

Abstract
Background
Gene duplication has often been invoked as a key mechanism responsible for evolution of new morphologies. The floral homeotic B-group gene family has undergone a number of gene duplication events, and yet the functions of these genes appear to be largely conserved. However, detailed comparative analysis has indicated that such duplicate genes have considerable cryptic variability in their functions. In the Solanaceae, two duplicate B-group gene lineages have been retained in three subfamilies. Comparisons of orthologous genes across members of the Solanaceae have demonstrated that the combined function of all four B-gene members is to establish petal and stamen identity, but that this function was partitioned differently in each species. These observations emphasize both the robustness and the evolvability of the B-system.
Scope
We provide an overview of how the B-function genes can robustly specify petal and stamen identity and at the same time evolve through changes in protein–protein interaction, gene expression patterns, copy number variation or alterations in the downstream genes they control. By using mathematical models we explore regulatory differences between species and how these impose constraints on downstream gene regulation.
Conclusions
Evolvability of the B-genes can be understood through the multiple ways in which the B-system can be robust. Quantitative approaches should allow for the incorporation of more biological realism in the representations of these regulatory systems and this should contribute to understanding the constraints under which different B-systems can function and evolve. This, in turn, can provide a better understanding of the ways in which B-genes have contributed to flower diversity.
Keywords: Robustness, evolvability, flower development, B-function, mathematical modelling
ROBUSTNESS, EVOLVABILITY AND TRANSCRIPTIONAL REGULATION IN PLANTS
Robustness versus evolvability
Most developmental systems are extraordinarily robust to genetic or environmental perturbations; nonetheless, such systems evolve into new, and potentially more complex, systems. Robustness allows for a developmental network to accumulate cryptic genetic variation, and thus provide the potential for evolutionary change (Rutherford and Lindquist, 1998; Specchia et al., 2010). One common form of such robustness is genetic redundancy; this is common in plant systems, with the Arabidopsis genome, for instance, displaying a considerable degree of redundancy (Martienssen and Irish, 1999). However, other types of genetic robustness can also promote the maintenance of a specific developmental outcome; this is often referred to as canalization (Waddington, 1957; Gibson and Wagner, 2000; Flatt, 2005). Evolvability, on the other hand, refers to the capacity of a system, and its inherent variation, to be adaptive under the right conditions. The processes of robustness and evolvability, then, appear to contradict each other, but a considerable literature has suggested that, paradoxically, robustness can promote evolvability (reviewed in Wagner, 2005). This can be understood by taking into account the many solutions possible to solve a single problem. Mechanisms that are functionally distinct at the molecular level can result in the same developmental outcome. This is the case for the B-class gene network in angiosperms. B-genes are known to be key regulators of petal and stamen identity in distantly related flowering plant species such as Arabidopsis, Antirrhinum, Petunia and Papaver, and this function appears to be conserved (Coen and Meyerowitz, 1991; Vandenbussche et al., 2004; Drea et al., 2007; Kramer et al., 2007; Benlloch et al., 2009). Yet it has now become clearer that related species of Solanaceae have evolved qualitatively different systems but that those systems have a conserved role in establishing petal and stamen identity (Geuten and Irish, 2010). Here we review, using the B-system as an example, the different ways such a system can be both robust and evolvable at the same time and we explore the constraints of such systems using theoretical models.
Evolution of transcriptional regulation in plants
The question of robustness versus evolvability is being addressed for several animal and microbial systems (Wagner, 2005). However, as plants utilize distinct molecular machinery to control their development and physiology, it appears that the specifics of how plants have responded to selective pressures may be different. The evolution of land plants, and in particular angiosperms, has been associated with MYB and MADS transcription factor family amplification (Riechmann et al., 2000). Moreover, flowering plant genome sizes vary over a much wider range than other taxonomic groups, which is probably a consequence of differences in ancestral polyploidy and the amplification of retrotransposons (Bennetzen, 2005; Vitte and Panaud, 2005; Barker et al., 2008). Although genomes can diminish in size after duplication (Devos et al., 2002), the requirement for a consistent stoichiometry of regulatory complexes potentially explains why transcriptional regulators are often retained (Blanc and Wolfe, 2004; Freeling and Thomas, 2006; Birchler et al., 2007). Because duplicate transcription factor genes can undergo sub- or neofunctionalization, they can lose redundancy, and so the absolute number of regulators per genome can be variable and can differ in complex ways between species.
Plants offer a number of interesting features pertinent to the study of the evolution of gene regulation, as compared with animal and microbial systems. First, the evidence for long-range ‘enhancers’, transcriptional control regions located at a distance from the coding region, is much more scarce for plants. To our knowledge only a few cases of distant cis-regulatory control elements have been found in plants; for instance, the maize tb1 gene possesses upstream enhancer elements that reside more than 41 kb away from the coding sequences (Clark et al., 2006). The presumed more restricted spatial localization of transcriptional control in plants could both facilitate their study as well as reflect salient differences in the ways in which gene regulation has evolved in plants. Second, the variation in genome sizes, usually seen as a difficulty, can also be an advantage in understanding regulatory evolution, because regulatory DNA may be relatively conserved between species, while non-functional DNA may accumulate mutations more rapidly (Peterson et al., 2009). The potential of these advantages remains to be explored more fully.
ROBUSTNESS IN CLASS B GENES THROUGH MULTIPLE COPIES
One commonly cited mechanism for robustness is gene redundancy. This is the idea that more than one gene performs the same function, and so any mutation in one gene is compensated for by the additional gene copy (Gu et al., 2003; Wagner, 2005). In the Solanaceae, for instance, duplicate copies of the MADS box B-gene lineages exist, and can, in some cases, compensate for the mutational loss of a redundant gene copy in specifying petal and stamen development (Fig. 1; Geuten and Irish, 2010). However, complete elimination of B-gene activity in these species indicates that this system is not robust against complete elimination in that evolutionarily unrelated developmental mechanisms cannot compensate for its absence. This is in contrast to other developmental genetic systems such as flowering time, for which backup regulatory mechanisms do exist. Flowering will occur even after a single pathway is eliminated as several other interacting but not fully redundant pathways can still promote the floral transition (Koorneef et al., 1998; Wang et al., 2009). However, the evolutionary history of class B genes throughout angiosperm diversification reveals another level of robustness in that multiple copies of these genes were generated and functions were partitioned throughout evolution.
Fig. 1.

Origin and evolution of B-function genes in Solanaceae. The four lineages, indicated in different colours, are present in tomato, Nicotiana and Petunia. Plausible relationships between these species are shown and their floral morphology is shown in the pictures. Below are a summary of yeast-two-hybrid studies by showing dimeric protein interactions. Most stringent are the interactions in tomato, followed by those in Petunia. Interactions showed least specificity in Nicotiana but N-terminally truncated constructs were used.
