

Abstract
The human genome is estimated to contain 23,000 to 33,000 retropseudogenes. To study the properties of genes giving rise to these retroelements, we compared the structure and expression of genes with or without known retropseudogenes. Four main features have emerged from the analysis of 181 genes associated to retropseudogenes: Reverse-transcribed genes are (1) widely expressed, (2) highly conserved, (3) short, and (4) GC-poor. The first two properties probably reflect the fact that genes giving rise to retropseudogenes have to be expressed in the germ-line. The two latter points suggest that reverse-transcription and transposition is more efficient for short GC-poor mRNAs. In addition, this analysis allowed us to reject previous hypotheses that widely expressed genes are GC rich. Rather, globally, genes with a wide tissue distribution are GC poor.
Retropseudogenes arise in evolution by reverse transcription of processed mRNAs and incorporation of the resulting cDNAs back into the genome (Vanin 1985). Hence, retropseudogenes can be distinguished from other types of pseudogenes by the fact that they lack introns, possess relics of the poly-(A) tail at their 3′ ends, and are flanked by target-site duplications. Generally, because they lack a promoter, these retropseudogenes are nonfunctional from the moment they are incorporated into the genome. Whereas the patterns of spontaneous substitutions, deletions, and insertions in retropseudogenes have been studied extensively (Gojobori et al. 1982; Graur et al. 1989; Gu and Li 1995; Ophir and Graur 1997), not much is known about the nature and structure of their functional homologs. Are they different from genes without known retropseudogenes?
To answer this question, we compared the expression pattern, the structure, and the evolutionary rate of human protein-coding genes with and without retropseudogenes. Our analysis revealed that the expression level, the selective pressure for amino-acid sequence conservation, and the structure of reverse-transcribed genes differ from genes without known retropseudogenes. Surprisingly, these differences vary according to the number of associated retropseudogenes.
RESULTS
We systematically searched for pseudogenes within the long (>50 kb) and generally nonannotated sequences that are produced in large amounts by the human genome sequencing project. For this purpose, we selected all complete and intron-containing human genes available in the databases (N = 2399), and we compared them with 451 Mb of genomic sequence to detect homologous nonfunctional retroelements. This approach allowed us to detect 109 retropseudogenes. To increase the data set, we also selected in GenBank all human sequences annotated as pseudogenes. Overall, we identified 249 sequence fragments that had the features of retropseudogenes: loss of introns, presence of a poly (A) tail at the 3′ end, and stop codons or frameshifts in the coding region. Pseudogenes associated with intronless genes have been excluded from the study because it is difficult to determine whether they result from duplication by DNA rearrangement, or from retrotranscription.
The 181 functional genes corresponding to these 249 retropseudogenes were compared to the reference data set of 2218 complete and intron-containing human genes that do not have retropseudogenes (as far as we can know with the human genomic sequences available at the time we performed the analyses). The properties of genes with or without retropseudogenes are described below.
Tissue Distribution of Gene Expression
The tissue distribution of human genes was estimated by comparison of their protein coding sequences (CDS) to a database of expressed sequence tags (ESTs) representing 19 tissues from three developmental stages: embryo, infant, and adult. These 19 tissues are expected to be representative of the whole organism. Hereafter, genes that are expressed in at least 15 tissues will be considered as widely expressed, whereas those that are detected in 0–1 tissue will be considered as tissue-specific. Widely expressed and tissue-specific genes make up 5% and 32% of the data set, respectively. The tissue-distribution breadth of genes shows a sharp difference between genes with and without retropseudogenes (Fig. 1). Indeed, genes without retropseudogenes are more tissue specific than widely expressed genes (34% vs. 3%), whereas genes with retropseudogenes are more widely expressed genes than tissue-specific (36% vs. 6%). Of the genes with one retropseudogene, 30% are expressed in at least 15 tissues, and this value exceeds 60% for genes with at least three retropseudogenes. In fact, there is a significant positive correlation between the number of retropseudogenes and the tissue–distribution breadth (R = 0.30, P < 10−4).
