

Abstract
Liquid chromatography-multiple reaction monitoring mass spectrometry of peptides using stable isotope dilution (SID) provides a powerful tool for targeted protein quantitation. However, the high cost of labeled peptide standards for SID poses an obstacle to multiple reaction monitoring studies. We compared SID to a labeled reference peptide (LRP) method, which uses a single labeled peptide as a reference standard for all measured peptides, and a label-free (LF) approach, in which quantitation is based on analysis of un-normalized peak areas for detected MRM transitions. We analyzed peptides from the Escherichia coli proteins alkaline phosphatase and β-galactosidase spiked into lysates from human colon adenocarcinoma RKO cells. We also analyzed liquid chromatography-multiple reaction monitoring mass spectrometry data from a recently published interlaboratory study by the National Cancer Institute Clinical Proteomic Technology Assessment for Cancer network (Addona et al. (2009) Nat. Biotechnol. 27: 633–641), in which unlabeled and isotopically labeled synthetic peptides or their corresponding proteins were spiked into human plasma. SID displayed the highest correlation coefficients and lowest coefficient of variation in regression analyses of both peptide and protein spike studies. In protein spike experiments, median coefficient of variation values were about 10% for SID and 20–30% for LRP and LF methods. Power calculations indicated that differences in measurement error between the methods have much less impact on measured protein expression differences than biological variation. All three methods detected significant (p < 0.05) differential expression of three endogenous proteins in a test set of 10 pairs of human lung tumor and control tissues. Further, the LRP and LF methods both detected significant differences (p < 0.05) in levels of seven biomarker candidates between tumors and controls in the same set of lung tissue samples. The data indicate that the LRP and LF methods provide cost-effective alternatives to SID for many quantitative liquid chromatography-multiple reaction monitoring mass spectrometry applications.
A rapidly evolving approach to protein quantitation is the targeted analysis of representative peptides by liquid chromatography-tandem mass spectrometry by multiple reaction monitoring (LC-MRM-MS)1 analysis (1–3). In this approach, peptides are quantified by monitoring several MRM transitions for each peptide with either a triple quadrupole or a quadrupole-ion trap instrument. Stable isotope dilution (SID), in which labeled peptides are used as internal standards is considered the gold standard for rigorous quantitation by LC-MRM-MS (1, 4, 5). In contrast to antibody-based quantitation, where antibody availability and specificity are often limiting, LC-MRM-MS enables configuration of an assay for essentially any protein. In practice, this approach has proven sensitive enough to apply to challenging protein quantitation problems. For example, proteins can be quantified at single-digit copy numbers in cells (6) and in plasma at levels approaching ng/ml (7, 8). With antibody-based enrichment, LC-MRM-MS can achieve even greater sensitivity (9–12).
Despite the power of the method, the use of SID is nevertheless limited practically by the cost of labeled standards, which are expensive (∼$1000 per milligram for labeled peptides of high purity). This issue is particularly important in considering LC-MRM-MS to evaluate candidate biomarkers for disease. Application of biomarker discovery platforms, such as shotgun proteomics or transcriptome profiling can yield hundreds of biomarker candidates. The next phase of analysis, termed “verification,” consists of configuring assays for the candidates and evaluating them in well-defined test cohorts (1). The cost of configuring SID-LC-MRM-MS assays for three representative peptides each for 50 proteins would be approximately $150,000.
An MRM-based approach for targeted protein quantitation with a more limited number of isotopically labeled standards could be particularly useful for biomarker candidate screening, in which the expense of labeled standards presents a real barrier to verification of large numbers of candidates. Although SID should outperform methods that do not employ labeled standards for each analyte, there are insufficient data available to evaluate the performance of alternative techniques or to determine appropriate contexts for their use.
Here we compared SID with two alternative methods. The first is a labeled reference peptide (LRP) method, which employs a single isotopically labeled peptide as the reference peptide for all of the other peptide analytes. The second is a label-free (LF) method that employs no standard and where quantitation is based only on the peak areas extracted from LC-MRM-MS product ion chromatograms. We compared these three approaches with datasets from analyses of defined peptide and protein mixtures on triple quadrupole LC-MS instruments. Test samples included synthetic peptides or their corresponding proteins spiked into a human cell lysate. We also analyzed LC-MRM-MS data from a recent study by the National Cancer Institute Clinical Proteomic Technology Assessment for Cancer (CPTAC) program (3), which analyzed human plasma spiked with peptide and protein standards. Finally, we compared the methods in analysis of several lung cancer biomarker candidate proteins in normal lung and lung-tumor tissues. Our studies document the performance of all three methods with the same datasets. The data establish the performance of the LRP and LF methods and provide a basis to select between all three methods for appropriate applications in quantitative proteomics.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Iodoacetamide and ammonium bicarbonate (>99.0% purity) were from Sigma (St. Louis, MO); d,l-1,4 dithiothreitol was from Bio-Rad (Hercules, CA); 2,2,2-trifluoroethanol was from Acros (Geel, Belgium). Two Escherichia coli proteins, alkaline phosphatase (AP) and β-galactosidase (BG) were purchased from Sigma. Six pairs of C-terminal isotopically labeled peptides containing U-13C6, U-15N4-arginine or U-13C6, U-15N2-lysine and corresponding unlabeled peptides derived from AP and BG were supplied by New England Peptide, LLC (Gardner, MA) at over 95% chemical purity according to amino acid analysis (shown in Table I). Three C-terminal isotopically labeled peptides containing U-13C6, U-15N4-arginine or U-13C6, U-15N2-lysine from human advanced glycosylation end product-specific receptor (AGER‖‖) (VLSPQGGGPWDSVA*R), from γ- and β-actin (referred to herein as ACTIN to denote both proteins) (GYSFTTTAE*R) and from annexin A1 (ANXA1) (VLDLEL*K) also were obtained from New England Peptide at 95% chemical purity. Mass spectrometry grade trypsin (Trypsin Gold) was purchased from Promega (Madison, WI). HPLC grade water and acetonitrile were from Mallinckrodt Baker (Phillipsburg, NJ).
Table I. AP and BG peptides and transitions selected for LC-MRM MS.
| Protein | Peptidea | Precursor m/z | Product m/z |
|---|---|---|---|
| β-galactosidase (BG) | LPSEFDLSAFLR (BG 698) | 697.9 | 593.34, 706.42, 821.45, 968.52 |
| LPSEFDLSAFL*R | 702.9 | 603.35, 716.43, 831.46, 978.53 | |
| LWSAEIPNLYR (BG 681) | 681.4 | 662.36, 775.45, 904.49, 1062.56 | |
| LWSAEIPNLY*R | 686.4 | 672.37, 785.45, 914.50, 1072.57 | |
| APLDNDIGVSEATR (BG 729) | 729.4 | 563.28, 719.37, 832.45, 1061.52 | |
| APLDNDIGVSEAT*R | 734.4 | 573.29, 729.38, 842.46, 1071.53 | |
| Alkaline phosphatase (AP) | AAQGDITAPGGAR (AP 593) | 592.8 | 457.25, 528.29, 629.34, 914.47 |
| AAQGDITAPGGA*R | 597.8 | 467.26, 538.30, 639.34, 924.48 | |
| APGLTQALNTK (AP 557) | 557.3 | 546.32, 674.38, 775.43, 945.54 | |
| APGLTQALNT*K | 561.3 | 554.34, 682.40, 783.44, 953.55 | |
| NYAEGAGGFFK (AP 581) | 580.8 | 555.29, 683.35, 812.39, 883.43 | |
| NYAEGAGGFF*K | 584.8 | 563.31, 691.36, 820.41, 891.44 |
a Peptides marked with * are isotopically labeled.
Cell Culture and Cell Lysate Preparation
The human colon adenocarcinoma cell line (RKO) was cultured at 37 °C in the McCoy's 5A medium (Mediatech, Herndon, VA) with 10% fetal bovine serum (Atlas Biologicals, Fort Collins, CO) in the presence of 5% CO2 and harvested at >85% confluence. Cells were washed twice with 10 ml phosphate-buffered saline, collected in 10 ml phosphate-buffered saline buffer, and then centrifuged at 2000 rpm for 5 min at 4 °C to obtain the cell pellet. Cells were lysed and proteins were extracted with ammonium bicarbonate and 2,2,2-trifluoroethanol as described previously (13). Protein concentration of cell lysate was measured using bicinchoninic acid assay with bovine serum albumin used as protein standard.
Analyses of E. coli AP and BG Synthetic Peptides into RKO Cell Lysate
Three peptides each from AP and BG were synthesized for spike studies and are listed in Table I together with their corresponding isotopically labeled standards. The AP and BG peptides were spiked into an RKO cell lysate, which then was processed by a workflow based on in-solution tryptic digestion. The protein concentration of RKO lysate or the digest was 0.5 μg/μl. The unlabeled AP and BG peptides were spiked in at equal molarity at 2, 10, 40, 100, or 200 fmol/μg protein. Each isotopically labeled standard peptide was spiked at a constant concentration of 60 fmol/μg protein. Two samples were prepared and processed for each experimental variation and concentration for LC-MRM-MS (see below).
The RKO cell lysate was subjected to tryptic digestion by a modification of a previously reported method (13). Briefly, proteins were reduced with 10 mm dithiotreitol for 30 min at 65 °C and then alkylated with 20 mm iodoacetamide for 30 min in the dark at room temperature, followed by the addition of trypsin with 1:50 enzyme to protein ratio. Digestion was performed at 37 °C overnight and was terminated with the addition of formic acid (>98% purity, EMD, Darmstadt, Germany) to a final concentration of 1%. The peptide mixture was desalted with an Oasis HLB extraction plate (Waters Corp., Milford, MA), which was prewashed with 1 ml of acetonitrile and then equilibrated with 2 ml of water. Following sample loading, plates were washed with 1 ml water and the peptides were eluted with 80% aqueous acetonitrile. The eluate then was evaporated in a SpeedVac concentrator (Thermo-Fisher, Waltham, MA) and the peptides were reconstituted in water containing 0.1% formic acid for LC-MRM-MS analysis.