The molecular evolution of the B-system illustrates its robustness against changes in copy number throughout angiosperm evolution
B-gene copy number can vary between species; phylogenetic analyses of such duplication events can also provide a relative timing for the evolutionary origin of paralogues (Kramer et al., 1998, 2006, 2007; Becker and Theissen, 2003; Aoki et al., 2004; Kim et al., 2004; Stellari et al., 2004; Hernández-Hernández et al., 2007; Jaramillo and Kramer, 2007; Mondragón-Palomino et al., 2009; Viaene et al., 2009). Although Antirrhinum and Arabidopsis appear each to possess only a single DEFICIENS (DEF) and GLOBOSA (GLO) lineage gene, phylogenetic studies have shown that a duplication in the DEF lineage at the base of the core eudicots has resulted in the presence of a third B-class gene belonging to the TOMATO MADS6 (TM6) lineage in most core eudicot species (Kramer et al., 1998). Because the genome of Arabidopsis is sequenced and the transcriptome of Antirrhinum has been thoroughly sampled (Bey et al., 2004), it is very likely that these two model species have independently lost their TM6 lineage gene. Similarly, it is now clear that the core eudicot GLOBOSA lineage also underwent a duplication event, probably early in asterid diversification (Vandenbussche et al., 2004; Viaene et al., 2009; Geuten and Irish, 2010). This means that the B-system in Arabidopsis and Antirrhinum does not represent the same derivation of an ancestral regulatory system. These observations of gain and loss are an indication of the robustness of the B-system against copy number variation, as their roles in petal and stamen formation are conserved in all these species, despite the different gene duplication and gene loss events.
The function of B-genes can be redistributed over multiple paralogues when comparing related species
The B-system could further obtain robustness against elimination by distributing its functions over multiple fully redundant genes. However, there is little evidence for such redundancy in which B-gene paralogues share completely identical functions. Rather, duplicate B-genes tend to diversify in their function through changes at the level of gene expression, in their protein interaction specificity or potentially in the set of downstream genes they control. Robustness in this sense would reflect the partitioning of an ancestral function over multiple partially redundant paralogues. This was first indicated by the comparison of the DEF and TM6 lineages in Petunia and tomato, two species belonging to the family Solanaceae (Fig. 2). Although the B-system as a whole is still responsible for petal and stamen development, similar to the situation in Arabidopsis and Antirrhinum, the DEF and TM6 lineage genes do not have the same loss-of-function phenotypes in Petunia as compared with tomato. While DEF has a function necessary for petal development in Petunia, it is required for both petal and stamen development in tomato (de Martino et al., 2006; Rijpkema et al., 2006). It appears that the TM6 gene is not required for petal or stamen development in Petunia as its loss of function produces no phenotype. However, the knock-down phenotype of TM6 in tomato affects stamen development. In Nicotiana benthamiana (also belonging to the Solanaceae), knock-down of both TM6 and DEF has been shown to affect both petal and stamen development (Geuten and Irish, 2010; Liu et al., 2004). Similar observations can be made for the two GLO lineages present in Solanaceae. While GLO1 and GLO2 knock-downs in N. benthamiana result in phenotypes in petals and stamens, knock-down of either one of these genes in tomato results only in defects in stamen development. Furthermore, knock-out of the Petunia GLO1 gene weakly affects petal and stamen development, while knock-out of the GLO2 gene has no characterized phenotype (Vandenbussche et al., 2004). Together, this indicates that B-genes when present in multiple copies can fairly rapidly partition their function over multiple paralogues yet overall maintain their joint function in establishing petal and stamen identity. So in tomato, Petunia and N. benthamiana a full conversion of petal and stamens is the result when the function of both DEF and TM6 or GLO1 and GLO2 paralogues are lost (Vandenbussche et al., 2004; de Martino et al., 2006; Rijpkema et al., 2006; Geuten and Irish, 2010). In core eudicots, it is possible that this is a consequence of the B-system being more constrained by its autoregulation and obligate heterodimerization. However, in the case of opium poppy (Papaver somniferum), basal to core eudicots, there is also a partially redundant role for DEF paralogues in establishing petal and stamen identity rather than in the recruitment of novel functions for this biological process (Drea et al., 2007). Functional analyses in more species will clarify whether this is the case across angiosperms.
Fig. 2.

Phenotypes observed in loss-of-function lines of three Solanaceae species, tomato, Nicotiana benthamiana and Petunia. Flowers appearing as per the wild-type show no colouring; whorls phenotypes are shown in green. Less severe phenotypes are indicated in transparent green.
ROBUSTNESS AND EVOLVABILITY OF CIS-REGULATION
Evolvability of B-gene expression
Several instances of expression divergence of B-genes have been described (reviewed in Zahn et al., 2005; Hileman and Irish, 2009; Litt and Kramer, 2010). The fact that B-genes show variation in expression between species may be an inherent property related to the architecture of their promoter sequences. Recent studies in yeast have suggested that not all genes possess the same inherent capacity to evolve in terms of their expression patterns and that promoter architecture would be one determining factor in explaining this differential evolvability (Landry et al., 2007; Tirosh et al., 2009). Genes with TATA boxes, genes occupied by more nucleosomes, genes having more trans-acting factors that regulate their expression or genes that show unstable tandem repeats in their cis-regulatory regions appear to be more evolvable in yeast. To understand the robustness and evolvability of cis-regulation of the B-system genes, it would be important to investigate whether the same expression pattern can result from different functional architectures of promoter sequences in different species or whether the functional architecture is similar throughout evolution. This would require the functional analysis of B-gene promoter sequences in more than one species.
Does evolvability of B-gene expression contribute to evolutionary novelties?
Although expression patterns of B-genes have revealed differences between species, it has not yet been demonstrated through knock-down approaches that this variation is relevant to the origin of the identity of novel organs in evolution. Some already well-documented cases could provide such evidence in the future. Both in Ranunculales and orchids the gene expression patterns of paralogous B-genes correlate with different types of petals (Kramer et al., 2007; Mondragón-Palomino and Theissen, 2008). In orchids, B-genes belonging to the DEF lineage have acquired different but overlapping expression patterns and this could indicate that the different DEF genes acquired complementary roles to establish the identity of the different petaloid organs (Tsai et al., 2004; Mondragón-Palomino and Theissen, 2008; Chang et al., 2009). Functional evidence to support this claim is hard to obtain, however, because transgenic strategies are not straightforward. Also in Ranunculales, diversification of DEF genes has been implicated in the diversification of different petal types. Specifically, the AP3-III lineage acquired a petal-specific expression pattern within the order (Kramer et al., 2007; Rasmussen et al., 2009). Further functional analysis may confirm a function of AP3-III to be restricted to petal development in Ranunculales. Although these B-genes may be involved in the development of such organs, it will be difficult to distinguish between mutations in cis-regulation of B-genes or whether mutations in trans-acting upstream regulators can best explain these altered expression patterns. An alternative view to interpret these observed expression patterns is that they reflect the partitioning of function after duplication through changes in expression patterns of B-genes to establish petal and stamen identity. Such interpretation emphasizes robustness rather than a role for B-genes in contributing to diverse flowering plant morphologies. Again, stable transformation techniques may help to clarify these issues but need to be developed in these species.
This area of research is important because variation in the organs that plants use to attract pollinators is a frequent observation for flowering plants and is of ecological importance. One of the first and seemingly obvious research strategies in plant evo-devo was to investigate these instances for heterotopic expression of B-genes (e.g. Albert et al., 1998). Several instances have now been studied, but for none of these cases was hard evidence for a role of B-genes obtained (reviewed in Kanno et al., 2007; Hileman and Irish, 2009; Litt and Kramer, 2010). Interesting cases for future research may be the petaloid bracts of dogwoods (Cornus sp.), the petaloid sepals of Impatiens (Geuten et al., 2006) or the first whorl tepals from Tricyrtis (Liliales). These species have indeed been reported to be stably transformable (Adachi et al., 2005; Nakano et al., 2007; Feng et al., 2009; Dan et al., 2010).