Figure 1.
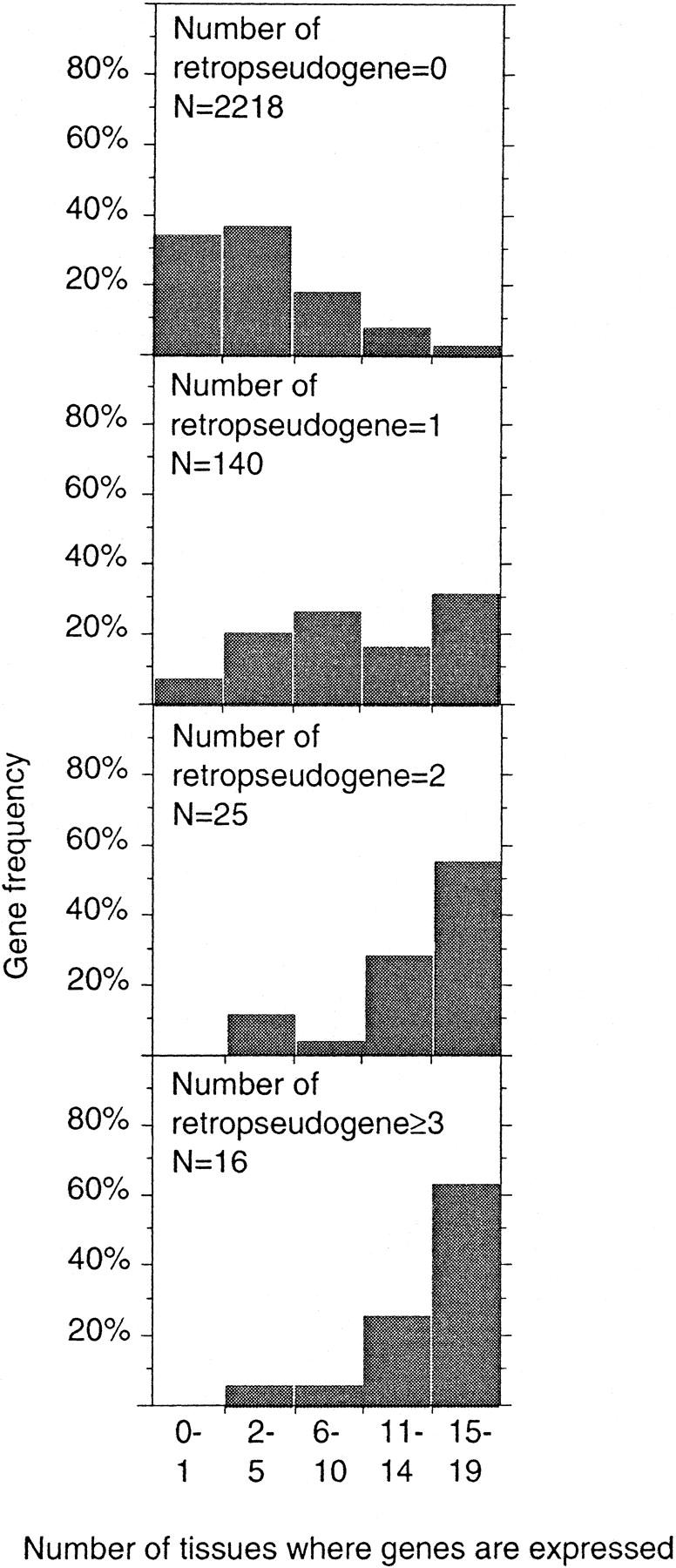
Frequency distribution of human genes according to their expression pattern and their number of retropseudogenes.