Analyses of AP and BG Proteins Spiked into RKO Cell Lysate
A second set of experiments employed AP and BG proteins spiked an RKO lysate (0.5 μg/μl protein) background. AP and BG were added at equimolar concentrations of 5, 20, 80, and 200 fmol/μg protein. Three synthetic isotopically labeled peptides each from AP and BG were spiked into each protein mixture to achieve a concentration of 60 fmol/μg protein and in-solution tryptic digestion and peptide recovery and desalting were performed as described above. Five replicates were prepared for each spike concentration and 2 μl was injected on-column for LC-MRM-MS.
Analysis of Synthetic Peptides Spiked Into Human Plasma
LC-MRM-MS analyses were done as part of a CPTAC interlaboratory study (3), in which both peptide-spike and protein-spike experiments were done. In the peptide-spike experiment (Study I), 11 synthetic peptides derived from seven proteins (bovine aprotinin, murine leptin, equine myoglobin, bovine myelin basic protein, human prostate specific antigen horseradish peroxidase, and human C-reactive protein) were spiked into digested human plasma (1 μg/μl) at concentrations ranging from 1 to 500 fmol/μl. An equimolar mixture of 11 stable-isotope labeled peptides corresponding to the seven proteins (three of the proteins were represented by multiple peptides) was spiked in at 50 fmol/μl as internal standards. In the protein-spike experiment (Study III), the seven intact proteins were spiked into undigested plasma and the mixture was then subjected to reduction, alkylation, and digestion, followed by addition of the internal standards. For LC-MRM-MS analyses, 1 μl was injected on-column. Three MRM transitions were recorded for each peptide and four replicates were collected for each concentration. Other details for sample preparation and experiment design of this work can be found in (3).
Analysis of Human Lung Tissues
Surgically resected human lung tumor samples (adenocarcinomas (ADC), squamous cell carcinomas (SCC), and normal lung tissues dissected at least 2 cm from each tumor) were collected at Vanderbilt University Medical Center, snap frozen, and kept in liquid nitrogen until use for individual analysis. Informed consent was obtained and the project was approved by the Vanderbilt University Medical Center Institutional Review Board. Tissues were cut into small pieces in a Petri dish on dry ice. Ice cold RIPA buffer was then added to tissue pieces at a ratio of 100 mg/ml (w/v). Tissues were homogenized with a Brinkmann Polytron Homogenizer (Brinkmann, Switzerland) in RIPA buffer on ice. Lysates were left on ice for 30 min followed by centrifugation at 20,000 × g for 15 min at 4 °C. Supernatants were stored at −80 °C. Protein concentrations of the supernatants were estimated using bichinchoninic acid assay (Thermo Scientific, Rockford, IL) with bovine serum albumin used as a standard.
Protein digests from lung tumor and normal tissue lysates were subjected to short (∼1 cm) SDS-PAGE separation, cleanup and in-gel digestion, as described previously (14). Tissue protein (20 μg) was prepared in 4× LDS buffer and loaded onto NuPAGE 10% Bis-Tris SDS-PAGE gel (Invitrogen, Carlsbad, CA). Isotopically labeled peptides from AGER, ANXA1, and ACTIN were spiked into solution at a concentration of 60 fmol/μg protein prior to in-gel digestion. Peptides were extracted as described above and reconstituted in 40 μl 0.1% formic acid for LC-MRM-MS analysis. Because of limited amount of available tissue, a single process replicate of each sample was analyzed.
Selection of Signature Peptides for LC-MRM-MS Analyses
The selection of signature peptides was based on criteria reported previously (5, 15, 16), which consider unique (proteotypic) peptide sequences and features that enhance chemical stability. Priority was given to those peptides that were previously identified in the shotgun data set with high MS/MS spectral quality. Previous work demonstrated a high correlation between intense product ions in ion trap MS/MS spectra and the most intense MRM transitions on triple quadrupole instruments (17, 18). Additional peptides were selected by in silico digestion and all peptides included in MRM analysis were required to have 7 to 25 amino acids in length, be fully tryptic (both N- and C termini are formed by cleavage at lysine or arginine) and contain no ragged ends or potential post translational modification motifs (e.g. NXT/S, for possible N-glycosylation). Peptides containing cysteine or methionine residues were not excluded and cysteines were present as carboxyamidomethylated derivatives following treatment with iodoacetamide during sample work-up. Peptide uniqueness was confirmed by searching against the International Protein Index human database (Version 3.56).
Criteria to include specific signature peptides and their MRM transitions for use in our experiments were as follows: (1) when peptide standards were used, chromatographic retention time alignment was required for both precursor signal and MRM transitions; (2) when standard peptides were available, the relative intensities of MRM transition signals were consistent with those observed in full scan MS/MS of the corresponding standards; (3) when no standard peptides were available, relative intensities of MRM transition signals were consistent with those observed previously in linear ion trap MS/MS spectra and were consistent between different samples; (4) in analyses of lung tissue samples, at least three of the specified MRM transitions with measured signal-to-noise greater than three were observed in either normal or tumor samples. Signal-to-noise was estimated as
where b is the signal intensity for the measured peak and a is the mean signal intensity over the intervals equal to ∼10 times peak width measured both immediately before and immediately following elution.
LC-MRM-MS Analyses
Analyses for AP/BG spike experiments were performed on a TSQ Quantum triple quadrupole mass spectrometer (Thermo-Fisher Scientific, Waltham MA) equipped with an Eksigent 1D Plus NanoLC pump (Eksigent Technologies, Dublin CA). The mobile phase consisted of solvent A, 0.1% aqueous formic acid and solvent B, acetonitrile with 0.1% formic acid. Peptides were separated on a capillary column (Polymicro Technologies, 100 μm × 11 cm) packed with Jupiter C18 resin (5 μm, 300 Å, Phenomenex) using an in-line solid-phase extraction column (100 μm × 6 cm) packed with the same C18 resin (using a frit generated with liquid silicate Kasil 1 similar to that previously described (19). Injections were 2 μl of a sample solution containing 0.5 mg/ml peptide mixture (based on protein concentration) and were followed by a 10 min wash period with 100% A, then by elution with a gradient of 2–25% solvent B in 25 min, 25–50% solvent B in 20 min, and followed by 50–90% solvent B in 10 min.
LC-MRM-MS analyses of the AP and BG peptide and protein spike samples were done with an electrospray voltage of 1200 V, capillary temperature 210 °C and skimmer offset −5 V. Both Q1 and Q3 were set at unit resolution (FWHM 0.7 Da) and collision gas (He) pressure in Q2 was held at 1.5 mTorr. Scan width was 0.004 m/z and scan time was 20 ms for the AP and BG peptide and protein analyses and 10 ms for lung tissue samples. Collision energy for each peptide was calculated based on the equation
in which the m/z is the mass to charge ratio of the precursor ion. Peak areas for each peptide were extracted and integrated using Skyline software v. 0.6.1.2168 (20).
Routine assessment of instrument and chromatographic performance was done with a quality control (QC) standard consisting of three synthetic peptides (TPepH (Ac-AVAGHAGAR), and TPepW (Ac-AVAGWAGAR), and the annexin peptide VLDLELK), which was prepared at a concentration of 10 fmol/μl in 0.1% formic acid. Analyses of this sample were done several times daily and QC samples were interspersed with analysis series. A QC instrument method monitored four to five transitions per peptide. Following each QC injection, the extracted total ion current and retention times for each peptide were assessed. Peptide signal intensities of >5e6 were required for initiation or continuation of sample analysis series.
Statistical Methods
Concentration-response data were analyzed by weighted, robust regression of the nontransformed data, which is similar to the method employed by Adonna et al. (3). The robust linear model function, rlm(), from the R MASS library was used (http://cran.rproject.org/web/packages/MASS/index.html). Fitting with the option “method = 'MM”' uses Tukey's biweight as a residual weighting function. Heuristically, large residuals from the regression are given less weight than small residuals. In this way, outliers are down-weighted in fitting the regression model, but not in computing the standard deviation or the r2 statistics. Measurement error was calculated according to
in which a denotes a constant (additive) measurement error in the concentrations range from 0 to x0. The second term k(x - x0)+ is zero when x is less than the threshold x0 and is linearly increasing when x is greater than x0. At low concentrations, additive components of measurement error because of imprecise estimation of signal baseline and uncertainty in numerical integration of peaks can dominate the measurement error, whereas these components are negligible at higher concentrations (21). Inspection of the data indicated that at concentrations between 0 and 10 fmol/μl, the standard deviation is approximately constant and then linearly increases at higher concentrations.
In some analyses, signal appears to be constant at low concentrations (i.e. the concentration-response relationship “flattens out”). To account for this phenomenon, we also employed a change-point (CP) model, where the slope may change at some designated point, xCP. In this model, the mean response has a slope of zero from 0 to xCP, but at concentrations greater than xCP, the response increases linearly. Because this behavior was observed only for some peptides, we employed a model selection procedure based on Akaike's information criterion (AIC), as implemented in R. This AIC approach balances model fit with a penalty for increasing number of model parameters. For plots with no clear change-point, the improvement in fit is outweighed by the penalty for the extra parameters used to apply the change point model thus, the simpler model yields a smaller AIC value. Plots with a significant flattening out at the low end produce a much better fit when the change-point model is applied—the AIC value is lower, even though the penalty for the extra model parameters is applied. The AIC procedure removes observer bias from the selection of model to apply to the data.