ROBUSTNESS THROUGH PROTEIN–PROTEIN INTERACTIONS AND GENE BALANCE
Diversity of protein–protein interactions suggests evolution towards robustness of the B-system
B-proteins are best known to function as heterodimers. For instance, the interaction of Arabidopsis B-proteins AP3 and PI is necessary to enter the nucleus (McGonigle et al., 1996). When reviewing protein–protein interactions of MADS-domain proteins in general, it is important to distinguish between the properties of protein–protein interaction and whether such interacting proteins also have the capacity to bind DNA. Obligate heterodimerization and DNA binding of B-proteins appears to be acquired by core eudicots and conserved throughout this taxonomic group (Davies et al., 1996; Riechmann et al., 1996; Egea-Cortinez et al., 1999; Hernández-Hernández et al., 2007; Immink et al., 2010). Outside these core eudicots, B-class proteins have been shown to form homodimers or heterodimers in monocot species, this process being termed facultative heterodimerization (Winter et al., 2002; Tzeng et al., 2004). Recent new data are now also available for gymnosperms, in which it was shown that the Gnetum gnemon B-proteins form homodimers to bind DNA (Winter et al., 2002), but have now been shown to equally heterodimerize and bind DNA with several other MADS-domain proteins (Wang et al., 2010). Even more unexpected is that Gnetum B-proteins have been shown to loop DNA in tetrameric complexes, but only for a dimer of two homodimers binding DNA. It remains to be tested whether the capacity of Gnetum B-proteins to form higher order complexes will be extended to complexes involving also other MADS-domain proteins, such as AGL6 homologues (Rijpkema et al., 2006; Ohmori et al., 2009; Viaene et al., 2010). This is important to understand how the interaction network of MADS-domain proteins was remodelled around the time the angiosperm flower originated (Melzer and Theißen, 2009). An important advance in this area would be to obtain an accurate picture of the amino acid changes that result in changes in protein interaction specificity of B-proteins. One possibility is to study interaction specificity in more species (e.g. Liu et al., 2010). This should ultimately result in the ability to predict interactions from sequence information. A technical problem that slows down such accumulation of data is the cross-comparison of protein–protein interaction data between experiments and labs. A second step would be to establish ways to investigate MADS-domain protein–protein interaction in vivo. Efforts towards this goal have for now been mostly directed at other MADS-domain protein complexes (reviewed in Immink et al., 2010).
The evolution of protein interaction specificity of B-proteins apparently involves specialization as interactions and DNA binding starts off to be promiscuous and evolves to be more specific (Winter et al., 2002). This could be a recurring theme as B protein–protein interactions also in Solanaceae seem to show different specificity between species (Fig. 1; Vandenbussche et al., 2004; de Martino et al., 2006; Leseberg et al., 2008; Geuten and Irish, 2010). The answer to why such specialization could be advantageous has recently been proposed to be robustness. When using a stochastic model, the case of obligate heterodimerization provides a more certain decision behaviour than the case in which heterodimerization is only facultative (Lenser et al., 2009).
Robustness and evolvability of B-genes can be understood through gene balance
The gene balance hypothesis was formulated to understand dosage effects of genes in genomes. Effects of aneuploidy are more severe than those of polyploidy on phenotypes and gene expression levels. This can be explained by the fact that gene products often function as units of multiprotein complexes, and mutations that affect the stoichiometry of such a complex affect its function (reviewed in Birchler and Veita, 2007, 2010). The fact that B-proteins dimerize and multimerize implies that for B-genes dosage effects can also be expected. Such effects have indeed been demonstrated for duplicate B-genes in Petunia (Vandenbussche et al., 2004). The gene balance hypothesis also explains why B-gene lineages have survived rounds of whole genome duplication (Blanc and Wolfe, 2004; Freeling and Thomas, 2006; Birchler and Vieta, 2007). Because they function in protein complexes and their interaction partners in the complex have also duplicated, B-genes have to be retained after duplication. As such, gene balance could provide longer periods of time in which adaptive evolution can act on B-genes after duplication (Birchler and Veita, 2010; Geuten and Irish, 2010).
The study of the B-system also provides interesting information relevant to questions implicated by the gene balance hypothesis. An important outstanding issue is the molecular details by which multiprotein complexes can evolve when under the constraint of dosage balance (Birchler and Veita, 2010). Data in recent years suggest that B-proteins indeed evolve adaptively and this may relate to protein–protein interaction specificity (Martinez-Castilla and Alvarez-Buylla, 2003; Hernández-Hernández et al., 2007; Mondragón-Palomino et al., 2009; Viaene et al., 2009; Geuten and Irish, 2010). Gene balance could provide the necessary time to evolve specific protein–protein interactions that install improved robustness in the system. This would require detailed study comparing protein–protein interaction specificity between many relatively closely related species using a single experimental system while tracing adaptive evolution to test this hypothesis.
COOPERATIVITY IS ACQUIRED THROUGH OTHER FACTORS AND PROBABLY CONTRIBUTES TO ROBUST GENE EXPRESSION
Several mechanisms at the level of transcriptional regulation can contribute to robustness of the B-system. One of these mechanisms is cooperativity, which is defined as the facilitated binding to additional binding sites by binding a first site. This provides robustness of gene expression because sharp transitions in expression level can thus be established with small changes in regulator concentration. B-proteins have been illustrated to bind cooperatively to their DNA binding sequences, although probably not alone but in combination with other MADS-domain proteins such as SEPALLATA and APETALA1 (Egea-Cortines et al., 1999; Melzer and Theißen, 2009; Melzer et al., 2009; Immink et al., 2010). Binding of MADS-domain proteins in general has been shown to show specificity for the CC-(A/T)6-GG sequence but no studies have revealed a specific preference of B-protein dimers for a subset of this canonical sequence. Binding site selection studies such as performed for AGL15, another MADS-domain protein, could help to understand binding specificity of B-proteins and this could aid in defining position weight matrices to explore binding specificity for sites in genomes (Stormo, 2000; Tang and Perry, 2003). One application of this type of data could be to complement experimental approaches in sequenced genomes to find downstream targets (Zik and Irish, 2003; Bey et al., 2004). Chromatin immunoprecipation combined with microarray or next-generation sequencing should further reveal binding sites experimentally as has recently been performed for SEPALLATA3 and APETALA1 (Kaufmann et al., 2009, 2010a, b).
The presence of multiple conserved CArG boxes in promoter sequences of B-genes suggests also that binding of upstream regulators to B-gene promoters can be cooperative (Hill et al., 1998; Tilly et al., 1998; Koch et al., 2001; Rijpkema et al., 2006). An indication of the possible extent of this upstream cooperativity can be derived from quantitative measurements for the binding of SEP3 quartets to perfectly spaced consensus CArG sites. The relative affinity for binding of a SEP3 dimer when a first dimer was already bound increases around 80-fold (Melzer et al., 2009). A promising additional tool in this respect could be surface plasmon resonance, which has been illustrated to work with longer cis-regulatory sequences and has the power of providing quantitative measurements (Moyroud et al., 2009).
