

Abstract
A method has been developed to introduce a dimethyleneoxy constraint joining the nitrogen atoms of two consecutive amino acids in the context of a polypeptide. Such a constraint can profoundly affect the tendency of a polypeptide to suffer aggregation and desolubilization. The constraint is readily removed under mild conditions.
Peptides built from proteogenic L-amino acids often possess high affinity and selectivity in binding to various biotargets. However, the value of peptides bearing strictly natural motifs in clinical medicine tends to be compromised by poor in vivo pharmacoproperties, particularly bioavailability.1 A widely considered strategy to upgrade the value of polypeptides envisions incorporation of strategically placed conformational constraints into such molecules. Experience has shown that systems with designed motifs can have useful properties, including increased resistance to protease-induced degradation,2 and decreased flexibility relative to native linear peptides. Well-designed constraints in peptidic structures may result in enhanced affinity of the polypeptide to target receptors.3 For instance, Kim and coworkers designed peptidomimetic inhibitors of HIV-1 infection that outperform analogous linear peptides lacking these conformational constraints.4 Such successes have provided incentive for the development of improved methods for the synthesis of constrained peptides. In a particularly elegant example, Grubbs and coworkers demonstrated that ring-closing metathesis can enable construction of constrained peptide-like structures by exploiting enantiohomogeneous tertiary amides bearing appropriate unsaturation projecting from the amidic nitrogens.5
In the research described below, we undertook the development of a logic, wherein α-amino acids in a regular polypeptide motif would serve to anchor the constraint. In principle, our chemistry could encompass inclusion of either natural or “unnatural” amino acid building blocks. In the first instance we chose to focus on strictly proteogenic amino acids with the additional proviso that the restraining device should be detachable, with restoration of the natural motif. The feature of “reversibility” could be of particular value in peptide synthesis. Thus, a molecular constraint might serve to attenuate aggregation tendencies of otherwise difficultly manageable pre-polypeptide building blocks.6,7,8 The constraint could be dismantled when a more stable (foldable) polypeptide is in hand.
For these purposes, we drew from the growing body of chemistry that emerged from our recent studies of isonitriles.9 We revisited a key reaction, which we discovered, namely the coupling of a carboxylic acid (a) with an isonitrile (b) to afford an N-formylamide (c), presumably via the intermediacy of the formimidate carboxylate mixed anhydride (FCMA),9a as shown. This line of conjecture led to the proposal shown in Scheme 1. Thus, an acid (1) would combine with an amino acid–derived isonitrile 2.9d Following the coupling reaction described above, and subsequent discharge of the protecting group (P), there would be obtained acid 3. Reiteration of this reaction with amino acid–derived isonitrile 4 would give rise, in principle, to 5, containing N-formyl groups on contiguous amidic nitrogens. It was hoped that these proximal, imide-like formyl groups could be exploited to generate a constrained tripeptide, 6. Elongation in both the N- and C-terminal directions would lead to site-specific constrained polypeptide 7. Upon cleavage of the molecular constraint there would be generated polypeptide 8 in a natural motif. While the concept is, in principle, applicable to many versions of “X,” in our initial explorations, described below, we examined the case where X = oxygen.
Scheme 1.

Synthetic strategy.
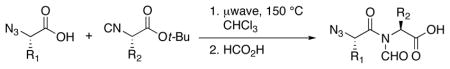
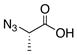
Our studies commenced with coupling of the valine-related azide 9 with leucine–derived isonitrile 109f to afford 11 in 82% yield (Scheme 2). Cleavage of the ester generated acid 12. We found it convenient to house the eventual N-terminii of our constructs in terminal azido linkages. Azides hold up well under the initial 1+1 acid+isonitrile coupling conditions, including the thermolytic O→N acyl migration step and, thereafter, in the 2+1 coupling step, which sets the stage for building the constrained tripeptide motifs. As will be seen (vide infra), the reductive cleavage of the azido group serves as an effective means of exposing the N-terminus (NH2) of the abbreviated constrained tripeptide to be elongated. In Table 1, we report three additional examples of this type of sequence.
Scheme 2.

Synthesis of azido precursors of dipeptide acids.a
aKey: (a) microwave, 150 °C, CHCl3, 82%; (b) HCO2H, 100%.
Table 1.