Coefficient of variation (CV) for each analysis method varied with analyte concentration, but were approximately constant at the higher analyte concentrations; thus, CV values were calculated at the highest concentration for each experiment.
Analytical sensitivity provides a useful representation of the concentration-response relationship for a method (22) and is defined as the instantaneous slope of the concentration-response curve divided by the standard deviation. The measurement methods were compared by computing the relative sensitivity (RS), which is the point-wise ratio of their sensitivities. For a comparison of SID and LRP methods
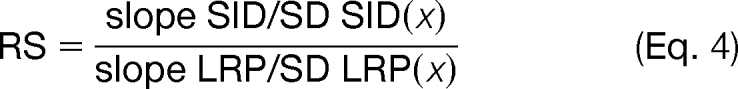 |
Because standard deviation is assumed to be proportional to concentration in the models used, the relative sensitivities are constant across concentrations for each peptide. Thus, RS for LRP and the LF methods were calculated as single values for each peptide concentration measurement series. The practical value of the RS parameter is that is that it indicates the degree to which the LF and LRP methods yield signal in proportion to concentration of each peptide.
Significance of measured differences for biomarker candidate proteins in paired lung tissues were calculated with one-tailed paired t test using Prism 5.0 (GraphPad Software, San Diego, CA).
RESULTS
Overview of Analytical Methods and Approach to Comparisons
This work compares three approaches to MRM-based quantitation of peptides and proteins. In SID, stable isotopically labeled peptide standards are synthesized for every peptide to be measured. The integrated peak areas for transitions for the unlabeled peptide analyte are summed and then normalized to the summed peak areas for transitions from the corresponding labeled peptide standard. The reported values for SID are “measured concentration,” although this assumes that digestion efficiency and peptide recovery is high and uniform across the concentration range; as in previous work (3, 4), this assumption has not been experimentally verified. Our LRP approach is analogous to LC-MS assays for small molecules that use chemically similar compounds, rather than isotope-labeled analogs as internal standards (see (23–31) for several recent examples). In the LRP method, a single isotopically labeled peptide is used as the normalization reference for all of the peptides in the analysis. The reference peptide is isotopically labeled to minimize interference from any endogenous, unlabeled peptide in the cell or plasma background. The integrated peak areas for transitions from the unlabeled peptide analyte are summed and then normalized to the summed peak areas for transitions from the labeled reference peptide. The reported values are “peak area ratio.” In the LF method, no peptide standards are employed, but samples are prepared and injected at equivalent concentrations based on starting protein concentration. The integrated peak areas for transitions from each peptide analyte are summed and reported as “peak area.” The LF method is analogous to MS1 profiling approaches to peptide quantitation (32–37), except that we have extended the concept from analysis of the MS1 signal to analysis of MRM transitions.
We compared the methods with a weighted robust regression model that included modeling of change points, which account for possible loss of signal response at low analyte concentrations (i.e. the response curve “flattens out” at low concentrations). We determined slopes of the regression curves, regression coefficients (r2) and the coefficients of variation (CV). We also calculated RS for the measurements, which enables comparison of the concentration-response relationships for the three methods. A detailed description of the statistical models and methods is provided under “Experimental Procedures.”
Analysis of AP and BG Peptides Spiked Into RKO Cell Lysates
To compare SID with the LRP and LF approaches to quantitation, we first employed a model system in which peptides from the E. coli AP and BG proteins were spiked into an RKO human colon adenocarcinoma cell lysate. Three peptides each from AP and BG were synthesized in isotopically labeled and unlabeled forms and were used as reference standards. The peptide sequences and transitions monitored are listed in Table I. Dilution curves for these six pairs of synthetic peptides were prepared in tryptic digests of cell lysates from RKO cells. To avoid unwanted side-reactions with the synthetic peptides, the lysate was first reduced with dithiotreitol and alkylated with iodoacetamide. Following addition of trypsin for enzymatic digestion, the six pairs of peptides were spiked into the mixture, which was then incubated overnight at 37 °C for protein digestion.
The data generated by analyses of RKO lysates spiked with the six AP and BG peptides were analyzed by the SID, LRP and LF quantitation methods, respectively and representative data for the AP593 peptide2 AAQGDITAPGGAR are shown in Figs. 1A–1C. For all AP and BG spike studies, the isotope-labeled form of the AP557 peptide (APGLTQALNT*K) was used as the LRP reference standard. This peptide was selected because it generated strong MRM transition signals, eluted near the midpoint of the chromatogram and displayed low variability in peak area. Slopes of the regression curves (labeled as “coeff”) and standard errors, r2, CV, and RS values for all of the AP and BG peptides are presented in Table S1 and concentration-response plots for all of the AP and BG peptides analyzed in this experiment by the SID, LRP and LF methods are presented in supplemental Figs. S1–S6. One peptide (BG698) yielded low, variable signal intensities and low slope and r2 values and a high CV for all three analysis methods.
Fig. 1.

Signal responses versus spike concentration plots for synthetic AP593 peptide (upper panels) and intact AP protein (lower panels) spiked into RKO lysates and quantified by SID (A and D), LRP (B and E) and LF (C and F) methods. AP593 peptide or intact AP protein were spiked at the indicated concentrations into 0.5 μg μl−1 RKO cell lysates and processed as described under “Experimental Procedures.” For all AP and BG spike studies, the isotope-labeled form of the AP557 peptide (APGLTQALNT*K) was used as the LRP reference standard. Symbols represent signals measured with each method. Solid curves represent regression fits to the data. Dotted lines represent a slope of unity.
The median slope for the SID analysis curves was 1.03 (range 0.6 to 2.0 (excluding the BG698 peptide)), which is similar to the median value and range reported by Addonna et al. for a similar experimental design (see below) (3). Slope values for the LRP and LF analyses compare different units on the y axis and x axis and would not be expected to be unity.
The SID analyses displayed the highest median r2 values (0.999) and the lowest median CV (0.019) (Figs. 2A and 2B and supplemental Table S1). LRP and LF analyses displayed lower median r2 values (0.920 and 0.9175, respectively) and higher median CV values (0.059 and 0.142, respectively). The median RS for the LRP and LF methods relative to SID was 0.324 and 0.140, respectively. Use of the other labeled AP and BG peptides as LRP standards yielded similar data (not shown).
Fig. 2.

Summaries of r2 (upper panels) and %CV values (lower panels) for the AP/BG peptide and protein spike experiments (A and B), the CPTAC peptide spike (Study I) experiments (C and D) and CPTAC protein spike (Study III) experiments (E and F). Sites 65 and 52 are the laboratory designations given in (3).
Essentially identical results were obtained in an experiment in which the AP and BG peptides were spiked into the RKO lysate following reduction and alkylation, but prior to addition of trypsin (not shown). We also obtained similar results in experiments in which the RKO lysate was subjected to SDS-PAGE and gel bands were excised and spiked with the AP or BG peptides and labeled standards either prior to or following in-gel tryptic digestion (not shown).
Analysis of AP and BG Proteins Spiked Into RKO Cell Lysates
A second set of studies compared the methods to analyze the intact AP and BG proteins spiked into the lysate prior to in-solution digestion. The AP and BG proteins were spiked into 0.5 μg/μl of RKO lysates at equimolar concentrations of 5, 20, 80, 200, and 400 fmol/μg and the six isotopically labeled AP and BG peptides (Table I) were spiked in at a concentration of 60 fmol/μg prior to digestion. Five replicate samples were prepared for each AP or BG protein concentration for LC-MRM-MS. Figs. 1D–1F shows the concentration-response plots for the AP593 peptide AAQGDITAPGGAR analyzed by the SID, LRP and LF methods. Slopes of the regression curves, r2, CV, and RS values for all of the AP and BG peptides are presented in supplemental Table S2 and concentration-response plots for all of the AP and BG peptides analyzed in this experiment by the SID, LRP, and LF methods are presented in supplemental Figs. S7–S12.
The median slope for the SID analysis curves was 0.645 (range 0.195 to 1.95), which is similar to values reported by Addonna et al. for a similar protein spike experimental design (see below)(3). As noted above, the LRP and LF analysis plots had different x axis and y axis values and the slopes would not be expected to be unity.
The LRP and LF methods displayed nearly identical performance to SID in the protein spike experiments (Figs. 2A and 2B and supplemental Table S2). The SID analyses yielded a median r2 of 0.9335 and a median CV of 0.18. LRP and LF analyses displayed similar median r2 values (0.918 and 0.8615, respectively) and similar median CV values (0.148 and 0.195, respectively). The RS of the LRP and LF methods also approached that for SID in the protein spike experiments, with median values of 0.958 and 0.928, respectively. Use of the other labeled AP and BG peptides as LRP standards yielded similar data (not shown).