MATHEMATICAL MODELS DESCRIBING THE GENE REGULATORY NETWORK OF FLOWER DEVELOPMENT
Models are commonly used in biology to describe the current understanding of a regulatory system explicitly and to indicate the gaps in our understanding. In flower development, two such models are the ABC model and the floral quartet model, which represent compatible genetic and molecular models of floral organ identity specification (Coen and Meyerowitz, 1991; Theißen and Saedler, 2001). Extensions to such descriptive models are now possible through mathematical modelling. Computerized modelling approaches have several advantages in that (1) they allow more complexity, (2) they can incorporate quantitative experimental data and (3) they allow in silico hypothesis testing through simulation.
To represent the gene regulatory networks to be analysed, a mathematical formalism needs to be chosen (Karlebach and Shamir, 2008). Different formalisms have been used that represent different trade-offs between the type of input data available and biological realism. The most often used formalism in biological modelling is the use of ordinary differential equations (ODEs), because these allow precise numerical prediction of the behaviour of a biological system (e.g. Conrad and Tyson, 2006; Alon, 2007).
ODE modelling assumes quantitative knowledge of the kinetics of the biological system, information that is mostly absent. Therefore, more qualitative approaches have been explored first in the modelling of flower development and can be sufficient to capture many aspects (e.g. De Jong and Ropers, 2006). One such more qualitative approach is the use of a Boolean logical network. These models use discrete representations of the biological data (such as expression absence or presence) and incorporate logical rules grounded in experiments to connect these data (Mendoza et al., 1999; Espinosa-Soto et al., 2004). These models have illustrated the robustness of flower development to different initial conditions and to stochastic noise. An extension of these models in the form of discrete abstraction of ODEs, Glass equations, has also been used (Alvarez-Buylla et al., 2008).
A second type of formalism has used Petri nets and its extensions to describe the Arabidopsis flower development gene regulatory network (Kaufmann et al., 2010a, b). Petri nets have been chosen to represent biological reactions because they allow an intuitive graphical representation of biological processes and a well-defined mathematical formalism. An important advantage is that this is implemented in the Cell Illustrator software, which facilitates the user-friendly construction, visualization and simulation of the network. A main contribution of this model is the complexity captured through the many genes and genetic interactions incorporated. Simulations of this model also highlight the importance of heteromeric higher-order complexes and autoregulatory feedback loops for the generation of robust organ-specific gene expression.
The classical ODE modelling strategy for biological systems has only recently been applied to understand flower development (Van Moorik et al., 2010). ODE modelling requires (1) a diagram to represent the different molecular interactions, (2) assigning a set of kinetic rate equations that relate the rate of a reaction to the concentrations of substrates and (3) values for the parameters in the rate reactions. The first two elements are not problematic, because diagrams can be derived from the literature and the kinetics of the reactions in gene regulatory networks such as cooperative gene regulation are well understood. Estimating parameters, however, is rarely possible although plausible guesses can be made or parameter ranges can be tested. Simulations of the Van Moorik model have equally stressed the importance of dimers in the gene regulatory network and generated predictions for mutations yet to be made, offering further tools for model validation.
Finally, simulation of evolutionary different B-systems has been undertaken using the Gillespie algorithm, which is a general and exact way to take into account stochasticity (Lenser et al., 2009). Stochastic behaviour is the random unpredictable behaviour that can occur when only a few molecules are involved and is relevant and inherent to many biological systems. A major conclusion of this study is that obligate heterodimerization of B-proteins, as observed in core eudicots, has the evolutionary advantage of being more robustly able to control cell-identity switching under stochastic conditions. This provides an explanation as to why in evolution obligate heterodimerization would have evolved.
Mathematical models to explore robustness and evolvability of B-systems
To explore the use of mathematical modelling for our understanding of the evolution of the B-system, we built gene regulatory models. The formalism used here is to automatically generate a set of ODEs for graphically designed models in CellDesigner4·1. Our goal is to examine, in theory, the possible effects of evolutionary changes to different regulatory systems, rather than to provide an encompassing model that is predictive and can be experimentally validated for a single species. This approach illustrates (1) how different theoretical regulatory systems show a different sensitivity to parameter changes and acquire robustness and (2) how user-friendly software permits biologists to incorporate mathematical modelling in thinking and experimenting.
We present three models that differ in the number of B-genes involved, their protein interactions and cross-regulatory interactions. They represent cases which may be similar to B-gene regulation in gymnosperms (Fig. 3A), Arabidopsis and Antirrhinum (Fig. 3B) or tomato (Fig. 3C; see Appendix for the methods employed). For each of these models, Hill functions were taken to represent transcriptional regulation and homo- and heterodimerization was taken as a reversible process modelled by mass action kinetics. Decay of molecules in the model was assumed to be linearly proportional to the species concentration. With the same parameters taken for the same kinetic equations in all three models, they produce a similar outcome for the activation of the gene downstream (Fig. 4). The third model results in double the absolute expression of downstream genes and this is because in this model downstream genes are activated by two heterodimers of B-proteins rather than one in the first two models and this activation is independent (Fig. 3), making this model more robust in the sense of downstream activation. To illustrate the robustness of these models, we will study the behaviour of each model in response to changes in parameter values, which can be interpreted as evolutionary perturbations. We varied activator concentration, cooperativity of transcriptional regulation as measured by the Hill coefficient and the rate constant for dimerization.
Fig. 3.

CellDesigner4·1 models for B-gene systems. Yellow blocks represent genes, pale green blocks proteins, parallelograms mRNA and pink discs as degradation. (A) Model in which a single activated B-gene of which the protein product homodimerizes and autoactivates. The homodimer regulates a single downstream gene. (B) Model in which a DEF and GLO gene produce protein products that obligately heterodimerize to autoactivate and regulate a downstream gene. (C) Model in which protein products of DEF, TM6, GLO1 and GLO2 genes obligately heterodimerize, cross-regulate and autoregulate and activate two downstream genes.
Fig. 4.
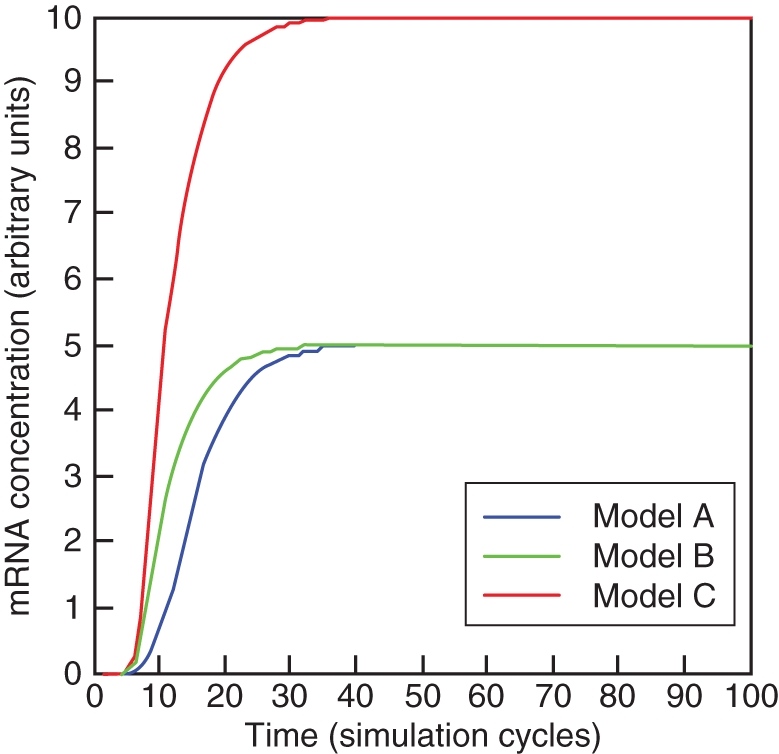
Activation over time of a single downstream gene by three different B-systems, corresponding to Fig. 3. The same parameters and initiating values were taken for each model.