The reaction of azido acids with isonitriles.
 | ||||
|---|---|---|---|---|
| Entry | Amino acid | Isonitrile | Product | Yield |
| 1 |
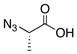
|
|
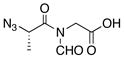 13 |
85% |
| 2 |
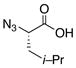
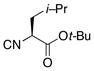
|
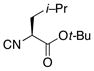
|
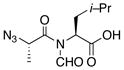 14 |
80% |
| 3 |
|
|
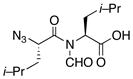 15 |
87% |
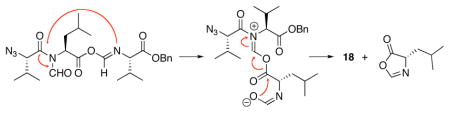
The next phase of the plan involved building tripeptidyl constructs containing contiguous N-formyl groups. The key reaction for accomplishing this end is illustrated in the 2+1 coupling of acid 12 with isonitrile ester 16 (Scheme 3). This reaction was conducted under our conditions of hindered thiophenol mediation.9g In this case, a 50% yield of the target construct 17 was obtained. Curiously, this reaction also gave rise to a 5% yield of dipeptide 18. An assortment of related mechanisms to rationalize formation of this “deletion” product can be proposed, but the actual pathway has not been pinned down.10 In this connection, we note that when the reaction of 12 and 16 was conducted in the absence of hindered thiophenol (requiring thermolysis at 150 °C, rather than 120 °C) the ratio of the unexpected 18:17 was as high as 5:1.
Scheme 3.

ArSH-mediated synthesis of bis-N-formyl tripeptides.a
aKey: (a) microwave, 120 °C, CHCl3, 2,6-dimethyl thiophenol.
The N-formyl group within 12 plays a key role in the critical second coupling reaction leading to diformyltripeptide (17), in that it stabilizes the intermediate FCMA arising from the reaction of 12+16 against oxazolone formation, thereby allowing the required O→N acyl tranfer to occur. In Table 2, we summarize five additional examples of this chemistry.11
Table 2.
Synthesis of tripeptides.a
 | ||||
|---|---|---|---|---|
| Entry | Dipeptide | Isonitrile | Product | Yield |
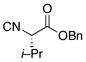
| 1 | 13 |
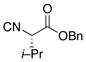 16 |
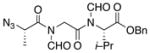 19 |
65% |
| 2 | 14 |
 16 |
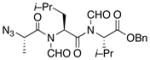 20 |
55% |
| 3 | 15 |
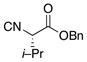 16 |
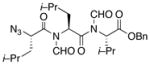 21 |
55% |
| 4 | 12 |
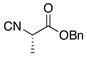 22 |
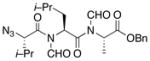 23 |
52% |
| 5 | 15 |
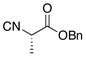 22 |
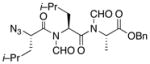 24 |
53% |
Key: (a) microwave, 120 °C, CHCl3, 2,6-dimethyl thiophenol.
With the bis-N-formyl tripeptides in hand, we turned to the synthesis of the constrained systems. Initial studies centered on reduction of the two N-formyl functions of the mixed imides, hoping to generate the corresponding diol derivatives (cf 26). These efforts are somewhat complicated by the sensitivity of the imide-like N-formyl group to bases and nucleophiles. Fortunately, after considerable experimentation, it was found that treatment of bis-N-formyltripeptides 17, 21, 23, and 24 with lithium borohydride in a mixed solvent system (CH2Cl2:n-PrOH = 20:1) at −60 °C in the presence of acetic anhydride furnished the desired methylols (Table 3).9l The presence of acetic anhydride apparently serves to suppress the formation of side products arising from deformylation, possibly by quenching the n-PrOLi generated during the course of the reaction.12 Moreover, careful tuning of workup conditions was crucial to ensure liberation of the diols from their presumed boronate esters (see Supporting Information).13 Happily, treatment of the crude diols with trifluoroacetic acid (TFA) in CH2Cl2 furnished the corresponding constrained tripeptides (28–31) in unoptimized but still manageable overall yields (Table 3).
Table 3.