Comparison of Quantitation Methods in Analysis of Synthetic Peptides Spiked Into Human Plasma (CPTAC study I)
The National Cancer Institute (NCI) CPTAC program reported a multisite assessment of LC-MRM-MS using SID to analyze a set of seven proteins and corresponding peptides in human plasma (3). The data produced by that study provide an additional opportunity to compare the performance of SID, the LRP, and LF methods with different peptides and in a different background matrix from the studies described above. We analyzed data from two participating laboratories (sites 65 and 52). First, we analyzed data from the first experiment in the CPTAC study (CPTAC study I), in which 11 synthetic peptides derived from seven proteins (bovine aprotinin, murine leptin, equine myoglobin, bovine myelin basic protein, human prostate specific antigen, horseradish peroxidase, and human C-reactive protein) were spiked into digested human plasma with different concentrations, together with an equimolar mixture of 11 stable-isotope-labeled peptides corresponding to the seven proteins (three of the proteins were represented by multiple peptides). The unlabeled peptides were spiked at concentrations ranging from 1 to 500 fmol/μl and the standard peptides were spiked at 50 fmol/μl. These LC-MRM-MS analyses recorded three MRM transitions for each peptide, and four replicates were collected for each concentration. The list of peptides and transitions selected for data LC-MRM-MS acquisition is shown in Table II. We applied the SID, LRP and LF data analysis methods in the same way as we described above. The isotope-labeled APR745 peptide AGLCQTF*VYGGCR was used as the reference standard for all other peptides in LRP analyses.
Table II. Proteins and peptides used in the CPTAC Study (3).
| Protein | Peptidea | Precursor m/z | Product m/z |
|---|---|---|---|
| APR | AGLCQTFVYGGCR | 744.8 | 711.32, 858.39, 959.44 |
| AGLCQTF*VYGGCR | 747.3 | 716.53, 863.41, 964.46 | |
| HRP | SSDLVALSGGHTFGK | 492.6 | 537.29, 651.35, 790.39 |
| SSDLVALSGGHTFG*K | 495.3 | 541.31, 655.37, 798.40 | |
| CRP | ESDTSYVSLK | 564.8 | 609.36, 696.39, 797.44 |
| ESDTSYVSL*K | 568.8 | 617.37,704.41, 805.45 | |
| GYSIFSYATK | 568.8 | 569.29, 716.36, 916.48 | |
| GYSIFSYAT*K | 572.8 | 577.30, 724.38, 924.49 | |
| YEVQGEVFTKPQLWP | 911.0 | 805.37, 1016.56, 1053.49 | |
| YEVQGEVFTKPQ*LWP | 914.0 | 805.37, 1022.58, 1053.49 | |
| LEP | INDISHTQSVSAK | 467.2 | 586.80, 643.82, 720.389 |
| INDISHTQSVSA*K | 469.9 | 590.81, 647.83, 728.40 | |
| MBP | HGFLPR | 363.7 | 455.241, 532.32, 589.35 |
| HGFLP*R | 366.7 | 455.24, 538.34, 595.37 | |
| YLASASTMDHAR | 441.5 | 629.28,730.33, 817.36 | |
| YLASASTMDHA*R | 443.5 | 635.30, 736.35, 823.38 | |
| MYO | LFTGHPETLEK | 424.6 | 506.26, 579.79, 716.38 |
| LFTGHPETLE*K | 427.2 | 510.27, 583.80, 724.40 | |
| PSA | IVGGWECEK | 539.3 | 808.33, 865.35, 964.42 |
| I*VGGWECEK | 541.7 | 808.33, 865.35, 969.44 | |
| LSEPAELTDAVK | 636.8 | 646.38, 846.46, 943.51 | |
| LSEPAELTDAV*K | 640.8 | 654.39, 854.47, 951.52 |
a Peptides marked with * are isotopically labeled.
Fig. 3 shows the concentration-response plots for the PSA637 peptide LSEPAELTDAVK analyzed by the SID, LRP, and LF methods for both the site 65 and site 52 datasets. Slopes of the regression curves, r2, CV, and RS values for all of the peptides are presented in supplemental Table S3. Concentration-response plots for all of the peptides analyzed in this experiment by the SID, LRP, and LF methods are presented in supplemental Figs. S13–S23. Data for most peptides yielded robust concentration-response relationships, although there were some differences in plots for some peptides between sites 65 and 52.
Fig. 3.

Signal responses versus spike concentration plots for PSA637 peptide in CPTAC peptide spike experiments. Upper panels represent data from site 65, whereas lower panels represent data from site 52. PSA637 peptide and other peptides were spiked at the indicated concentrations into digested human plasma and then processed and analyzed as described (3). Plotted data represent summed MRM transitions quantified by SID (A and D), LRP (B and E) and LF (C and F) methods. Symbols represent signals measured with each method. Solid curves represent regression fits to the data. Dotted lines represent a slope of unity.
In the site 65 and site 52 data sets, the median slopes for the SID analysis curves were 1.105 and 1.241, respectively, which are similar to the values calculated from the same data by Addona et al. (3). Thus, our statistical analysis model fit the data similarly to the model used in the original CPTAC evaluation.
The median r2 values for SID measurements were 0.967 (site 65) and 0.995 (site 52), whereas the LRP method yielded lower median values of 0.915 (site 65) and 0.955 (site 52) (Fig. 2). The LF method yielded similar median r2 values of 0.945 (site 65) and 0.951 (site 52). The median CV for SID measurements from sites 65 and 52 were nearly identical (0.027 and 0.039, respectively), whereas median CVs were ∼three- to fivefold higher for both LRP (0.163 for site 65 and 0.0875 for site 52) and for LF (0.126 (site 65) and 0.093 (site 52)). RS for LRP (0.300 for site 65 and 0.3375 for site 52) and for LF (0.321 for site 65 and 0.353 for site 52) were comparable to the values measured for the AP and BG peptide spike experiments (see above).
The range of measured r2 and CV values for all three methods was greater in the CPTAC datasets than in the AP and BG peptide spike datasets. However, the same differences in method performance were apparent—the LRP and LF methods exhibited nearly identical performance and median CVs were ∼two- to fivefold higher than for SID.
Comparison of Quantitation Methods in Analysis of Seven Proteins Spiked Into Human Plasma (CPTAC study III)
We noted above that the LRP and LF methods exhibited nearly identical performance to SID in the AP and BG protein spike experiments (based on CV and RS values). This suggested that the performance advantage of SID over the other methods is diminished when the analysis workflow incorporates protein digestion. To further examine this possibility, we analyzed data from CPTAC study III, in which seven intact proteins were spiked into human plasma. The samples then were digested by the participating laboratories, spiked with isotope-labeled standard peptides and analyzed by LC-MRM-MS as described above. Again, we analyzed data from sites 65 and 52. The isotope-labeled APR745 peptide was again used as the reference peptide for the LRP analyses.
Fig. 4 shows the concentration-response plots for the PSA637 peptide LSEPAELTDAVK analyzed by the SID, LRP, and LF methods for both the site 65 and site 52 data sets. Slopes of the regression curves, r2, CV, and RS values for all of the peptides are presented in supplemental Table S4. Concentration-response plots for all of the peptides analyzed in this experiment by the SID, LRP, and LF methods are presented in supplemental Figs. S24–S34. As with the CPTAC peptide spike experiments, there were some differences in plots for some peptides between sites 65 and 52. Both sites failed to detect sufficient signal for peptide MBP442 (see supplemental Fig. S31), as was previously noted (3); data for this peptide were not included in our assessment of the methods.
Fig. 4.

Signal responses versus spike concentration plots for PSA637 peptide in CPTAC protein spike experiments. Upper panels represent data from site 65, whereas lower panels represent data from site 52. PSA and other intact proteins were spiked at the indicated concentrations into undigested human plasma and then processed and analyzed as described (3). Plotted data represent summed MRM transitions quantified by SID (A and D), LRP (B and E) and LF (C and F) methods. Symbols represent signals measured with each method. Solid curves represent regression fits to the data. Dotted lines represent a slope of unity.
In the site 65 and site 52 datasets, the median slopes for the SID analysis curves were 0.545 and 0.571, respectively, which are similar to the values calculated from the same data by Addona et al. for the same data (3). As noted above, this indicates that our statistical analysis model fit the data similarly to the model used in the original CPTAC evaluation.
The median r2 values for SID measurements were 0.980 (site 65) and 0.984 (site 52), whereas the LRP method yielded lower median values of 0.537 (site 65) and 0.882 (site 52) (Fig. 2E). Median r2 values for the LF method were 0.751 (site 65) and 0.931 (site 52). The median CV for SID measurements from sites 65 and 52 were nearly identical (0.086 and 0.084, respectively) (Fig. 2F), whereas median CVs were ∼two- to threefold higher for both LRP (0.279 for site 65 and 0.165 for site 52) and for LF (0.243 (site 65) and 0.132 (site 52) (supplemental Table S4). RS for LRP (0.323 for site 65 and 0.436 for site 52) and for LF (0.290 for site 65 and 0.526 for site 52) were lower than the values measured for the AP and BG protein spike experiments (see above).
Overall, the analyses of the CPTAC protein spike data showed that SID exhibited performance superior to that of the LRP and LF methods. Aside from lower CV values, the major advantage of SID was a higher median r2 and a narrower range of CV values. However, the two sites differed markedly in ranges for these parameters.
Comparison of LC-MRM-MS Quantitation Methods for Analysis of Cancer Biomarker Candidate Proteins in Human Lung Tissues
A key rationale for this work is the need for alternatives to SID, which can be excessively costly when applied to screening large numbers of biomarker candidates. We therefore evaluated the SID, LRP, and LF approaches to analyze the relative expression level of three housekeeping proteins and seven biomarker candidate proteins in ten pairs of human normal lung and lung tumor tissue samples. The selection of candidate biomarkers is based on reported gene expression analyses and our recent shotgun proteome analyses of normal lung and lung tumor tissues (Kikuchi, T. et al., manuscript submitted). Signature peptides for target proteins were selected and refined according to the criteria described above (Table III). For each protein, 1 to 3 signature peptides were selected and four MS/MS transitions were selected for each peptide. Three isotopically labeled standard peptides, GYSFTTTAE*R (with U-13C6, U-15N4-arginine), from ACTIN; VLSPQGGGPWDSVA*R (with U-13C6, U-15N4-arginine) from AGER and VLDLEL*K (with U-13C6, U-15N2-lysine) from ANXA1, were spiked into tissue digests at a concentration of 60 fmol/μg prior to LC-MRM-MS analysis. Selected transitions from all candidate peptides were optimized in trial LC-MRM-MS analyses of normal, ADC, and SCC tissue samples. Transitions with high peak intensity and no interferences from background matrix were selected for further data collection. The selected peptides and their MRM transitions for those biomarker candidates were listed in Table III, which includes a total of 10 proteins, 27 peptides, and 108 transitions.