The simplest model represents an autoregulatory circuit in which a stable activator concentration is responsible for initiating transcription of a single B-gene. The protein product homodimerizes and autoregulates its own expression and the expression of a single downstream gene (Fig. 3A). The parameters that have the strongest influence over the switching behaviour of the downstream gene are the activator concentration (Fig. 5A), the rate constant for homodimerization (Fig. 5B) and the cooperativity of direct regulation of the downstream gene (Fig. 5C). The cooperativity of activation of the single B-gene and autoregulation have only minimal impact on the switch to ON of the downstream gene (data not shown).
Fig. 5.

Activation of a single downstream gene by B-systems. (A,D,G) Model A, (B,E,H) model B, (C,F,I) model C. (A–C) Activator concentration varies over a 100-fold range. (A) Model A can activate a downstream gene over a wider range of activator concentrations. (B,C) Models B and C require stronger concentrations of activator to activate a downstream gene. (D–F) Hill coefficient of cooperativity in the activation of the downstream gene as shown in the figure varies over a 4-fold range. (D) In model A the downstream gene is most sensitive to variation of the cooperativity coefficient. Model C is most robust because a second complex can activate the same downstream gene independently. (G,H) Rate constant of association for homodimerization (G), heterodimerization (H) or heterodimerization of one or both heterodimers varies over a 100-fold range. Model A using homodimerization is more sensitive to this parameter than model B or C.
The second model uses obligate heterodimerization of two B-proteins rather than homodimerization (Fig. 5B). This implies that two B-genes need to be activated, which then can only autoregulate their expression and the expression of a downstream gene through heterodimerization. Transcription of the downstream gene in this model is more sensitive to low activator concentrations than is the case in the first model (Fig 5D). Thus, in species such as Arabidopsis and Antirrhinum, which may represent real-life examples of this model, one would expect a more elaborate control of the activation of the B-system than in species which use a conceptually simpler system of homodimerization of a single B-protein. Transcription of the downstream gene is also somewhat less sensitive to variations in association rate between the two different B-proteins (compare Fig. 5B and 5E). This becomes intuitive when considering that two independently regulated genes provide the proteins to interact, rather than a single gene in the homodimerization model. Apparently, using two different genes that obligately heterodimerize can be more robust to changes in interaction strength of the proteins. Similar to the previous model, cooperativity in activating the B-genes or in autoregulating the B-genes has only a minor impact on downstream transcription, while cooperativity of the direct regulation of the downstream gene does affect its transcription (Fig. 5F). This illustrates the intuitive point that the transcription of the downstream genes is more robust to changes in cooperativity of activation when these changes occur upstream in the regulation cascade, but is most sensitive to changes in its direct regulation, such as mutations in the cis-regulatory binding sites in its promoter.
The third model incorporates four different genes that form two types of heterodimer (Fig. 3C). Such regulation may be similar to that found in tomato (Leseberg et al., 2008), but may be different in related species such as tobacco and Petunia for which interaction specificity was not found to be as exclusive (Vandenbussche et al., 2004; de Martino et al., 2006; Rijpkema et al., 2006; Geuten and Irish, 2010). In this model, activator concentration affecting downstream genes varies over the same range as in the second model (compare Fig. 3D and 3G). This may be somewhat surprising as activation molecules now have to activate four genes rather than two, but this effect may be compensated for by the fact that each B-gene is both autoregulated and cross-regulated by a second B-dimer. This third model is also most robust to changes in the association rate of one of the dimers and to changes in the cooperativity of direct activation of the downstream gene (Fig. 5H, I). This is trivial as a second and largely independent system is redundant. In the absence of one of both B-protein interactions, expression of downstream genes reduces to only half, further illustrating that being regulated by two fully redundant dimers makes downstream gene expression partially robust when this direct regulation is independent (data not shown).
CONCLUSIONS
Transcriptional regulation of the B-system is only well characterized in a single model species, Arabidopsis thaliana. Comparative studies including more species indicate that the evolution of B-proteins is characterized by duplication, retention through gene balance and adaptive evolution that may result in specific protein–protein interactions or altered cis-regulation. Overall, the experimental evidence demonstrates that the B-system can be robust in different functional ways. It is unclear whether the molecular evolution of the B-system has been instrumental in the origin of novelties in plant evolution. Although expression suggests that the B-system is involved in establishing the identity of organs that are evolutionarily derived from petals, this function has yet to be experimentally demonstrated. In addition, it will be difficult to test whether mutations in B-genes were causative in the origin of these organs or whether B-genes were recruited through changes in upstream regulation.
To understand in detail the constraints of evolvability and the different ways in which the B-system can be robust will require more extensive functional characterization in more species. In addition, quantitative measurements will add more biological realism into models that try to capture the molecular details of transcriptional regulation. These details are important to fully test our understanding of the B-system and the way it is constrained in evolution.
APPENDIX
To model the gene regulatory interactions involving B-genes as depicted in Fig. 3, we used the CellDesigner4·1 software. This software has as the main advantage that it allows the easy graphical construction of a gene regulatory network and supports the SBML format (Systems Biology Markup Language). This in turn allows communication and reuse of models in different software programs. Automatic generation and simulation of ODEs associated with the network kinetic rate equations is possible through the SBML ODE Solver (Machne et al., 2006). All reactions were taken as irreversible, except for protein dimerization. The kinetics of transcriptional activation were modelled using a Hill function. The Hill equation has three parameters n, K and β. Cooperativity n was initiated as n = 2, the activation coefficient K was initiated as K = 0·1 and the maximal rate of expression was initiated as β = 1. The integration of multiple input signals was achieved by taking the weighted sum of multiple input signals with different regulators taking equal weights. Protein dimerization was assumed to be reversible and followed mass action kinetics. Translation and degradation was assumed to be not affected by regulation and followed zero-order mass-action kinetics. All kinetic parameters were initiated as k = 0·1.