Synthesis of constrained tripeptides.a
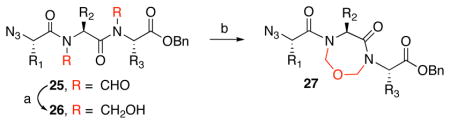 | |||
|---|---|---|---|
| Entry | Bis N–formyl tripeptide | Constrained peptide | Yield |
| 1 |
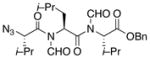 17 |
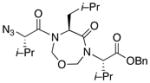 28 |
60% |
| 2 |
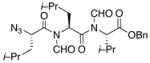 21 |
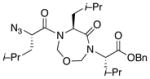 29 |
55% |
| 3 |
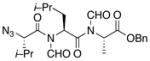 23 |
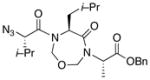 30 |
53% |
| 4 |
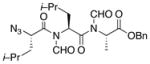 24 |
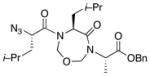 31 |
45% |
Key: (a) LiBH4, CH2Cl2, n-PrOH, Ac2O, −60 °C. (b) TFA, CH2Cl2.
Armed with a reliable method for obtaining these novel constrained tripeptides, we turned to the task of synthesizing larger polypeptides with site-specific constraining motifs. In this way, we hoped to probe the effect of the constraint on peptide conformation and on resulting properties (Scheme 4). In a typical example, reductive cleavage of tripeptide 28 with triphenylphosphine (Ph3P) followed by coupling with Boc–Lys–Gly–COOH employing HATU and DIPEA as condensing agents, provided the desired pentapeptide 32 in 75% yield, over the two steps. Hydrogenolytic cleavage of the benzyl ester of 32 followed by coupling of the resultant C-terminal acid with dipeptide H2N–Gly–Ala–OBn afforded heptapeptide 33 in 80% yield (over two steps). Finally, global deprotection of 33 furnished the desired heptapepeptide 34 in 80% yield over two steps. Following this general protocol, the same precursor 28 was converted to constrained system 35.
Scheme 4.

Synthesis of large constrained peptides.a
aKey: (a) PPh3, THF, H2O. (b) Boc–Lys–Gly–CO2H, HATU, DIPEA, DMF. (c) Pd/C, MeOH, H2. (d) HCl·H2N–Gly–Ala–OBn, HATU, DIPEA, DMF. (e) 10% Pd/C, MeOH, H2. (f) TFA, CH2Cl2.
In line with our original plan (vide supra), we hoped to remove the methylene bridging constraint to generate the native peptide. Indeed, treatment of 34 with 0.1 M HCl and 1,3-propanedithiol in trifluoroethanol at RT furnished the corresponding water-soluble linear heptapeptide 36 in 95% yield (Scheme 5).14 When 35 was subjected to the same conditions, the reaction proceeded to ca. 90% conversion (by LCMS analysis), but most intriguingly, no product could be isolated due to the formation of an intractable precipitate, presumably due to aggregation of linearized 37.15 Efforts to solubilize the residue in H2O, DMSO, DMF, CH3CN, MeOH, and CH2Cl2 proved fruitless. The apparent insolubility of 37 in aqueous solution is especially striking considering that its constrained counterpart 35 is perfectly soluble in H2O at concentrations of 10 mg/mL, at least. This dramatic contrast suggests that the concept of using constraints to enforce intramolecular proximities, thereby attenuating proclivities to aggregation, may well have merit. As noted above, such a capability could also be of advantage in synthesis. A particularly important application would be that of stabilizing aggregation-prone sequences of β-amyloids.16
Scheme 5.

Deprotection of constrained peptides 34 and 35.a
aKey: (a) 0.1M HCl, CF3CH2OH, HSCH2CH2CH2SH.
Extensive NMR studies provided more searching insights as to the conformational consequences of our constrained 7-mers 34 and 35. Systematic inspection of the NOESY and ROESY data for each constrained peptide in pure D2O and 9:1 H2O/D2O at 4 °C did not reveal suggestive through-space interactions between any of the backbone αH or amide NH protons. However, in both cases, an NOE correlation was observed between one of the α-protons of Gly-2 and a sidechain γ-methyl of Val-5 (shown for 34 in Figure 1a). In contrast, the de-constrained linear system 36 did not exhibit any discernable NOEs between protons of noncontiguous residues, as would be expected (Figure 1b). Taken together, these data signify that the CH2–O–CH2 constraint induces a conformational bias wherein the peptide backbone is somehow folded such that residues 2 and 5 are brought into proximity. It could well be that this effect is responsible for the abatement of aggregation in 35. The constraint may promote solubility by providing a scaffold that juxtaposes the hydrophobic surfaces of the molecule, enabling them to achieve energetically favorable non-covalent interactions at the intramolecular level.17 Linear peptide 37, lacking the constraint, achieves its noncovalent interactions by intermolecular means—namely, aggregation.