Table III. Lung cancer protein biomarker candidates.
| Protein | Peptidea | Precursor m/z | Product m/z |
|---|---|---|---|
| YWHAQ | VISSIEQK | 452.3 | 517.30, 604.33, 691.36, 804.45 |
| NLLSVAYK | 454.3 | 480.28, 567.31, 680.40, 793.48 | |
| AVTEQGAELSNEER | 766.9 | 634.28, 876.41, 947.44, 1004.46 | |
| AGR2 | LPQTLSR | 407.7 | 375.23, 476.28, 604.34, 701.39 |
| HLSPDGQYVPR | 634.8 | 662.36, 719.38, 834.41, 931.46 | |
| LAEQFVLLNLVYETTDK | 998.5 | 464.24, 756.34, 855.41, 968.49 | |
| AGER | VLSPQGGGPWDSVAR | 763.4 | 613.80, 657.32, 944.46, 1001.48 |
| VLSPQGGGPWDSVA*R | 768.4 | 442.26, 897.45, 954.47, 1011.49 | |
| ANXA1 | VLDLELK | 415.3 | 389.24, 502.32, 617.35, 730.43 |
| VLDLEL*K | 419.3 | 397.25, 510.34, 625.36, 738.45 | |
| ACTIN | GYSFTTTAER | 566.8 | 476.25, 678.34, 825.41, 912.44 |
| GYSFTTTAE*R | 571.8 | 486.25, 688.35, 835.42, 922.45 | |
| CEACAM1 | TTVTGDK | 361.2 | 319.16, 420.21, 519.28, 620.32 |
| LQLSNGNR | 451.2 | 346.18, 547.26, 660.34, 788.40 | |
| TLTLLSVTR | 502.3 | 462.27, 575.35, 688.44, 789.48 | |
| NAPSA | VDGILSEDK | 488.3 | 478.21, 591.30, 704.38, 876.43 |
| FAIQYGTGR | 506.8 | 553.27, 681.33, 794.42, 865.45 | |
| VGPGLTLCAK | 508.3 | 592.31, 705.40, 762.42, 916.49 | |
| PDIA6 | GSFSEQGINEFLR | 742.4 | 678.36, 791.44, 848.46, 976.52 |
| LAAVDATVNQVLASR | 764.4 | 787.44, 886.51, 987.56, 1058.60 | |
| GSTAPVGGGAFPTIVER | 808.4 | 714.41, 861.48, 989.54, 1046.56 | |
| PARK7 | ALVILAK | 364.3 | 331.23, 444.32, 543.39, 656.47 |
| VTVAGLAGK | 408.3 | 445.28, 516.31, 615.38, 716.43 | |
| DGLILTSR | 437.8 | 363.20, 476.28, 589.37, 702.45 | |
| TPD52 | LGINSLQELK | 557.8 | 389.24, 517.30, 630.38, 717.41 |
| ASAAFSSVGSVITK | 662.9 | 604.37, 703.43, 877.50, 1024.57 | |
| GWQDVTATSAYK | 663.8 | 569.29, 640.33, 840.45, 1083.53 |
a Peptides marked with * are isotopically labeled.
Portions of five human lung ADC and five SCC, together with paired adjacent normal tissues were lysed and the proteins were subjected to short (∼1 cm) SDS-PAGE separation (14). The entire protein mixture was digested in-gel with trypsin and LC-MRM-MS of the digests was performed as described above. The isotopically labeled AGER, ACTIN and ANXA1 peptide standards were spiked into each sample prior to in-gel digestion. The stability of these three isotopically labeled peptides during in-gel digestion was evaluated in a trial experiment in which RKO cell lysate was used as protein background (data not shown). Good signal reproducibility for peptide peak areas was observed for all three isotopically labeled peptides (CV <20% in five replicates).
The three LC-MRM-MS data analysis approaches were first evaluated using the three target proteins for which unlabeled and isotopically labeled peptide pairs were available-AGER, ANXA1, and ACTIN (Fig. 5). Although MRM data were acquired for multiple peptides per protein, data for a single representative peptide per protein were analyzed. For SID, the normalization peptides were the labeled isotopomers for each target sequence. For the LRP method, the AGER768 peptide VLSPQGGGPWDSVA*R was used as the reference standard for the other target peptides. The ratios of peptide peak areas for the unlabeled peptides and the corresponding spiked normalization peptides were plotted for each tissue sample. LF analysis was done directly from the peak areas of the target peptides.
Fig. 5.

Comparison of three proteins in 10 pairs of normal lung and matched lung tumor tissues by three quantitation approaches. ADC and matched normal controls are compared in the left panels and SCC and matched normal controls are compared in the right panels. The proteins and representative peptides analyzed are shown at the top of each column of plots. Peptide quantitation was performed by SID, LRP (with AGER peptide VLSPQGGGPWDSVA*R as reference peptide), and by the LF approach. Y-axis label for SID is the log10 of the ratio of summed MRM transition peak areas for the target peptide and its labeled standard. Y-axis label for LRP is the log10 of the ratio of summed MRM transition peak areas for the target peptide and the LRP standard Y-axis label for LF is the log10 of the summed MRM transition peak area for the target peptide. Statistically significant differences between tumor and control values (*p < 0.05) are indicated.
Comparisons of expression of AGER, ACTIN, and ANXA1 in each pair of SCC and ADC tumor and corresponding normal tissues were demonstrated with SID (Fig. 5A), LRP (Fig. 5B), and LF methods (Fig. 5C), respectively. It was clear that for AGER, the expression level was significantly reduced in both ADC and SCC tissues compared with their corresponding normal lung tissues (p < 0.01 for all measurements), as has been observed by previous immunohistochemistry and mRNA expression analyses (38). In addition, ACTIN did not show significant expression difference between SCC and normal tissues. However, its expression level significantly increased in ADC compared with normal tissue type (p < 0.05 with all three quantitation approaches). This indicated that ACTIN expression maybe differ with tumor phenotype. In contrast, ANXA1 did not show a consistent and significant expression difference between tumor-normal pairs for either tumor type. Although tumor-normal differences were not statistically significant, the same patterns of ANXA1 were detected by all three data analysis methods.
The expression levels of seven candidate lung cancer biomarkers in ADC and SCC tumor-normal pairs were compared using the LRP method (with AGER768 peptide VLSPQGGGPWDSVA*R as internal standard) and LF quantitation. For these analyses, one to three peptides were analyzed for each protein (Table III) and the peak area for a representative peptide from each was used as the basis for comparison of the target proteins. When multiple peptides per protein were analyzed, the trends for tumor and normal expression were consistent. However, we did occasionally observe inconsistencies, which reflected relatively small differences that were within the range of measurement error. For example, the PARK7 protein was measured by three peptides (Table III). Of these, the PARK7 peptides ALVILAK and VTVAGLAGK gave consistent trends for all tumor-normal pairs with both LRP and LF analyses, whereas the peptide DGLILTSR yielded different trends for LRP and LF for one tumor-normal pair (supplemental Fig. S36). This example illustrates the value of measuring multiple peptides for each target protein wherever possible.
Both the LRP and LF approaches yielded similar results and consistently detected the same expression differences between tumor-normal pairs (Fig. 6). Significant expression differences between tumors and corresponding adjacent normal tissues were observed for NAPSA, TPD52, and PDIA6 in SCC (Fig. 6A and 6B) and for CEACAM1, TPD52, PARK7, PDIA6, SFN, and AGR2 in ADC (Fig. 6C and 6D). To test the consistency of the LRP with different standards, the normalization calculations were repeated using either the ANXA1 VLDLEL*K peptide (supplemental Fig. S37) and the ACTIN GYSFTTTAE*R peptide (supplemental Fig. S38) as LRP standards. All of the differences detected with the AGER peptide as LRP standard were also detected with the other two standards. In the analyses with the ANXA1 and ACTIN peptide LRP standards, the expression difference for CEACAM1 in SCC versus normal tissue achieved statistical significance.
Fig. 6.

Relative quantitation of seven lung cancer biomarker candidates in 10 pairs of lung tumor and matched normal control lung tissues by the LRP and LF methods. The proteins and representative peptides analyzed are shown at the top of each column of plots. Comparisons of SCC tumors and matched normals using the LRP method and LF method are shown in the top two rows. Comparisons of ADC tumors and matched normals by the LRP method and LF method are shown in the bottom two rows. The AGER peptide VLSPQGGGPWDSVA*R was used as the reference peptide for the LRP method. Y-axis labels for LRP and LF plots are as shown in Fig. 5. Statistically significant differences between tumor and control values (*p < 0.05) are indicated.
Our measurements of protein biomarkers in lung tumors are consistent with previous literature reports (39–42). All of the significant expression differences were detected by both the LRP and LF methods. The value of screening individual tumor-normal pairs is indicated by the fact that combined analysis of all tumors versus all normal (not shown) indicated significant differences only for TPD52, PDIA6, and CEACAM1, but would not have detected the tumor-type-specific differences for candidate markers.