LITERATURE CITED
- Adachi Y, Mori S, Nakano M. Okubo W, Miller B, Chastagner GA, editors. Agrobacterium-mediated production of transgenic plants in Tricyrtis hirta (Liliaceae) IX International Symposium on Flower Bulbs. Acta Horticulturae. 2005;673 [Google Scholar]
- Albert V, Gustafsson M, Di Laurenzio L. Ontogenetic systematics, molecular developmental genetics and the angiosperm petal. In: Soltis D, Soltis P, Doyle J, editors. Molecular systematics of plants II. The Netherlands: Kluwer Academic Publishers; 1998. [Google Scholar]
- Alon U. An introduction to systems biology. Boca Raton, FL: Chapman and Hall; 2007. [Google Scholar]
- Alvarez-Buylla E, Chaos A, Aldana M, et al. Floral morphogenesis: stochastic explorations of a gene network epigenetic landscape. Plos One. 2008;3:e3626. doi: 10.1371/journal.pone.0003626. doi:10.1371/journal.pone.0003626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki S, Uehara K, Imafuku M, Hasebe M, Ito M. Phylogeny and divergence of basal angiosperms inferred from APETALA3- and PISTILLATA-like MADS-box genes. Journal of Plant Research. 2004;117:229–244. doi: 10.1007/s10265-004-0153-7. [DOI] [PubMed] [Google Scholar]
- Barker M, Kane N, Matvienko M, et al. Multiple paleopolyploidizations during the evolution of the Compositae reveal parallel patterns of duplicate gene retention after millions of years. Molecular Biology and Evolution. 2008;25:2445–2455. doi: 10.1093/molbev/msn187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A, Theissen G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Molecular Phylogenetics and Evolution. 2003;29:464–489. doi: 10.1016/s1055-7903(03)00207-0. [DOI] [PubMed] [Google Scholar]
- Benlloch R, Roque E, Ferrandiz C, et al. Analysis of B function in legumes: PISTILLATA proteins do not require the PI motif for floral organ development in Medicago truncatula. Plant Journal. 2009;60:102–111. doi: 10.1111/j.1365-313X.2009.03939.x. [DOI] [PubMed] [Google Scholar]
- Bennetzen J. Transposable elements, gene creation and genome rearrangement in flowering plants. Current Opinion in Genetics and Development. 2005;15:621–627. doi: 10.1016/j.gde.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Bey M, Stuber K, Fellenberg K, et al. Characterization of antirrhinum petal development and identification of target genes of the class B MADS box gene DEFICIENS. Plant Cell. 2004;16:3197–3215. doi: 10.1105/tpc.104.026724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J, Veitia R. The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell. 2007;19:395–402. doi: 10.1105/tpc.106.049338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J, Veitia R. The gene balance hypothesis: implications for gene regulation, quantitative traits and evolution. New Phytologist. 2010;186:62–64. doi: 10.1111/j.1469-8137.2009.03087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J, Yao H, Chudayalandi S. Biological consequences of dosage dependent gene regulatory systems. Biochimica et Biophysica Acta. 2007;1769:422–428. doi: 10.1016/j.bbaexp.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Wolfe K. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16:1667–1678. doi: 10.1105/tpc.021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-Y, Kao N-H, Li Y-Y, et al. Characterization of the possible roles for B class MADS box genes in regulation of perianth formation in orchid. Plant Physiology. 2009;152:837–853. doi: 10.1104/pp.109.147116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R, Nussbaum Wagler T, Quijada P, Doebley J. A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nature Genetics. 2006;38:394–397. doi: 10.1038/ng1784. [DOI] [PubMed] [Google Scholar]
- Coen E, Meyerowitz E. The war of the whorls: genetic interactions controlling flower development. Nature. 1991;353:31–37. doi: 10.1038/353031a0. [DOI] [PubMed] [Google Scholar]
- Conrad E, Tyson J. Modeling molecular interaction networks with nonlinear ordinary differential equations. In: Szallasi Z, Stelling J, Periwal V, editors. System modeling in cellular biology. Cambridge, MA: The MIT Press; 2006. pp. 97–125. [Google Scholar]
- Dan Y, Baxter A, Zhang S, Pantazis CJ, Veuilleux RE. Development of efficient plant regeneration and transformation system for Impatiens using Agrobacterium tumefaciens and multiple bud cultures as explants. BMC Plant Biology. 2010;10:165. doi: 10.1186/1471-2229-10-165. doi:10.1186/1471-2229-10-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B, Egea-Cortines M, de Andrade-Silva E, Saedler H, Sommer H. Multiple interactions amongst floral homeotic MADS box proteins. EMBO Journal. 1996;15:4330–4343. [PMC free article] [PubMed] [Google Scholar]
- De Jong H, Ropers D. Qualitative approaches to the analysis of genetic regulatory networks. In: Szallasi Z, Stelling J, Periwal V, editors. System modeling in cellular biology. Cambridge: The MIT Press; 2006. pp. 125–149. [Google Scholar]
- Devos K, Brown J, Bennetzen J. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Research. 2002;12:1075–1079. doi: 10.1101/gr.132102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drea S, Hileman L, de Martino G, Irish V. Functional analyses of genetic pathways controlling petal specification in poppy. Development. 2007;134:4157–4166. doi: 10.1242/dev.013136. [DOI] [PubMed] [Google Scholar]
- Egea-Cortines M, Saedler H, Sommer H. Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO Journal. 1999;18:5370–5379. doi: 10.1093/emboj/18.19.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Soto C, Padilla-Longoria P, Alvarez-Buylla E. A gene regulatory network model for cell-fate determination during Arabidopsis thaliana flower development that is robust and recovers experimental gene expression profiles. Plant Cell. 2004;16:2923–2939. doi: 10.1105/tpc.104.021725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Qu R, Zhou L, Xie D, Xiang Q. Shoot regeneration of dwarf dogwood (Cornus canadensis L.) and morphological characterization of the regenerated plants. Plant Cell, Tissue and Organ Culture. 2009;97:27–37. [Google Scholar]
- Flatt T. The evolutionary genetics of canalization. Quarterly Review of Biology. 2005;80:287–316. doi: 10.1086/432265. [DOI] [PubMed] [Google Scholar]
- Freeling M, Thomas B. Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Research. 2006;16:805–814. doi: 10.1101/gr.3681406. [DOI] [PubMed] [Google Scholar]
- Geuten K, Irish V. Hidden variability of floral homeotic B genes in Solanaceae provides a molecular basis for the evolution of novel functions. Plant Cell. 2010;22:2562–2578. doi: 10.1105/tpc.110.076026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuten K, Becker A, Kaufmann K, et al. Petaloidy and petal identity MADS-box genes in the balsaminoid genera Impatiens and Marcgravia. Plant Journal. 2006;47:501–518. doi: 10.1111/j.1365-313X.2006.02800.x. [DOI] [PubMed] [Google Scholar]
- Gibson G, Wagner G. Canalization in evolutionary genetics: a stabilizing theory? Bioessays. 2000;22:372–380. doi: 10.1002/(SICI)1521-1878(200004)22:4<372::AID-BIES7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Gu Z, Steinmetz L, Gu X, Davis R, Li W. Role of duplicate genes in genetic robustness against null mutations. Nature. 2003;421:63–66. doi: 10.1038/nature01198. [DOI] [PubMed] [Google Scholar]