Figure 1.

Selected 600 MHz NOESY data for (a) constrained peptide 34 and (b) linear peptide 36. Data obtained for 4.7 mM peptide samples at 4 °C in D2O, pH 3.6–3.8 (not corrected for deuterium) with a 250 ms mixing time.
In summary, we have developed a method that is enabled by our recently developed isonitrile coupling technology for assembling conformationally biased polypeptides. Consecutive 2-component coupling reactions of isonitriles exploiting a strategic N-terminal azide linkage in the first union afforded a novel tripeptide containing contiguous N-formyl amidic linkages. These N-formyl groups were used to establish a tripeptide bearing a CH2–O–CH2 bridge spanning contiguous amides. This system was further elaborated to construct 7-mer peptides containing this bridge, which can then be dismantled under mild treatment. Studies of the constrained and linear systems, so produced, have underscored the possibility of modulating protein conformations that influence proclivities to aggregation. The broader implications of these findings are the subject of considerable current interest in our laboratory.
Supplementary Material
Acknowledgments
This work was supported by the NIH (CA28824 to S.J.D.). P.K.P. is supported by a postdoctoral fellowship (PF-11-014-01-CDD) from the American Cancer Society. The authors thank Dr. George Sukenick, Ms. Hui Fang, and Ms. Sylvi Rusli of the NMR Core Facility at SKI for MS and NMR assistance. Dr. John Decatur of the NMR facility at Columbia University is gratefully acknowledged for NMR assistance. We also thank Professors Ann McDermott, Tom Muir, and Pavel Nagorny for advice and helpful discussions. We thank Ms. Rebecca Wilson for valuable advice and suggestions.
Footnotes
Supporting Information Available: Experimental procedures and analytical data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Marx V. Chem Eng News. 2005;83(11):17–24. [Google Scholar]; (b) Nestor JJ., Jr Curr Med Chem. 2009;16:4399–4418. doi: 10.2174/092986709789712907. [DOI] [PubMed] [Google Scholar]
- 2.Gentilucci L, De Marco R, Cerisoli L. Curr Pharm Des. 2010;16:3185–3203. doi: 10.2174/138161210793292555. [DOI] [PubMed] [Google Scholar]
- 3.Hruby VJ. Nat Rev Drug Discov. 2002;1:847–858. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 4.Sia SK, Carr PA, Cochran AG, Malashkevich VN, Kim PS. Proc Natl Acad Sci USA. 2002;99:14664–14669. doi: 10.1073/pnas.232566599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Miller SJ, Grubbs RH. J Am Chem Soc. 1995;117:5855–5856. [Google Scholar]; (b) Miller SJ, Blackwell HE, Grubbs RH. J Am Chem Soc. 1996;118:9606–9614. [Google Scholar]
- 6.The incorporation of “reversible” perturbations to the peptide backbone in the form of pseudo-Pro has proved to be a valuable strategy for the synthesis of “difficult” sequences: Haack T, Mutter M. Tetrahedron Lett. 1992;33:1589–1592.Wöhr T, Mutter M. Tetrahedron Lett. 1995;36:3847–3848.Mutter M, Nefzi A, Sato T, Sun X, Wahl F, Wöhr T. Pept Res. 1995;8:145–153.Wöhr T, Wahl F, Nefzi A, Rohwedder B, Sato T, Sun X, Mutter M. J Am Chem Soc. 1996;118:9218–9227.
- 7.Protection of the peptide backbone with the N-(2-hydroxy-4-methoxybenzyl) (Hmb) group has also been shown to aid the synthesis of aggregation-prone peptide sequences: Johnson T, Quibell M, Owen D, Sheppard RC. J Chem Soc, Chem Commun. 1993:369–372.Hyde C, Johnson T, Owen D, Quibell M, Sheppard RC. Int J Peptide Prot Res. 1994;43:431–440.Quibell M, Turnell WG, Johnson T. J Org Chem. 1994;59:1745–1750.Quibell M, Packman LC, Johnson T. J Am Chem Soc. 1995;117:11656–11668.