Structure of Genes with Retropseudogenes
We found striking differences in the size and GC content of CDSs between genes with and without known retropseudogenes (Fig. 2). First, the average size of CDSs of genes with retropseudogenes is two times shorter than that of genes without retropseudogenes (Mann-Whitney test: P < 10−4). Moreover, among genes with retropseudogenes, the CDS size decreases with the number of pseudogenes (R = −0.20; P < 0.01). Second, there is a significant difference in GC content between the two groups of genes: the CDSs of genes with retropseudogenes are more GC poor (Mann-Whitney test: P < 10−4). Once again, among genes with retropseudogenes, we found a slight decrease of the GC content with the number of retropseudogenes (R = −0.16; P < 0.05). This last result reflects the different distributions of the two groups of genes among the isochore classes (Table 1). Genes belonging to the GC-poor isochores are two times more frequent among genes with at least three retropseudogenes than among genes without retropseudogenes. This effect is independent of the size of the CDS as no significant correlation has been observed between the size (log-scaled) and the GC level of coding sequences (R = 0.03; P = 0.32).
Figure 2.

Average CDS length and average CDS GC content of genes according to their number of retropseudogenes. Error bars, 95% confidence interval.
Table 1.
Gene Distribution in Different Isochore Classes According to the Number of Retropseudogenes
| No. of retropseudogenes | Isochore (%) | ||
|---|---|---|---|
| L1 + L2 | H1 + H2 | H3 | |
| 0 | 31 | 39 | 30 |
| 1 | 47 | 35 | 18 |
| 2 | 56 | 40 | 4 |
| ≥3 | 69 | 31 | 0 |
The isochore class into which genes are located was predicted according to their GC content at third codon positions (GC3): GC3 <57% for L1 + L2 isochores, GC3 <75% for H1 + H2 isochores and GC3 ≥75% for H3 isochores (Bernardi 1993).
Evolutionary Rates of Genes with Retropseudogenes
Evolutionary rates were measured by calculating the number of substitutions at synonymous (Ks) and nonsynonymous sites (Ka) between human genes and their orthologs in a rodent (mouse or rat). We found that Ka values are on average two times higher in genes without known retropseudogenes than in genes with one retropseudogene and four times higher than in genes with several retropseudogenes (Kruskal-Wallis test: P < 10−4, Table 2). Ks values show the same trend (Kruskal-Wallis test: P < 10−3, Table 2). However, Ks and Ka have been shown to be correlated, probably because of neighboring effects between synonymous and nonsynonymous sites (Wolfe and Sharp 1993; Mouchiroud et al. 1995; Makalowski and Boguski 1998; Duret and Mouchiroud 2000). To test whether there was a difference in the synonymous substitution rate between genes with or without retropseudogenes independent of Ka variations we computed Ks_nd, that is, Ks after removal of all codons (or codon pairs) in which doublet substitutions (in positions 1–2, 2–3, and 3–1 of codons) occurred. We found no difference of Ks_nd between genes with and without retropseudogenes (Kruskal-Wallis test: P = 0.19), which suggests that there is no variation in the mutation rate between these two classes of genes.
Table 2.
Average Nonsynonymous (Ka) and Synonymous (Ks) Substitution Rate in Orthologous Genes Between Human and Murids According to their Number of Pseudogenes
| No. of retropseudogenes | Ka | Ks | Ks_nd |
|---|---|---|---|
| 0 (n = 349) | 0.109 ± 0.095 | 0.519 ± 0.149 | 0.396 ± 0.103 |
| 1 (n = 96) | 0.044 ± 0.063 | 0.470 ± 0.241 | 0.421 ± 0.236 |
| 2 (n = 22) | 0.022 ± 0.042 | 0.470 ± 0.134 | 0.436 ± 0.099 |
| ≥3 (n = 13) | 0.026 ± 0.045 | 0.437 ± 0.135 | 0.399 ± 0.13 |
Rate according to no. of pseudogenes = mean value ± s.d. Ks_nd:Ks computed after removing doublet substitutions (see Duret and Mouchiroud 2000). The number of genes is indicated for each sample.