- Hernández-Hernández T, Martinez-Castilla L, Alvarez-Buylla E. Functional diversification of B MADS-box homeotic regulators of flower development: adaptive evolution in protein–protein interaction domains after major gene duplication events. Molecular Biology and Evolution. 2007;24:465–481. doi: 10.1093/molbev/msl182. [DOI] [PubMed] [Google Scholar]
- Hileman L, Irish V. More is better: the uses of developmental genetic data to reconstruct perianth evolution. American Journal of Botany. 2009;96:83–95. doi: 10.3732/ajb.0800066. [DOI] [PubMed] [Google Scholar]
- Hill T, Day C, Zondlo S, Thackeray A, Irish V. Discrete spatial and temporal cis-acting elements regulate transcription of the Arabidopsis floral homeotic gene APETALA3. Development. 1998;125:1711–1721. doi: 10.1242/dev.125.9.1711. [DOI] [PubMed] [Google Scholar]
- Immink R, Kaufmann K, Angenent G. The ‘ABC’ of MADS domain protein behaviour and interactions. Seminars in Cell Development and Biology. 2010;21:87–93. doi: 10.1016/j.semcdb.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Jaramillo M, Kramer E. Molecular evolution of the petal and stamen identity genes, APETALA3 and PISTILLATA, after petal loss in the Piperales. Molecular Phylogenetics and Evolution. 2007;44:598–609. doi: 10.1016/j.ympev.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Kanno M, Nakada Y, Akita Y, Hirai M. Class B gene expression and the modified ABC model in nongrass monocots. TSW Development and Embryology. 2007;2:17–28. doi: 10.1100/tsw.2007.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlebach G, Shamir R. Modeling and analysis of gene regulatory networks. Nature. 2008;9:770–780. doi: 10.1038/nrm2503. [DOI] [PubMed] [Google Scholar]
- Kaufmann K, Muino J, Jauregui R, et al. Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biology. 2009;7:e1000090. doi: 10.1371/journal.pbio.1000090. doi:10.1371/journal.pbio.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann K, Nagasaki M, Jauregui R. Modelling the molecular interactions in the flower developmental network of Arabidopsis thaliana. In silico Biology. 2010a;10:0008. doi: 10.3233/ISB-2010-0414. http://www.bioinfo.de/isb/2010/10/0008/ [DOI] [PubMed] [Google Scholar]
- Kaufmann K, Wellmer F, Muino J, et al. Orchestration of floral initiation by APETALA1. Science. 2010b;328:85–89. doi: 10.1126/science.1185244. [DOI] [PubMed] [Google Scholar]
- Kim S, Yoo M-J, Albert V, Farris J, Soltis P, Soltis D. Phylogeny and diversification of B-function MADS-box genes in angiosperms: evolutionary and functional implications of a 260 million-year-old duplication. American Journal of Botany. 2004;91:2102–2118. doi: 10.3732/ajb.91.12.2102. [DOI] [PubMed] [Google Scholar]
- Koch M, Weisshaar B, Kroymann J, Haubold B, Mitchell-Olds T. Comparative genomics and regulatory evolution: conservation and function of the Chs and Apetala3 promoters. Molecular Biology and Evolution. 2001;18:1882–1891. doi: 10.1093/oxfordjournals.molbev.a003729. [DOI] [PubMed] [Google Scholar]
- Koorneef M, Alonso-Blanco C, Peeters A, Soppe W. Genetic control of flowering time in Arabidopsis. Annual Review of Plant Physiology and Plant Molecular Biology. 1998;49:345–370. doi: 10.1146/annurev.arplant.49.1.345. [DOI] [PubMed] [Google Scholar]
- Kramer E, Dorit R, Irish V. Molecular evolution of genes controlling petal and stamen development: duplication and divergence within the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics. 1998;149:765–783. doi: 10.1093/genetics/149.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer E, Su H-J, Wu C-C, Hu J-M. A simplified explanation for the frameshift mutation that created a novel C-terminal motif in the APETALA3 gene lineage. BMC Evolutionary Biology. 2006;6:30. doi: 10.1186/1471-2148-6-30. doi:10.1186/1471-2148-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer E, Holappa L, Gould B, Jaramillo M, Setnikov D, Santiago P. Elaboration of B gene function to include the identity of novel floral organs in the lower eudicot Aquilegia. Plant Cell. 2007;19:750–766. doi: 10.1105/tpc.107.050385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry C, Lemos B, Rifkin S, Dickinson W, Hartl D. Genetic properties influencing the evolvability of gene expression. Science. 2007;317:118. doi: 10.1126/science.1140247. [DOI] [PubMed] [Google Scholar]
- Lenser T, Theißen G, Dittrich P. Developmental robustness by obligate interaction of class B floral homeotic genes and proteins. PLoS Computers and Biology. 2009;5:e1000264. doi: 10.1371/journal.pcbi.1000264. doi:10.1371/journal.pcbi.1000264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leseberg C, Eissler C, Wang X, Johns M, Duvall M, Mao L. Interaction study of MADS-domain proteins in tomato. Journal of Experimental Botany. 2008;59:2253–2265. doi: 10.1093/jxb/ern094. [DOI] [PubMed] [Google Scholar]
- Litt A, Kramer E. The ABC model and the diversification of floral organ identity. Seminars in Cell Development and Biology. 2010;21:129–137. doi: 10.1016/j.semcdb.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Liu C, Zhang J, Zhang N, et al. Interactions among proteins of floral MADS-box genes in basal eudicots: implications for evolution of the regulatory network for flower development. Molecular Biology and Evolution. 2010;27:1598–1611. doi: 10.1093/molbev/msq044. [DOI] [PubMed] [Google Scholar]
- Liu Y, Nakayama N, Schiff M, Irish V, Dinesh-Kumar S. Virus induced gene silencing of a DEFICIENS ortholog in Nicotiana benthamiana. Plant Molecular Biology. 2004;54:701–711. doi: 10.1023/B:PLAN.0000040899.53378.83. [DOI] [PubMed] [Google Scholar]
- Martienssen R, Irish V. Copying out our ABCs: the role of gene redundancy in interpreting genetic hierarchies. Trends in Genetics. 1999;15:435–437. doi: 10.1016/s0168-9525(99)01833-8. [DOI] [PubMed] [Google Scholar]
- Martinez-Castilla L, Alvarez-Buylla E. Adaptive evolution in the Arabidopsis MADS-box gene family inferred from its complete resolved phylogeny. Proceedings of the National Academy of Sciences USA. 2003;11:13407–13412. doi: 10.1073/pnas.1835864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Martino G, Pan I, Emmanuel E, Levy A, Irish V. Functional analyses of two tomato APETALA3 genes demonstrate diversification in their roles in regulating floral development. Plant Cell. 2006;18:1833–1845. doi: 10.1105/tpc.106.042978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machne R, Finney A, Müller S, Lu J, Widder S, Flamm C. The SBML ODE Solver Library: a native API for symbolic and fast numerical analysis of reaction networks. Bioinformatics. 2006;22:1406–1407. doi: 10.1093/bioinformatics/btl086. [DOI] [PubMed] [Google Scholar]
- McGonigle B, Bouhidel K, Irish V. Nuclear localization of the Arabidopsis APETALA3 and PISTILLATA homeotic gene products depends on their simultaneous expression. Genes and Development. 1996;10:1812–1821. doi: 10.1101/gad.10.14.1812. [DOI] [PubMed] [Google Scholar]
- Melzer R, Theißen G. Reconstitution of ‘floral quartets’ in vitro involving class B and class E floral homeotic proteins. Nucleic Acids Research. 2009;37:2723–2736. doi: 10.1093/nar/gkp129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer R, Verelst W, Theißen G. The class E floral homeotic protein SEPALLATA3 is sufficient to loop DNA in ‘floral quartet’-like complexes in vitro. Nucleic Acids Research. 2009;37:144–157. doi: 10.1093/nar/gkn900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza L, Thieffry D, Alvarez-Buylla E. Genetic control of flower morphogenesis in Arabidopsis thaliana: a logical analysis. Bioinformatics. 1999;15:593–606. doi: 10.1093/bioinformatics/15.7.593. [DOI] [PubMed] [Google Scholar]