DISCUSSION
We undertook these studies to explore less costly alternatives to SID for quantitation by LC-MRM-MS. Our studies compared the LRP and LF approaches with SID in both peptide and protein spike experiments using cell lysates and plasma backgrounds—use contexts typical of many quantitative proteomics studies. SID is clearly the best of the three methods, based on all performance metrics. The LRP and LF methods displayed greater measurement variability, as reflected by higher median CVs, lower r2 values, and lower RS values. In protein spike experiments—the most “real world” use context for MRM quantitation—median CV values were about 10% for SID and 20–30% for LRP and LF methods. The impact of these differences in measurement precision can be modeled with power calculations, as we describe below. In lung tumor and normal tissues, all three methods detected the same significant differences in protein expression, which demonstrates that moderate differences in measurement precision have less impact on real measurements than biological variation. In the following sections, we summarize the performance characteristics of the SID, LRP, and LF methods, we consider sources of error and how they affect the methods and we provide a guide to appropriate implementation contexts for each.
All quantitative measurements are characterized by precision and accuracy. The precision of MRM analyses can be characterized easily. Accuracy is harder to assess and is complicated by uncontrolled variability in protein digestion and recovery (3). Full-length, labeled protein internal standards would be required for truly accurate quantitation, but they are generally unavailable. Accuracy is thus uncertain for all MRM analysis methods. However, the goal of most MRM applications is comparison of protein levels, rather than assessment of absolute amounts. The main assumption in such quantitative comparisons is that deviations from accuracy are evenly distributed across the measurements in all sample sets. This assumption seems plausible, at least when applied to sample sets of the same tissue type.
SID analyses provide a performance benchmark for evaluating the LRP and LF methods and our SID analyses yielded data consistent with previous reports. We used the summed peak areas for four transitions from each peptide as our primary unit of comparison. This approach is based on the observations of Anderson et al. (2), who demonstrated that measurement based on the combined signal from multiple transitions reduced CV values. Our SID analyses of the AP/BG and CPTAC peptide spike experiments yielded CV values typically well below 10%, with a global median of 2.9% (Fig. 2 and supplemental Table S5)). In the CPTAC study, measurements of the spiked peptides were based on the ratios for individual transitions and yielded intralaboratory CVs of <5% across the concentration range studied (3). In our analysis of the AP/BG protein spike experiments, CVs ranged from 10–20%, with a median of 18% (Fig. 2 and supplemental Table S5). Our analyses of the CPTAC protein spike data from two sites yielded CVs ranging from 3–21% with a median of 8.5%. These results also are consistent with previous evaluations of SID (2, 3, 7, 8).
In peptide spike experiments, SID was clearly the best method and yielded the highest median r2 values and the lowest median CV values (Fig. 2 and supplemental Table S5). In protein spike experiments, the advantage of SID over the other two methods was diminished. Median CV values across all of the protein spike datasets for SID, LRP, and LF methods were 9.9%, 17.7%, and 16.5%, respectively (supplemental Table S5). This more modest advantage of SID over the LRP and LF methods in protein spike experiments suggests that protein digestion and extraction steps introduce variability that is not well controlled by any of the methods.
LRP and LF analyses of the CPTAC data indicated large differences in the range and median CV (Fig. 2D and 2F), but little variability in SID analyses. Inspection of the chromatograms corresponding to measurements displaying high variability indicated retention time variations and split peaks for some of the peptides, which especially impacted LRP and LF measurements. Co-elution of isotope labeled standards with the target peptides in SID largely eliminated this source of variation. However, the retrospective application of LRP and LF analyses to the CPTAC data provide “worst case” examples for these methods, as we were restricted to the available data and we did not have the opportunity to select peptides and optimize performance for LRP and LF. We were able to do so for the AP and BG spike studies, which yielded data much more comparable to SID (compare Figs. 2E and 2A).
Another consideration in comparing CVs for the three methods is the choice of analyte concentration at which CVs are compared. In our model, CVs are calculated from the regression plots at the highest analyte concentration, based on our observation that the CVs showed the lowest variation. However, one may ask whether this comparison is valid for measurements that frequently are used to measure peptides at relatively low concentrations. To address this point, we reanalyzed the AP and BG protein spike data to determine CVs at all concentrations. Although calculation based on datapoints from each concentration yields ∼1.7-fold higher CVs than does estimation from the regression fits, this calculation allows comparison of relative CVs at different concentrations. The plots for the six AP and BG peptides are shown in supplemental Fig. S35 and indicate that all three methods display elevated CVs at the lowest spike concentrations. Between 40 and 200 fmol, CVs for all three methods display similar variation and their relative magnitude is consistent.
Despite the superiority of SID over the other methods in the spike experiments, all three methods distinguished the same significant protein expression differences in analyses of lung tissue specimens. In Fig. 5, each paired tumor-normal comparison for each of the four proteins analyzed yielded the same differential comparison by all three methods. The p values for all of the measured differences were nearly identical. In the additional studies where only LRP and LF analyses were done (no SID standards were available), the two methods yielded consistent differences for the target proteins in each of the tumor-normal pairs (Fig. 6). In these analyses, p values calculated from LRP measurements were usually lower than those from LF measurements. Thus, although SID is the most precise method, the LRP and LF methods can also detect biologically relevant protein differences in tissue specimens.
The ability of methods to detect differences can be represented by power calculations, which consider both measurement variability and the magnitude of measured differences (43). The CV for a measured biological signal reflects both biological variability and measurement error. The law of propagation of errors (22) yields the following relationship:
where z is the measured signal, x is the biological variability and e is the measurement error. Consider measurement of a protein with typical biological variability in expression (CV of 40%). Analysis by SID (measurement error CV of 10%) yields a measured CV of 41%. Analysis of the same protein by LRP (measurement error CV of 20%) yields a measured CV of 45%. Thus, the effect of measurement error on total measured variation is relatively small, but becomes more significant as biological variation decreases. These relationships are illustrated in Table IV, which indicates that SID (median CV 10%) can detect true differences as small as 1.5-fold in analyses of only 3 samples. On the other hand, LRP and LF analysis (median CV 20%) can detect the same differences in analyses of 5 samples. All three methods thus can detect protein expression differences in sets of biological samples, although application of the LRP and LF methods would require more measurements than SID.
Table IV. Sample sizes required to detect actual fold differences in protein expression as a function of measurement CV (43).
| Actual fold-difference | ||||
|---|---|---|---|---|
| %CV | 1.5 | 2 | 2.5 | 3 |
| 10 | 3 | 3 | 3 | 4 |
| 20 | 5 | 3 | 3 | 3 |
| 30 | 10 | 4 | 3 | 3 |
| 40 | 16 | 6 | 4 | 4 |
| 50 | 23 | 9 | 6 | 5 |
| 100 | 68 | 24 | 14 | 11 |
All MRM analyses are subject to several sources of error, many of which are minimized in SID by a co-eluting labeled peptide standard. These sources of error should be considered in implementing LRP and LF methods. The first is erroneous assignment of an unrelated signal to a target peptide—i.e. a “false positive quantitation.” SID essentially removes this problem because the labeled standard unambiguously establishes chromatographic retention and correct MRM transitions. In the absence of standards, association of MRM transitions to peptides is typically done with MS/MS spectral libraries (17); intense signals in ion trap MS/MS spectra—particularly for y-ions—reliably predict high intensity MRM transitions obtained with triple quadrupole instruments (18). The use of multiple ions and transitions to select target signals increases confidence that the correct target is measured. (We provide four examples of the correspondence between ion trap MS/MS spectra and MRM transitions for peptides analyzed in this work in supplemental Figs. S39–S40). Optimization of LRP analyses can employ specific types of biological samples where other analyses (e.g. shotgun proteomics) has established the presence of the target proteins and peptides. For example, we used preliminary analyses of lung tumor tissues to select and evaluate candidate peptides for our analyses of lung cancer biomarker proteins.
The second source of error is interference from contaminating substances that generate signal in the transitions measured. This has been noted in previous work and demonstrated in detail by Addona et al. (3). In most cases, interference from contaminants can be recognized, although not removed, by inspecting the relative intensities and co-elution of peaks corresponding to the MRM transitions. A third source of error is variation in system sensitivity (e.g. the performance of detectors, ionization sources, and system electronics) within the time frame of an individual LC-MRM-MS run. SID provides the greatest protection against such problems, as commonly used 13C/15N-labeled standards co-elute with their unlabeled analogs, thus correcting for moment-to-moment variation. However, current generation LC-MS systems are actually quite stable, which accounts for the ability of both the LRP and LF methods to produce median CV values from 12–15% across the peptide spike experiments. Median CV values for the LRP and LF methods were 15.3% and 13.4%, respectively, compared with 2.9% for SID across the peptide spike experiments, which indicates the degree to which co-eluting, isotopically labeled standards correct for within-run variation. A fourth source of error is ion suppression, which may vary during an LC run, thus affecting different peptides to different extents. Ion suppression may also vary with sample matrix. A fifth source of error is gradual system drift, which is the change in system sensitivity from run to run, particularly over longer time periods (days to weeks). SID and LRP should reduce error because of gradual system drift by providing normalizing signals from the standards. LF measurements would be expected to be most susceptible to error because of system drift. The LC-MRM-MS analyses for our AP and BG spike studies were done over a time frame of 1–4 weeks and the performance of the system in our laboratory was monitored by periodic analyses of a QC peptide mixture.