The difference in average Ka values between genes with and without known retropseudogenes is highly significant (Mann-Whitney test: P < 10−4). This effect is independent of CDS size as no significant relationship has been observed between size and Ka values for widely expressed genes (R = 0.05; P = 0.70). This effect is partly explained by the difference of tissue-distribution breadth between these genes. Indeed, the nonsynonymous substitution rate (Ka) is sharply negatively correlated with the tissue-distribution breadth (Duret and Mouchiroud 2000). This difference reflects a stronger selective pressure for amino acid sequence conservation on widely expressed genes. However, comparison of genes of similar expression patterns shows that the low Ka values of genes with retropseudogenes is not entirely due to their wide tissue distribution (Fig. 3a). Indeed, whatever their tissue distribution, genes with retropseudogenes show smaller Ka values than genes without known retropseudogenes. This difference is significant for all expression classes, except the tissue-specific class, which contains very few (eight) genes with retropseudogenes. Thus, independently of their tissue-distribution breath, the selective pressure for amino acid sequence conservation is stronger for genes with retropseudogenes.
Figure 3.

Relationships between gene expression pattern and the nonsynonymous substitution rate (Ka; a), the size (b), and the GC content (c) of CDSs for genes with and without retropseudogenes. Error bars, 95% confidence interval. (Stars) Expression classes for which the difference between genes with and without retropseudogenes is significant (Wilcoxon test: P < 0.05).
Impact of the Tissue-Distribution Breadth on the Structure of Genes with Retropseudogenes
One way to explain the differences in structure between genes with and without known retropseudogenes is to assume that not only the selective pressure for amino acid sequence conservation but also the CDS size and the GC content vary according to the gene tissue-distribution breadth (Table 3). Analysis of CDS size revealed a relationship with the tissue-distribution breadth. Widely expressed genes (expressed in at least 15 tissues) exhibit the shortest CDSs (Kruskal-Wallis test: P < 10−4). A weak negative correlation has also been observed between the GC level and the number of tissues in which genes are expressed (R = −0.13; P < 10−4). So, widely expressed genes are shorter and tend to be GC poorer than genes with a narrow tissue distribution. Thus, the fact that genes with retropseudogenes are mostly widely expressed can partly explain the plots of Figure 2. Nevertheless, once again, the particular structural features of genes with retropseudogenes are not totally due to their wide expression pattern (Fig. 3). In the majority of expression classes, genes with retropseudogenes show significantly shorter CDSs (Fig. 3b) and poorer GC levels (Fig. 3c) than genes without known retropseudogenes.
Table 3.
Relationship Between the Size and the GC Content of CDS According to their Tissue Distribution Breadth
| No. of tissues where gene are expressed | CDS | |
|---|---|---|
| GC level (%) | Size (bp) | |
| 0–1 (n = 765) | 56.22 ± 8.26 | 1254 ± 1065 |
| 2–5 (n = 853) | 54.69 ± 7.75 | 1478 ± 1305 |
| 6–10 (n = 451) | 54.04 ± 7.22 | 1541 ± 1638 |
| 11–14 (n = 203) | 54.15 ± 6.89 | 1523 ± 1709 |
| 15–19 (n = 127) | 52.68 ± 6.63 | 930 ± 619 |
Relationship according to tissue distribution breadth = mean value ± s.d.. The number of genes is indicated for each sample.
Database Biases
Before discussing the results, we have to make certain that they are not due to biases in the data. First, it can be argued that the number of retropseudogenes observed for each functional gene reflects the interest of the scientific community for the gene family and not the propensity of the gene to give retropseudogenes. To test this point, we analyzed separately the 82 genes associated with 109 retropseudogenes found in nonannoted human genomic sequences. We found that these genes are short genes (829 ± 513 bp for genes with one retropseudogene and 717 ± 312 bp for genes with several retropseudogenes), are expressed in more than 10 tissues, and are GC poor. More than 59% of these genes are located in L1 + L2 versus only 10% in H3. So, even if the number of retropseudogenes reflects partly the interest of the scientific community for these genes, genes with known retropseudogenes in the databases are representative of reverse-transcribed genes in the human genome.
