAGCTCGTGAGGAAGAACATCGAG
CGI-17R: GCAGATATGGGCGAGGCCTTC
CGI-19F: GCTGGTACCTTACCTTAGTGCAG
CGI-19R: GGGCCAATCACTGTCTATACAGC
CGI-27F: GATGTCCAACCGAGTGGTCTGC
CGI-27R: CTTCAGTGGACCGTGAGTGCTC
CGI-40F: GTATGAGTTCCCTGAAGGCGTGG
CGI-40R: CAAACGATGCAGAGCAGGGGGATG
CGI-41F: GGGTTGATGGTGAAGAGCATCG
CGI-41R: GTAGAGCCAGGCTGAAGAAGG
CGI-42F: CCTTCACGTTGGGTTCGCCAAGC
CGI-42R: AGTTCTCCTTTCCTCATCAGGGC
We used two pairs of primers for YJ12405 gene but did not obtain any PCR products.
YJ12405F: CCAACACCTACTCCTACCACAAAG
YJ12405R: GTTTATTTGCCACCTCCGCCTCCT
YJ12405F2: GGGTACTCAGAAGTGATCTACGG
YJ12405R2: CTTCCTTCAGCAGGTGTTC
cDNA was prepared from a human gastric cancer cell line (Lin et al. 1998). Briefly, reverse transcription was carried out with 2 μg total mRNA, oligo (dT)15 and Moloney murine leukemia virus (MMLV) reverse transcriptase obtained from Promega (Madison, WI). The quality of RT products was examined by agarose gel electrophoresis and by PCR reaction with GAPDH-specific primers. The PCR primer pairs were used to amplify the gap regions. The PCR reactions were conducted at 94°C for 5 minutes, 35 cycles of 94°C for 15 seconds, 58°C for 30 seconds, and 70°C for 2 minutes, and final extension phase at 72° C for 10 minutes with a Perkin Elmer 2400 PCR thermocycler and Takara Taq polymerase (Shiga, Japan). The final PCR products were analyzed with 1% agarose gel electrophoresis. The amplified fragments were eluted from the gel and subcloned into pCR2.1 T/A cloning vector from Invitrogen (Carlsbad, CA). Following the cloning procedure, several clones were randomly selected, purified and their sequence determined by an ABI 377 autosequencer in our Institute's core facility.
Tissue Distribution Analysis
For tissue expression and distribution analysis, multiple tissue mRNA blots (MTNI-III, and MTN 12 lane) and RNA human master blots were obtained from Clontech (Palo Alto, CA). Fragments that were 32P-labeled from verified gap-closure clones (CGI-1, 2, 7, 13, 17, 19, 27) were used for hybridization reactions following the recommended hybridization protocols from the supplier.
Acknowledgments
We thank Drs. Jeou-Yuan Chen and Chang-Jen Huang for providing the human RNA master blots and multi-tissue blots; and Drs. Lloyd A. Culp and Kenneth K. Wu for critical comments on this manuscript and helpful suggestions. We also acknowledge The Institute for Genomic Research for providing the HGI and MGI datasets. This research was supported in part by grant 5202401023–4 to L-Y. Ch'ang from the Academia Sinica.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL wenlin@ibms.sinica.edu.tw; FAX 886–2–2782–9142.