- Mondragón-Palomino M, Theissen G. MADS about the evolution of orchid flowers. Trends in Plant Science. 2008;13:51–59. doi: 10.1016/j.tplants.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Mondragón-Palomino M, Hiese L, Harter A, Koch M, Theissen G. Positive selection and ancient duplications in the evolution of class B floral homeotic genes of orchids and grasses. BMC Evolution and Biology. 2009;9:81. doi: 10.1186/1471-2148-9-81. doi:10.1186/1471-2148-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyroud E, Reymond M, Hames C, Parcy F, Scutt C. The analysis of entire gene promoters by surface plasmon resonance. Plant Journal. 2009;59:851–858. doi: 10.1111/j.1365-313X.2009.03903.x. [DOI] [PubMed] [Google Scholar]
- Nakano M, Umehara H, Hara Y, et al. Flower form alteration by genetic transformation with the class B MADS-box genes of Agapanthus praecox spp. orientalis in transgenic dicot and monocot plants. Molecular Breeding. 2007;20:425–429. [Google Scholar]
- Ohmori S, Kimizu M, Sugita M, et al. MOSAIC FLORAL ORGANS1, an AGL6-like MADS-box gene, regulates floral organ identity and meristem fate in rice. Plant Cell. 2009;21:3008–3025. doi: 10.1105/tpc.109.068742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson B, Hare E, Lyer V, et al. Big genomes facilitate the comparative identification of regulatory elements. PLoS ONE. 2009;4:e4688. doi: 10.1371/journal.pone.0004688. doi:10.1371/journal.pone.0004688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen D, Kramer E, Zimmer E. One size fits all? Molecular evidence for a commonly inherited petal identity program in Ranunculales. American Journal of Botany. 2009;96:96–109. doi: 10.3732/ajb.0800038. [DOI] [PubMed] [Google Scholar]
- Riechmann J, Wang M, Meyerowitz E. DNA-binding properties of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA and AGAMOUS. Nucleic Acids Research. 1996;24:3134–3141. doi: 10.1093/nar/24.16.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann J, Heard J, Martin G, et al. Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–2110. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
- Rijpkema A, Royaert S, Zethof J, van der Weerden G, Gerats T, Vandenbussche M. Analysis of the Petunia TM6 MADS box gene reveals functional divergence within the DEF/AP3 lineage. Plant Cell. 2006;18:1819–1832. doi: 10.1105/tpc.106.042937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford RL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- Specchia V, Piacentini L, Tritto P, et al. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature. 2010;463:662–665. doi: 10.1038/nature08739. [DOI] [PubMed] [Google Scholar]
- Stellari G, Jaramillo M, Kramer E. Evolution of the APETALA3 and PISTILLATA lineages of MADS-box-containing genes in the basal angiosperms. Molecular Biology and Evolution. 2004;21:506–519. doi: 10.1093/molbev/msh044. [DOI] [PubMed] [Google Scholar]
- Stormo G. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- Tang W, Perry S. Binding site selection for the plant MADS domain protein AGL15: an in vitro and in vivo study. Journal of Biological Chemistry. 2003;278:28154–28159. doi: 10.1074/jbc.M212976200. [DOI] [PubMed] [Google Scholar]
- Theißen G, Saedler H. Floral quartets. Nature. 2001;409:469–471. doi: 10.1038/35054172. [DOI] [PubMed] [Google Scholar]
- Tilly JJ, Allen DW, Jack T. The CArG boxes in the promoter of the Arabidopsis floral organ identity gene APETALA3 mediate diverse regulatory effects. Development. 1998;125:1647–1657. doi: 10.1242/dev.125.9.1647. [DOI] [PubMed] [Google Scholar]
- Tirosh I, Barkai N, Verstrepen K. Promoter architecture and the evolvability of gene expression. Journal of Biology. 2009;8:95. doi: 10.1186/jbiol204. doi:10.1186/jbiol204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WC, Kuoh C, Chuang M, Chen W, Chen H. Four DEF-like MADS box genes displayed distinct floral morphogenetic roles in Phalaenopsis orchid. Plant Cell Physiology. 2004;45:831–844. doi: 10.1093/pcp/pch095. [DOI] [PubMed] [Google Scholar]
- Tzeng TY, Liu Hc, Yang CH. The C-terminal sequence of LMADS1 is essential for the formation of homodimers for B function proteins. Journal of Biological Chemistry. 2004;279:10747–55. doi: 10.1074/jbc.M311646200. [DOI] [PubMed] [Google Scholar]
- Vandenbussche M, Zethof J, Royaert S, Weterings K, Gerats T. The duplicated B-class heterodimer model: whorl-specific effects and complex genetic interactions in Petunia hybrida flower development. Plant Cell. 2004;16:741–754. doi: 10.1105/tpc.019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Moorik S, van Dijk A, de Gee M, et al. Continuous-time modeling of cell fate determination in Arabidopsis flowers. BMC Systems Biology. 2010;4:101. doi: 10.1186/1752-0509-4-101. doi:10.1186/1752-0509-4-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaene T, Vekemans D, Irish V, et al. Pistillata-duplications as a mode for floral diversification in (Basal) asterids. Molecular Biology and Evolution. 2009;26:2627–2645. doi: 10.1093/molbev/msp181. [DOI] [PubMed] [Google Scholar]
- Viaene T, Vekemans D, Becker A, Melzer S, Geuten K. Expression divergence of the AGL6 MADS domain transcription factor lineage after a core eudicot duplication suggests functional diversification. BMC Plant Biology. 2010;10:148. doi: 10.1186/1471-2229-10-148. doi:10.1186/1471-2229-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitte C, Panaud O. LTR retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenetics and Genome Research. 2005;110:91–107. doi: 10.1159/000084941. [DOI] [PubMed] [Google Scholar]
- Waddington C. The strategy of the genes. New York: MacMillan; 1957. [Google Scholar]
- Wagner A. Robustness and evolvability in living systems. Princeton, NJ: Princeton University Press; 2005. [Google Scholar]
- Wang J-W, Czech B, Weigel D. miR156-Regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell. 2009;138:738–749. doi: 10.1016/j.cell.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Wang Y-Q, Melzer R, Theißen G. Interaction patterns of orthologues of floral homeotic proteins from the gymnosperm Gnetum gnemon provide a clue to the origin of ‘floral quartets. Plant Journal. 2010;64:177–190. doi: 10.1111/j.1365-313X.2010.04325.x. [DOI] [PubMed] [Google Scholar]
- Whipple C, Ciceri P, Padilla C, Ambrose B, Bandong S, Schmidt R. Conservation of B-class floral homeotic gene function between maize and Arabidopsis. Development. 2004;131:6083–6091. doi: 10.1242/dev.01523. [DOI] [PubMed] [Google Scholar]
- Winter K, Weiser C, Kaufmann K, et al. Evolution of class B floral homeotic proteins: obligate heterodimerization originated from homodimerization. Molecular Biology and Evolution. 2002;19:587–596. doi: 10.1093/oxfordjournals.molbev.a004118. [DOI] [PubMed] [Google Scholar]
- Zahn L, Leebens-Mack J, dePamphilis C, Ma H, Theissen G. To B or Not to B a flower: the role of DEFICIENS and GLOBOSA orthologs in the evolution of the angiosperms. Journal of Heredity. 2005;96:225–240. doi: 10.1093/jhered/esi033. [DOI] [PubMed] [Google Scholar]
- Zik M, Irish V. Global identification of target genes regulated by APETALA3 and PISTILLATA floral homeotic gene action. Plant Cell. 2003;15:207–222. doi: 10.1105/tpc.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]