By comparison with complete CDSs of genes with at least one internal intron, we found that retrotranscribed genes were short and predominantly located in GC-poor isochores. However, complete genes presently available in databases may not be representative of all human CDS. By comparing the distribution of GC-content of CDSs sequenced from mRNAs or from genomic DNA, it has been shown that large genes from GC-poor isochores are under-represented among human genomic sequences of GenBank (Duret et al. 1995). This bias is due to the fact that genes located in GC-poor isochores contain large introns and hence are more costly to sequence than those from GC-rich isochores. mRNA (cDNA) sequences, which are technically easier to obtain, are probably more representative of the whole genes present in a genome than are genomic DNA sequences. Despite the recent progresses in genome sequencing, there is still a bias among human genes annotated in GenBank: The distribution of genes sequenced from genomic DNA in the different isochore classes (Table 1) is significantly different from that expected according to mRNA data (43% in L1 + L2, 36% in H1 + H2, and 21% in H3; χ2 = 139.5; P < 10−3). Could this bias affect the results presented previously? Compared with mRNAs, the distribution of genes with retropseudogenes still shows an excess in the GC-poor isochores (χ2 = 6.04; P < 0.05). Moreover, the fact that genes without known retropseudogenes are in reality longer than we estimated from our data suggests that the difference of size observed between genes with and without known retropseudogenes may indeed be underestimated. Thus, database biases do not seem to be responsible for the observed patterns.
DISCUSSION
The human genome is ∼3400 Mb long, and contains ∼70,000–100,000 genes (Fields et al. 1994). Sequencing projects have revealed that retropseudogenes are very common in mammalian genomes. For example, in the complete sequence of human chromosome 22 (33.4 Mb), 110 retropseudogenes have been identified (i.e., about three copies per megabase; Dunham et al. 1999). This frequency is an underestimate because the procedure used to identify retropseudogenes was not exhaustive. The systematic comparison of 2399 complete human genes to 451 Mb of genomic sequence allowed us to detect 109 retropseudogenes. Therefore, if these 2399 genes for which we screened pseudogenes are a representative sample, the human genome might contain 23,000–33,000 copies (i.e., 6–10 copies per megabase) of such nonfunctional retroelements.
To be fixed in the genome, the insertion of a retroelement has to occur in the germ line. Therefore, genes giving retropseudogenes have to be expressed, at least at some stage, in the germinal tissue. Moreover, the probability of an mRNA to give rise to a retroelement depends on the efficiency of the following steps: transfer to the nucleus, reverse-transcription, and integration into a chromosome. Finally, the fixation of this new allele in the species depends on the strength of the selection against the possible deleterious effects of the new insertion. The particular properties of genes giving rise to retropseudogenes reflect these different factors.
A Great Majority of Genes with Retropseudogenes are Widely Expressed
Retrotranscribed genes, whose functions are known, are predominately housekeeping genes involved in metabolism (28%) or in protein and RNA synthesis (24%) that are probably expressed in all tissues. Indeed, analysis of EST expression data showed that genes with retropseudogenes have a wide tissue-distribution breadth (Fig. 1). Of the 19 EST libraries analyzed, 18 are somatic and 1 (testis) includes some germ-line tissues. Therefore, these data are not directly representative of gene expression in the germ line. However, whereas most tissue-specific genes are somatic (the number of germ-line-specific genes is probably small compared with other tissue-specific genes), it is likely that a significant fraction of widely expressed genes are also active in the germ line (and thus are susceptible to giving rise to retropseudogenes). Thus, the fact that, on average, retrotranscribed genes are widely expressed can be explained by the fact that such genes are more likely to be expressed in the germ line than tissue-specific ones. Interestingly, there is a positive correlation between the number of retropseudogenes and the tissue-distribution breadth (R = 0.30; P < 10−4). The number of retropseudogenes is expected to be correlated with gene expression level in the germ line, because abundant mRNAs are more likely to be taken as a substrate by reverse-transcriptases. But a priori, there is no reason why it should be correlated with tissue-distribution breadth. However, the measure of tissue distribution is not independent of the expression level of genes: Because the sensitivity of the detection is limited, the tissue-distribution breadth of genes expressed at low levels is underestimated. Therefore, the positive correlation between the number of retropseudogenes and the tissue-distribution breadth may be due to the fact that genes detected to be widely expressed are also expressed in larger amounts than those considered to be tissue specific. Indeed, the average number of EST per tissue is 2.7 times larger for genes with three or more retropseudogenes compared with those with only one.