References
- Aaronson JS, Eckman B, Blevins RA, Borkowski JA, Myerson J, Imran S, Elliston KO. Toward the development of a gene index to the human genome: an assessment of the nature of high-throughput EST sequence data. Genome Res. 1996;6:829–845. doi: 10.1101/gr.6.9.829. [DOI] [PubMed] [Google Scholar]
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, et al. Complementary DNA sequencing: expressed sequence tags and human genome project. Science. 1991;252:1651–1656. doi: 10.1126/science.2047873. [DOI] [PubMed] [Google Scholar]
- Adams MD, Kerlavage AR, Fleischmann RD, Fuldner RA, Bult CJ, Lee NH, Kirkness EF, Weinstock KG, Gocayne JD, White O, et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature. 1995;377:3–17. [PubMed] [Google Scholar]
- Andrade MA, Daruvar A, Casari G, Schneider R, Termier M, Sander C. Characterization of new proteins found by analysis of short open reading frames from the full yeast genome. Yeast. 1997;13:1363–1374. doi: 10.1002/(SICI)1097-0061(199711)13:14<1363::AID-YEA182>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Bailey LC, Jr, Searls DB, Overton GC. Analysis of EST-driven gene annotation in human genomic sequence. Genome Res. 1998;8:362–376. doi: 10.1101/gr.8.4.362. [DOI] [PubMed] [Google Scholar]
- Banfi S, Borsani G, Bulfone A, Ballabio A. Drosophila-related expressed sequences. Hum Mol Genet. 1997;6:1745–1753. doi: 10.1093/hmg/6.10.1745. [DOI] [PubMed] [Google Scholar]
- Banfi S, Borsani G, Rossi E, Bernard L, Guffanti A, Rubboli F, Marchitiello A, Giglio S, Coluccia E, Zollo M, et al. Identification and mapping of human cDNAs homologous to Drosophila mutant genes through EST database searching. Nat Genet. 1996;13:167–174. doi: 10.1038/ng0696-167. [DOI] [PubMed] [Google Scholar]
- Boguski MS, Lowe TM, Tolstoshev CM. dbEST–database for “expressed sequence tags.”. Nat Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- Boguski MS, Schuler GD. ESTablishing a human transcript map. Nat Genet. 1995;10:369–371. doi: 10.1038/ng0895-369. [DOI] [PubMed] [Google Scholar]
- Buetow KH, Edmonson MN, Cassidy AB. Reliable identification of large numbers of candidate SNPs from public EST data. Nat Genet. 1999;21:323–325. doi: 10.1038/6851. [DOI] [PubMed] [Google Scholar]
- Burke J, Wang H, Hide W, Davison DB. Alternative gene form discovery and candidate gene selection from gene indexing projects. Genome Res. 1998;8:276–290. doi: 10.1101/gr.8.3.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Lane CR, Lim EP, Kalayanaraman N, Nemesh J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Chervitz SA, Aravind L, Sherlock G, Ball CA, Koonin EV, Dwight SS, Harris MA, Dolinski K, Mohr S, Smith T, et al. Comparison of the complete protein sets of worm and yeast: orthology and divergence. Science. 1998;282:2022–2028. doi: 10.1126/science.282.5396.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claverie JM. Computational methods for the identification of genes in vertebrate genomic sequences. Hum Mol Genet. 1997;6:1735–1744. doi: 10.1093/hmg/6.10.1735. [DOI] [PubMed] [Google Scholar]
- Collins F, Galas D. A new five-year plan for the U.S. Human Genome Project. Science. 1993;262:43–46. doi: 10.1126/science.8211127. [DOI] [PubMed] [Google Scholar]
- Collins FS, Patrinos A, Jordan E, Chakravarti A, Gesteland R, Walters L. New goals for the U.S. Human Genome Project: 1998–2003. Science. 1998;282:682–689. doi: 10.1126/science.282.5389.682. [DOI] [PubMed] [Google Scholar]
- Dujon B. The yeast genome project: what did we learn? Trends Genet. 1996;12:263–270. doi: 10.1016/0168-9525(96)10027-5. [DOI] [PubMed] [Google Scholar]
- Eisen JA. Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998;8:163–167. doi: 10.1101/gr.8.3.163. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. Life with 6000 genes. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Greller LD, Tobin FL. Detecting selective expression of genes and proteins. Genome Res. 1999;9:282–296. [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Thijssen KL, Werner P, van der Horst M, Hazendonk E, Plasterk RH. The complete family of genes encoding G proteins of Caenorhabditis elegans. Nat Genet. 1999;21:414–419. doi: 10.1038/7753. [DOI] [PubMed] [Google Scholar]