We suggest that analyses using the LRP or LF methods should incorporate controls to detect or minimize these sources of error. First, technical replicate LC-MS injections should be incorporated into experimental designs, not only to provide statistical power (by reducing measurement variability), but also to enable better assessment of variability in a measurement series. Second, the run order for a sample series should distribute sample groups equally into “blocks,” in which a block is composed of samples from each group. In this arrangement, drift does not disproportionately affect any individual sample group. Third, comparisons of peak areas for LRP standards across a run series can indicate drift in assay performance. Similarly, in LF analyses, monitoring of MRM transitions for endogenous peptides known to have stable expression across the sample series can identify drift. Fourth, peak area CVs can be calculated for replicate QC analyses to assess changes in system performance.
For use in the LRP method, a reference peptide should have good chemical stability, ionization efficiency, and chromatographic properties. To avoid interference from native sequences, we used isotopically labeled reference peptides, although sequences from other organisms could also be employed. We used a single reference peptide to normalize all analyte signals in our analyses, but the LRP method could accommodate multiple reference peptides. These could either be chosen to distribute across the chromatographic separation, thus possibly offering standards for early, medium and late-eluting peptide analytes. Alternatively, the different reference peptides could be spiked at different concentrations in the samples to facilitate normalization of signals for high, medium and low abundance analytes. In addition to serving as normalization standards, the reference peptides also can serve as quality control spikes for use in evaluating system performance variation and chromatographic retention time shift between different LC runs. We have chosen labeled reference peptides that correspond to sequences that are commonly expressed in the tissues analyzed (e.g. mammalian housekeeping proteins), because these peptides also provide an SID measurement for the corresponding protein in each analysis, which also may be used for QC purposes.
Our LF quantitation approach is analogous to commonly used methods for MS1 signal profiling, in which a standardized LC-MS analysis and peak areas derived from peptide ions are used as the basis for sample comparisons (33, 36, 37, 44). Our method differs from such MS1 profiling in that peak areas are extracted from MRM transitions, which are summed to generate peptide or protein peak areas. Our LF approach yielded lower linearity for target peptide quantitation than the SID or LRP methods, as measured by r2 values (see supplementary Tables S1–S4). This difference reflects the lack of a normalization standard to correct for run-to-run variations in LC-MS instrument performance, which may include variation in injection volume, electrospray stability, ion transfer efficiency, fluctuation of LC solvent flow rate and other factors. Nevertheless, these sources of variation collectively had a modest impact, which was reflected primarily in the higher CV values measured in LF analyses. These higher CV values for LF analyses may compromise detection of subtle differences in protein levels. Nevertheless, for detection of differences greater than twofold, the LF method could be appropriate for preliminary screening of biomarker candidates in tissues or biofluid samples. Indeed, the LF method could be used in largely the same way immunoblotting is currently used—for routine exploration of protein level differences in applications where high precision is not required. Moreover, the power calculations summarized in Table IV can guide the design of LF MRM analyses.
A possible variation of the LF approach is the use of signal from endogenous housekeeping peptides (e.g. the ACTIN571 GYSFTTTAER peptide) for normalization of other measured peptide or protein peak areas. We explored this approach in preliminary studies, but determined that this normalization did not improve the linearity or precision of measurements. Indeed, the unnormalized LF approach we describe here displayed greater precision and measurement sensitivity than did LF measurements with normalization to housekeeping proteins (data not shown). We also noted that levels of some housekeeping proteins displayed systematic differences between biological phenotypes (for example, the ACTIN expression difference between normal lung versus ADC tumor tissues in our current study) (Fig. 5), which could complicate attempts to use this normalization strategy to evaluate cancer biomarker candidates.
Given our findings, when can LRP and LF methods be used instead of SID? The appropriate use contexts for the three methods overlap significantly, but the choice should be based on fitness for purpose and cost. A major advantage of LF and LRP analyses is that they can be quickly configured and applied without the cost and delays involved in obtaining labeled peptide standards. The LRP and LF methods are well suited to estimate differences in expression for a few proteins in a small number of samples, as is frequently done in biochemistry and cell biology studies. These analyses typically do not require high precision and are done by immunoblotting, which yields CVs in the range from 20–40% (45, 46).
LRP is well-suited to screen biomarker candidate proteins in larger collections of biospecimens corresponding to multiple phenotypes. We recently employed this approach to confirm protein expression differences found by shotgun proteomics of head and neck tumors and normal tonsillar tissues (16). In a typical discovery proteomics study, 50–200 protein differences may be identified by shotgun proteomic analyses of pooled samples corresponding to multiple specimens of two different tissue phenotypes. An LRP-based LC-MRM-MS screen then can be configured to confirm differential expression of the proteins in the individual specimens that comprised the pools. This analysis would help to identify biomarker candidates to pursue in subsequent studies. The advantage of an LRP-based analysis in this context is particularly clear when cost is considered. To configure SID assays for 50 differential proteins with three labeled standard peptides for each would cost approximately $150,000. If SID were the only option for quantitative analysis, this cost factor would significantly constrain the evaluation of candidates.
Despite the advantages of the LRP and LF methods, we consider SID the most appropriate choice for analyses that require the highest analytical precision and where the analyses will be done over an extended period of time or across multiple laboratories. SID provides the greatest protection against system drift and chromatographic instability, which are major contributors to measurement variation in interlaboratory studies (3). In the context of the LRP-based study described immediately above, we would configure SID assays for subsequent, larger-scale studies of a smaller number of best performing candidates.
In summary, we have introduced two alternatives to SID and compared their performance to that of SID-based LC-MRM-MS in different experimental contexts. Our results indicate that the LRP and LF approaches display greater measurement variability, yet all three can detect significant differences in protein expression in tissue specimens. We conclude that the LRP method provides a viable alternative to SID for many proteomics applications, particularly when costs of SID standards might be viewed as prohibitive. When circumstances require the most robust and precise LC-MRM-MS assay, SID remains the best choice.
Acknowledgments
We thank Dr. Steve Skates (Harvard University and Massachusetts General Hospital) for helpful discussions.
Footnotes
* This work was supported by the Clinical Proteomic Technology Assessment for Cancer (CPTAC) program (U24CA126479) and SPORE in Lung Cancer (P50CA090949) from the National Cancer Institute.
 This article contains supplemental Figs. S1 to S40 and Tables S1 to S7.
This article contains supplemental Figs. S1 to S40 and Tables S1 to S7.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material, including Tables S1 to S7 and Figs. S1 to S40. The data associated with this manuscript may be downloaded from ProteomeCommons.org Tranche, https://proteomecommons.org/tranche/, using the following hash: PzfhsUNk6+yU4d1Jsgkpe9I1d5VEV6uqqnMT+nKM/PCZ2EoK/srS1GY0RFOMofe5OHy8S4ymAjrjrUNEvAAImGMhItQAAAAAAAAqLg==
Human proteins are referred to by their corresponding Entrez gene names, as listed in the UniProt Knowledgebase (http://www.uniprot.org/).
We employ here a peptide naming shorthand based on the protein name abbreviation and the precursor m/z of the unlabeled peptide ion targeted for MRM analysis. Thus, the peptide AAQGDITAPGGAR from the BG protein with doubly charged precursor m/z 593 is referred to as “BG593”. The peptide sequences and precursor m/z are defined in Tables I, 2 and 3.
1 The abbreviations used are:
- LC-MRM-MS
- liquid chromatography-tandem mass spectrometry by multiple reaction monitoring
- SID
- stable isotope dilution
- LRP
- labeled reference peptide
- LF
- label-free
- CPTAC
- Clinical Proteomic Technology Assessment for Cancer
- ADC
- adeno carcinoma
- SCC
- squamous cell carcinoma
- QC
- quality control
- AIC
- Akaike's information criterion
- CV
- coefficient of variance
- RS
- relative sensitivity.