Properties of Genes with Retropseudogenes
Genes with retropseudogenes have a lower GC content, shorter CDSs, and lower Ka values than genes without known retropseudogenes. These features are partly explained by the fact that these genes are widely expressed. Indeed, widely expressed genes have small CDSs and, as has been shown previously, lower Ka values (Duret and Mouchiroud 2000). Moreover, contrary to what has been proposed by Bernardi (1993), widely expressed genes have a lower GC content that tissue-specific ones. The proportion of tissue-specific genes found in L1 + L2 (GC poor) is 27% whereas this proportion reaches 40% for genes expressed in at least 15 tissues. Moreover, 35% of tissue-specific genes are located in H3 (GC rich) versus only 20% for widely expressed genes.
However, the wide tissue distribution of genes with known retropseudogenes cannot totally explain their poverty in G + C nucleotides, the small size of their CDSs, and the stronger selective pressure for amino-acid sequence conservation. Indeed, for a same tissue-distribution breadth, genes with retropseudogenes are GC poorer, shorter, and have lower Ka values than genes without known retropseudogene (Fig. 3). There is no clear explanation for these observations. However, several hypotheses can be put forward.
It has been shown that the reverse-transcriptase involved in the reverse-transcription of genes giving retropseudogenes is probably a LINE reverse-transcriptase (Dhellin et al. 1997). This observation suggests that the process of retropseudogene insertion might be similar to that of L1 retrotransposition. The proposed mechanism of L1 retrotransposition (Kazazian and Moran 1998) is that an active L1 is transcribed in the nucleus and its RNA is subsequently transported to and translated in the cytoplasm. The L1 transcript and its protein products (including L1 reverse-transcriptase) form a ribonucleoprotein particle that is transported to the nucleus (it is not yet clear whether this complex is imported into the nucleus or reaches chromatin passively after nuclear breakdown in mitosis). Once in the nucleus, L1 RNA undergoes target-primed reverse-transcription to carry out retrotransposition. Interestingly, like genes that give retropseudogenes, mammalian LINE elements are GC poor (Korenberg and Rykowski 1988). Thus, the low GC content of genes giving rise to retropseudogenes might be due to the higher efficiency of reverse-transcription by LINE reverse-transcriptase of GC-poor compared with GC rich mRNAs.
The small CDS size of gene with retropseudogenes is independent of their compositions , as we found no significant correlation between the sizes and the GC contents of CDSs. It is possible that transfer between the cytoplasm and the nucleus and/or the reverse-transcription process and/or the insertion mechanism into the genome are more efficient for short mRNAs. Alternatively, it is possible that insertions of long cDNAs are more often counterselected because they are more likely to have deleterious effects.
Finally, the stronger selective pressure on genes with retropseudogenes can be explained by the fact that these genes are expressed in the germ line. Notably, it is likely that some of these genes are expressed during gametogenesis, a stage during which germ cells become haploid and where gametic selection may occur. Therefore, mutations in genes expressed in these cells are more likely to be counterselected than in genes expressed only in diploid cells.