- Lin J-S, Lu C-W, Huang C-J, Wu P-F, Robison D, Kung H-J, Chi C-W, Wu C-W, Yang W-K, Whang-Peng JJK, Lin W-C. Protein-tyrosine kinase and protein-serine/threonine kinase expression in human gastric cancer cell lines. J Biomed Sci. 1998;5:101–110. doi: 10.1007/BF02258363. [DOI] [PubMed] [Google Scholar]
- Lin W-C, Lai C-H, Tang C-JC, Huang C-J, Tang TK. Identification and gene structure of a novel human PLZF-related transcription factor gene, TZFP. Biochem Biophys Res Comm. 1999;264:789–795. doi: 10.1006/bbrc.1999.1594. [DOI] [PubMed] [Google Scholar]
- Marchese A, Sawzdargo M, Nguyen T, Cheng R, Heng HH, Nowak T, Im DS, Lynch KR, George SR, O'Dowd F. Discovery of three novel orphan G-protein-coupled receptors. Genomics. 1999;56:12–21. doi: 10.1006/geno.1998.5655. [DOI] [PubMed] [Google Scholar]
- Marra M, Hillier L, Kucaba T, Allen M, Barstead R, Beck C, Blistain A, Bonaldo M, Bowers Y, Bowles L, et al. An encyclopedia of mouse genes. Nat Genet. 1999;21:191–194. doi: 10.1038/5976. [DOI] [PubMed] [Google Scholar]
- Miller G, Fuchs R, Lai E. IMAGE cDNA clones, UniGene clustering, and ACeDB: an integrated resource for expressed sequence information. Genome Res. 1997;7:1027–1032. doi: 10.1101/gr.7.10.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Takagi T. Gene recognition by combination of several gene-finding programs. Bioinformatics. 1998;14:665–675. doi: 10.1093/bioinformatics/14.8.665. [DOI] [PubMed] [Google Scholar]
- Oliver SG, van der Aart QJ, Agostoni-Carbone ML, Aigle M, Alberghina L, Alexandraki D, Antoine G, Anwar R, Ballesta JP, Benit P, et al. The complete DNA sequence of yeast chromosome III. Nature. 1992;357:38–46. doi: 10.1038/357038a0. [DOI] [PubMed] [Google Scholar]
- Olson MV. The human genome project. Proc Natl Acad Sci USA. 1993;90:4338–4344. doi: 10.1073/pnas.90.10.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picoult-Newberg L, Ideker TE, Pohl MG, Taylor SL, Donaldson MA, Nickerson DA, Boyce-Jacino M. Mining SNPs from EST databases. Genome Res. 1999;9:167–174. [PMC free article] [PubMed] [Google Scholar]
- Pietu G, Mariage-Samson R, Fayein NA, Matingou C, Eveno E, Houlgatte R, Decraene C, Vandenbrouck Y, Tahi F, Devignes MD, et al. The Genexpress IMAGE knowledge base of the human brain transcriptome: a prototype integrated resource for functional and computational genomics. Genome Res. 1999;9:195–209. [PMC free article] [PubMed] [Google Scholar]
- Rawlings CJ, Searls DB. Computational gene discovery and human disease. Curr Opin Genet Dev. 1997;7:416–423. doi: 10.1016/s0959-437x(97)80158-x. [DOI] [PubMed] [Google Scholar]
- Rossi DL, Vicari AP, Franz-Bacon K, McClanahan TK, Zlotnik A. Identification through bioinformatics of two new macrophage proinflammatory human chemokines: MIP-3alpha and MIP-3beta. J Immunol. 1997;158:1033–1036. [PubMed] [Google Scholar]
- Schuler GD. Pieces of the puzzle: expressed sequence tags and the catalog of human genes. J Mol Med. 1997;75:694–698. doi: 10.1007/s001090050155. [DOI] [PubMed] [Google Scholar]
- Sluder AE, Mathews SW, Hough D, Yin VP, Maina CV. The nuclear receptor superfamily has undergone extensive proliferation and diversification in nematodes. Genome Res. 1999;9:103–120. [PubMed] [Google Scholar]
- Tao W, Zhang S, Turenchalk GS, Stewart RA, St. John MA, Chen W, Xu T. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat Genet. 1999;21:177–181. doi: 10.1038/5960. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Wasinger VC, Humphery-Smith I. Small genes/gene-products in Escherichia coli K-12. FEMS Microbiol Lett. 1998;169:375–382. doi: 10.1111/j.1574-6968.1998.tb13343.x. [DOI] [PubMed] [Google Scholar]
- Watson JD. The human genome project: past, present, and future. Science. 1990;248:44–49. doi: 10.1126/science.2181665. [DOI] [PubMed] [Google Scholar]
- White O, Kerlavage AR. TDB: new databases for biological discovery. Methods Enzymol. 1996;266:27–40. doi: 10.1016/s0076-6879(96)66004-2. [DOI] [PubMed] [Google Scholar]
- Wu CH, Shivakumar S, Shivakumar CV, Chen SC. GeneFIND web server for protein family identification and information retrieval. Bioinformatics. 1998;14:223–224. doi: 10.1093/bioinformatics/14.2.223. [DOI] [PubMed] [Google Scholar]
- Yuan YP, Eulenstein O, Vingron M, Bork P. Towards detection of orthologues in sequence databases. Bioinformatics. 1998;14:285–289. doi: 10.1093/bioinformatics/14.3.285. [DOI] [PubMed] [Google Scholar]