REFERENCES
- 1. Rifai N., Gillette M. A., Carr S. A. (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 [DOI] [PubMed] [Google Scholar]
- 2. Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 3. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gerber S. A., Rush J., Stemman O., Kirschner M. W., Gygi S. P. (2003) Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U.S.A. 100, 6940–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kirkpatrick D. S., Gerber S. A., Gygi S. P. (2005) The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods 35, 265–273 [DOI] [PubMed] [Google Scholar]
- 6. Picotti P., Bodenmiller B., Mueller L. N., Domon B., Aebersold R. (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Keshishian H., Addona T., Burgess M., Mani D. R., Shi X., Kuhn E., Sabatine M. S., Gerszten R. E., Carr S. A. (2009) Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell Proteomics 8, 2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whiteaker J. R., Zhao L., Anderson L., Paulovich A. G. (2009) An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell Proteomics. 10: M110.005645, E-pub [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson N. L., Anderson N. G., Haines L. R., Hardie D. B., Olafson R. W., Pearson T. W. (2004) Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 3, 235–244 [DOI] [PubMed] [Google Scholar]
- 11. Anderson N. L., Jackson A., Smith D., Hardie D., Borchers C., Pearson T. W. (2009) SISCAPA peptide enrichment on magnetic beads using an in-line bead trap device. Mol. Cell Proteomics 8, 995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hoofnagle A. N., Becker J. O., Wener M. H., Heinecke J. W. (2008) Quantification of thyroglobulin, a low-abundance serum protein, by immunoaffinity peptide enrichment and tandem mass spectrometry. Clin. Chem. 54, 1796–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slebos R. J., Brock J. W., Winters N. F., Stuart S. R., Martinez M. A., Li M., Chambers M. C., Zimmerman L. J., Ham A. J., Tabb D. L., Liebler D. C. (2008) Evaluation of strong cation exchange versus isoelectric focusing of peptides for multidimensional liquid chromatography-tandem mass spectrometry. J. Proteome Res. 7, 5286–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lapierre L. A., Avant K. M., Caldwell C. M., Ham A. J., Hill S., Williams J. A., Smolka A. J., Goldenring J. R. (2007) Characterization of immunoisolated human gastric parietal cells tubulovesicles: identification of regulators of apical recycling. Am. J. Physiol. Gastrointest Liver Physiol 292, G1249–1262 [DOI] [PubMed] [Google Scholar]
- 15. Keshishian H., Addona T., Burgess M., Kuhn E., Carr S. A. (2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li M., Gray W., Zhang H., Chung C. H., Billheimer D., Yarbrough W. G., Liebler D. C., Shyr Y., Slebos R. J. (2010) Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J. Proteome Res. 9, 4295–4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prakash A., Tomazela D. M., Frewen B., Maclean B., Merrihew G., Peterman S., Maccoss M. J. (2009) Expediting the development of targeted SRM assays: using data from shotgun proteomics to automate method development. J. Proteome Res. 8, 2733–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sherwood C. A., Eastham A., Lee L. W., Risler J., Vitek O., Martin D. B. (2009) Correlation between y-type ions observed in ion trap and triple quadrupole mass spectrometers. J. Proteome Res. 8, 4243–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Licklider L. J., Thoreen C. C., Peng J., Gygi S. P. (2002) Automation of nanoscale microcapillary liquid chromatography-tandem mass spectrometry with a vented column. Anal. Chem. 74, 3076–3083 [DOI] [PubMed] [Google Scholar]
- 20. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Durbin B. P., Hardin J. S., Hawkins D. M., Rocke D. M. (2002) A variance-stabilizing transformation for gene-expression microarray data. Bioinformatics. 18 Suppl 1:S105–10, S105–S110 [DOI] [PubMed] [Google Scholar]
- 22. Mandel J. (1984) The Statistical Analysis of Experimental Data, Dover Publications, Mineola, NY [Google Scholar]
- 23. Yan W., Byrd G. D., Brown B. G., Borgerding M. F. (2010) Development and validation of a direct LC-MS-MS method to determine the acrolein metabolite 3-HPMA in urine. J. Chromato. Sci. 48, 194–199 [DOI] [PubMed] [Google Scholar]
- 24. Singh S. P., Wahajuddin, Yadav D. K., Rawat P., Maurya R., Jain G. K. (2010) Quantitative determination of formononetin and its metabolite in rat plasma after intravenous bolus administration by HPLC coupled with tandem mass spectrometry. J. Chromatog. 878, 391–397 [DOI] [PubMed] [Google Scholar]
- 25. Helander A., Kenan N., Beck O. (2010) Comparison of analytical approaches for liquid chromatography/mass spectrometry determination of the alcohol biomarker ethyl glucuronide in urine. Rapid Commun. Mass Spectrom. 24, 1737–1743 [DOI] [PubMed] [Google Scholar]
- 26. Wiegand R., Wu J., Sha X., LoRusso P., Li J. (2010) Simultaneous determination of ABT-888, a poly (ADP-ribose) polymerase inhibitor, and its metabolite in human plasma by liquid chromatography/tandem mass spectrometry. J. Chromatog. 878, 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mendu D. R., Soldin S. J. (2010) Simultaneous determination of levetiracetam and its acid metabolite (ucb L057) in serum/plasma by liquid chromatography tandem mass spectrometry. Clin. Biochem. 43, 485–489 [DOI] [PubMed] [Google Scholar]
- 28. Korpi-Steiner N. L., Netzel B. C., Seegmiller J. C., Hagan J. B., Singh R. J. (2010) Liquid chromatography-tandem mass spectrometry analysis of urinary fluticasone propionate-17beta-carboxylic acid for monitoring compliance with inhaled-fluticasone propionate therapy. Steroids 75, 77–82 [DOI] [PubMed] [Google Scholar]
- 29. Zhang W., Dutschman G. E., Li X., Ye M., Cheng Y. C. (2009) Quantitation of Irinotecan and its two major metabolites using a liquid chromatography-electrospray ionization tandem mass spectrometric. J. Chromatog. 877, 3038–3044 [DOI] [PubMed] [Google Scholar]
- 30. Anacardio R., Mullins F. G., Hannam S., Sheikh M. S., O'Shea K., Aramini A., D'Anniballe G., D'Anteo L., Ferrari M. P., Allegretti M. (2009) Development and validation of an LC-MS/MS method for determination of methanesulfonamide in human urine. J. Chromatog. B. Analyt. Technol. Biomed. Life Sci. 877, 2087–2092 [DOI] [PubMed] [Google Scholar]
- 31. Shen H. W., Jiang X. L., Yu A. M. (2009) Development of a LC-MS/MS method to analyze 5-methoxy-N,N-dimethyltryptamine and bufotenine, and application to pharmacokinetic study. Bioanalysis 1, 87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. May D., Fitzgibbon M., Liu Y., Holzman T., Eng J., Kemp C. J., Whiteaker J., Paulovich A., McIntosh M. (2007) A platform for accurate mass and time analyses of mass spectrometry data. J. Proteome Res. 6, 2685–2694 [DOI] [PubMed] [Google Scholar]
- 33. Wang W., Zhou H., Lin H., Roy S., Shaler T. A., Hill L. R., Norton S., Kumar P., Anderle M., Becker C. H. (2003) Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal. Chem. 75, 4818–4826 [DOI] [PubMed] [Google Scholar]
- 34. Li X. J., Yi E. C., Kemp C. J., Zhang H., Aebersold R. (2005) A software suite for the generation and comparison of peptide arrays from sets of data collected by liquid chromatography-mass spectrometry. Mol. Cell Proteomics 4, 1328–1340 [DOI] [PubMed] [Google Scholar]
- 35. Bellew M., Coram M., Fitzgibbon M., Igra M., Randolph T., Wang P., May D., Eng J., Fang R., Lin C., Chen J., Goodlett D., Whiteaker J., Paulovich A., McIntosh M. (2006) A suite of algorithms for the comprehensive analysis of complex protein mixtures using high-resolution LC-MS. Bioinformatics 22, 1902–1909 [DOI] [PubMed] [Google Scholar]
- 36. Fang R., Elias D. A., Monroe M. E., Shen Y., McIntosh M., Wang P., Goddard C. D., Callister S. J., Moore R. J., Gorby Y. A., Adkins J. N., Fredrickson J. K., Lipton M. S., Smith R. D. (2006) Differential label-free quantitative proteomic analysis of Shewanella oneidensis cultured under aerobic and suboxic conditions by accurate mass and time tag approach. Mol. Cell Proteomics 5, 714–725 [DOI] [PubMed] [Google Scholar]
- 37. Finney G. L., Blackler A. R., Hoopmann M. R., Canterbury J. D., Wu C. C., MacCoss M. J. (2008) Label-free comparative analysis of proteomics mixtures using chromatographic alignment of high-resolution muLC-MS data. Anal. Chem. 80, 961–971 [DOI] [PubMed] [Google Scholar]
- 38. Kobayashi S., Kubo H., Suzuki T., Ishizawa K., Yamada M., He M., Yamamoto Y., Yamamoto H., Sasano H., Sasaki H., Suzuki S. (2007) Endogenous secretory receptor for advanced glycation end products in non-small cell lung carcinoma. Am. J. Respir. Crit. Care Med. 175, 184–189 [DOI] [PubMed] [Google Scholar]
- 39. Chuman Y., Bergman A., Ueno T., Saito S., Sakaguchi K., Alaiya A. A., Franzén B., Bergman T., Arnott D., Auer G., Appella E., Jörnvall H., Linder S. (1999) Napsin A, a member of the aspartic protease family, is abundantly expressed in normal lung and kidney tissue and is expressed in lung adenocarcinomas. FEBS Lett. 462, 129–134 [DOI] [PubMed] [Google Scholar]
- 40. Plunkett T. A., Ellis P. A. (2002) CEACAM1: a marker with a difference or more of the same? J. Clin. Oncol. 20, 4273–4275 [DOI] [PubMed] [Google Scholar]
- 41. Kim R. H., Peters M., Jang Y., Shi W., Pintilie M., Fletcher G. C., DeLuca C., Liepa J., Zhou L., Snow B., Binari R. C., Manoukian A. S., Bray M. R., Liu F. F., Tsao M. S., Mak T. W. (2005) DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell 7, 263–273 [DOI] [PubMed] [Google Scholar]
- 42. Qi W., Liu X., Qiao D., Martinez J. D. (2005) Isoform-specific expression of 14–3-3 proteins in human lung cancer tissues. Int. J. Cancer 113, 359–363 [DOI] [PubMed] [Google Scholar]
- 43. Van Belle G., Martin D. C. (1993) Sample size as a function of coefficient of variation and ratio of means. Am. Stat. 47, 165–167 [Google Scholar]
- 44. Jaffe J. D., Mani D. R., Leptos K. C., Church G. M., Gillette M. A., Carr S. A. (2006) PEPPeR, a platform for experimental proteomic pattern recognition. Mol. Cell Proteomics 5, 1927–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Struglics A., Larsson S., Hansson M., Lohmander L. S. (2009) Western blot quantification of aggrecan fragments in human synovial fluid indicates differences in fragment patterns between joint diseases. Osteoarthr. Cartilage 17, 497–506 [DOI] [PubMed] [Google Scholar]
- 46. Aldridge G. M., Podrebarac D. M., Greenough W. T., Weiler I. J. (2008) The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semiquantitative immunoblotting. J. Neurosci. Methods 172, 250–254 [DOI] [PMC free article] [PubMed] [Google Scholar]