METHODS
Sequence Data
Pseudogenes were extracted from GenBank (release 110, January 1999; Benson et al. 1998) in two ways. First, we selected sequences annotated as pseudogenes. Each pseudogene was associated with its functional homolog by a similarity search with the program BLASTN2 (Altschul et al. 1997). BLASTN2 hits showing at least 80% identity over 100 nucleotides or more were considered as a sequence match. Then, we visualized, with the software LalnView (Duret et al. 1996), the alignments between each pseudogene and its functional homolog (which shows the maximum identity). We kept pseudogenes that lack introns and possesses oligo (A) sequences at their 3′ ends as it reflects their processed origin. Pseudogenes associated with intronless genes were excluded from the study because it is difficult to test whether they result from duplication by DNA rearrangement or from mRNA reverse-transcription. Second, to increase the data set, we searched pseudogenes in long nonannotated genomic human sequences (>50 kb). In this latter strategy, we used BLASTN2, with the same parameters as above, to find homologies between these long sequences and the complete CDSs of genes with at least one internal intron. These CDSs were selected in the HOVERGEN database (release 34, February 1999; Duret et al. 1994), using the ACNUC retrieval system (Gouy et al. 1985). We considered as retropseudogenes sequences showing similarities with at least two exons of a gene and no similarity with the introns of this gene. We checked that these sequences were not functional either because of the presence of stop codons or because of a frameshift.
When several retropseudogenes are associated with the same functional gene, they can either result from independent events of reverse-transcription of this gene or from the duplication of one retropseudogene by DNA rearrangement. We used the method described by Ophir and Graur (1997), which is based on the analysis of the phylogenetic tree of the functional gene and its retropseudogenes, to keep only the retropseudogenes that directly diverged from the functional gene.
For each human coding gene, we selected, when available, the orthologous gene in a murid genome (Mus musculus or Rattus norvegicus) from HOVERGEN to compute the number of substitutions at the synonymous (Ks) and nonsynonymous sites (Ka; Li 1993). Ka and Ks were computed with the Java application JaDis (Gonçalves et al. 1999).
Expression Profiles
We selected from GenBank (release 110, January 1999) 679,286 ESTs from 19 tissues: placenta, fetal liver, fetal heart, fetal lung, fetal brain, infant brain, brain, breast, colon, testis, eye, uterus, liver, lymphocyte, muscle, prostate, lung, pancreas, and neuron. cDNA libraries from cell culture, tumors, pooled organs, unidentified tissues or that had been sampled with less than 10,000 ESTs were excluded.
Expression profiles of human CDSs were determined by counting of the number of tissues in which they are represented by at least one EST, as described previously (Duret and Mouchiroud 2000). CDSs were first filtered with the XBLAST program (Claverie and States, 1993) to mask repetitive elements (Alu, L1, MIR, microsatellites, etc.). CDS were then compared with the EST data set by use of BLASTN2. BLASTN2 hits showing at least 95% identity over 100 nucleotides or more were counted as a sequence match. This criteria has been chosen low enough to allow the detection of most ESTs despite sequencing errors, but stringent enough to distinguish, in most cases, different members of highly conserved gene families.
GenBank accession numbers of selected genes and their number of associated retropseudogenes, as their expression pattern are available upon request from the authors (E-mail: goncalve@biomserv.univ-lyon1.fr) or at http://pbil.univ_lyon1.fr/datasets/Goncalves_1999.html.
Acknowledgments
This work was supported by the MinistÈre de la Recherche and the Centre National de la Recherche Scientifique.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL goncalve@biomserv.univ-lyon1.fr; FAX 33 4 78 89 27 19.
REFERENCES
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Boguski MS, Lipman DJ, Ostell J, Ouellette BF. GenBank. Nucleic Acids Res. 1998;26:1–7. doi: 10.1093/nar/26.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi G. The isochore organization of the human genome and its evolutionary history—a review. Gene. 1993;135:57–66. doi: 10.1016/0378-1119(93)90049-9. [DOI] [PubMed] [Google Scholar]
- Claverie JM, States DJ. Information enhancement methods for large scale sequence analysis. Computers Chem. 1993;17:191–201. [Google Scholar]
- Dhellin O, Maestre J, Heidmann T. Functional differences between the human LINE retrotransposon and retroviral reverse transcriptases for in vivo mRNA reverse transcription. EMBO J. 1997;16:6590–6602. doi: 10.1093/emboj/16.21.6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Determinants of substitution rates in mammalian genes: Expression pattern affects selection intensity but not mutation rate. Mol Biol Evol. 2000;17:68–74. doi: 10.1093/oxfordjournals.molbev.a026239. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D, Gouy M. HOVERGEN: A database of homologous vertebrate genes. Nucleic Acids Res. 1994;22:2360–2365. doi: 10.1093/nar/22.12.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D, Gautier C. Statistical analysis of vertebrates sequences reveals that long genes are scarce in GC-rich isochores. J Mol Evol. 1995;40:308–317. doi: 10.1007/BF00163235. [DOI] [PubMed] [Google Scholar]
- Duret L, Gasteiger E, Perriere G. LALNVIEW: A graphical viewer for pairwise sequence alignments. Comput Appl Biosci. 1996;12:507–510. doi: 10.1093/bioinformatics/12.6.507. [DOI] [PubMed] [Google Scholar]
- Fields C, Adams MD, White O, Venter JC. How many genes in the human genome? Nature Genet. 1994;7:345–346. doi: 10.1038/ng0794-345. [DOI] [PubMed] [Google Scholar]
- Gojobori T, Li WH, Graur D. Patterns of nucleotide substitution in pseudogenes and functional genes. J Mol Evol. 1982;18:360–369. doi: 10.1007/BF01733904. [DOI] [PubMed] [Google Scholar]
- Goncalves I, Robinson M, Perriere G, Mouchiroud D. JaDis: Computing distances between nucleic acid sequences. Bioinformatics. 1999;15:424–425. doi: 10.1093/bioinformatics/15.5.424. [DOI] [PubMed] [Google Scholar]
- Gouy M, Gautier C, Attimonelli M, Lanave C, Di Paola G. ACNUC—a portable retrieval system for nucleic acid sequence databases: Logical and physical designs and usage. Comput Appl Biosci. 1985;1:167–172. doi: 10.1093/bioinformatics/1.3.167. [DOI] [PubMed] [Google Scholar]
- Graur D, Shuali Y, Li WH. Deletions in processed pseudogenes accumulate faster in rodents than in humans. J Mol Evol. 1989;28:279–285. doi: 10.1007/BF02103423. [DOI] [PubMed] [Google Scholar]
- Gu X, Li WH. The size distribution of insertions and deletions in human and rodent pseudogenes suggests the logarithmic gap penalty for sequence alignment. J Mol Evol. 1995;40:464–473. doi: 10.1007/BF00164032. [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Moran JV. The impact of L1 retrotransposons on the human genome. Nature Genet. 1998;19:19–24. doi: 10.1038/ng0598-19. [DOI] [PubMed] [Google Scholar]
- Korenberg JR, Rykowski MC. Human genome organization: Alu, lines, and the molecular structure of metaphase chromosome bands. Cell. 1988;53:391–400. doi: 10.1016/0092-8674(88)90159-6. [DOI] [PubMed] [Google Scholar]
- Li WH. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- Makalowski W, Boguski MS. Evolutionary parameters of the transcribed mammalian genome: An analysis of 2,820 orthologous rodent and human sequences. Proc Natl Acad Sci. 1998;95:9407–9412. doi: 10.1073/pnas.95.16.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud D, Gautier C, Bernardi G. Frequencies of synonymous substitutions in mammals are gene-specific and correlated with frequencies of nonsynonymous substitutions. J Mol Evol. 1995;40:107–113. doi: 10.1007/BF00166602. [DOI] [PubMed] [Google Scholar]
- Ophir R, Graur D. Patterns and rates of indel evolution in processed pseudogenes from humans and murids. Gene. 1997;205:191–202. doi: 10.1016/s0378-1119(97)00398-3. [DOI] [PubMed] [Google Scholar]
- Vanin EF. Processed pseudogenes: Characteristics and evolution. Annu Rev Genet. 1985;19:253–272. doi: 10.1146/annurev.ge.19.120185.001345. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Sharp PM. Mammalian gene evolution: Nucleotide sequence divergence between mouse and rat. J Mol Evol. 1993;37:441–456. doi: 10.1007/BF00178874. [DOI] [PubMed] [Google Scholar]