- 8.Another widely used approach to the problem of “difficult” sequences involves the synthesis of the corresponding O-acyl isopeptides: Sohma Y, Sasaki M, Hayashi Y, Kimura T, Kiso Y. Chem Commun. 2004:124–125. doi: 10.1039/b312129a.Mutter M, Chandravarkar A, Boyat C, Lopez J, Dos Santos S, Mandal B, Mimna R, Murat K, Patiny L, Saucède L, Tuchscherer G. Angew Chem Int Ed. 2004;43:4172–4178. doi: 10.1002/anie.200454045.Carpino LA, Krause E, Sferdean CD, Schümann M, Fabian H, Bienert M, Beyermann M. Tetrahedron Lett. 2004;45:7519–7523.Sohma Y, Hayashi Y, Kimura M, Chiyomori Y, Taniguchi A, Sasaki M, Kimura T, Kiso Y. J Peptide Sci. 2005;11:441–451. doi: 10.1002/psc.649.Dos Santos S, Chandravarkar A, Mandal B, Mimna R, Murat K, Saucède L, Tella P, Tuchscherer G, Mutter M. J Am Chem Soc. 2005;127:11888–11889. doi: 10.1021/ja052083v.Taniguchi A, Sohma Y, Kimura M, Okada T, Ikeda K, Hayashi Y, Kimura T, Hirota S, Matsuzaki K, Kiso Y. J Am Chem Soc. 2006;128:696–697. doi: 10.1021/ja057100v.Coin I, Dölling R, Krause E, Bienert M, Beyermann M, Sferdean CD, Carpino LA. J Org Chem. 2006;71:6171–6177. doi: 10.1021/jo060914p.
- 9.(a) Li X, Danishefsky SJ. J Am Chem Soc. 2008;130:5446–5448. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jones GO, Li X, Hayden AE, Houk KN, Danishefsky SJ. Org Lett. 2008;10:4093–4096. doi: 10.1021/ol8016287. [DOI] [PubMed] [Google Scholar]; (c) Li X, Yuan Y, Berkowitz WF, Todaro LJ, Danishefsky SJ. J Am Chem Soc. 2008;130:13222–13224. doi: 10.1021/ja8047078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li X, Yuan Y, Kan C, Danishefsky SJ. J Am Chem Soc. 2008;130:13225–13227. doi: 10.1021/ja804709s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li X, Danishefsky SJ. Nat Protoc. 2008;3:1666–1670. doi: 10.1038/nprot.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhu J, Wu X, Danishefsky S. Tetrahedron Lett. 2009;50:577–579. doi: 10.1016/j.tetlet.2008.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Wu X, Li X, Danishefsky SJ. Tetrahedron Lett. 2009;50:1523–1525. doi: 10.1016/j.tetlet.2009.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yuan Y, Zhu J, Li X, Wu X, Danishefsky S. Tetrahedron Lett. 2009;50:2329–2333. doi: 10.1016/j.tetlet.2009.02.205. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Wu X, Yuan Y, Li X, Danishefsky S. Tetrahedron Lett. 2009;50:4666–4669. doi: 10.1016/j.tetlet.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Stockdill J, Wu X, Danishefsky S. Tetrahedron Lett. 2009;50:5152–5155. doi: 10.1016/j.tetlet.2009.06.129. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Rao Y, Li X, Danishefsky S. J Am Chem Soc. 2009;131:12924–12926. doi: 10.1021/ja906005j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Wu X, Stockdill J, Wang P, Danishefsky S. J Am Chem Soc. 2010;132:4098–4100. doi: 10.1021/ja100517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
10.The structure of 18 was confirmed by 1H NMR and MS evidence. A plausible mechanism for its formation might involve acyl transfer from the N-formyl nitrogen to the formimidate nitrogen, with expulsion of the formyl-Leu residue in the form of an oxazolone:
- 11.Although the 2+1 coupling afforded synthetically useful yields of bis-N-formyl tripeptides containing bulky amino acids such as Leu and Val, we noted a precipitous decline in efficiency (yields < 5%) with couplings involving isoleucine-containing substrates.
- 12.Thomas EW, Rynbrandt RH, Zimmermann DC, Bell LT, Muchmore CR, Yankee EW. J Org Chem. 1989;54:4535–4543. [Google Scholar]
- 13.When tripeptides containing glycine at any position were subjected to reduction conditions, decomposition into multiple species was observed. Analysis of the mixture by MS indicated the presence of side products derived from deformylation as well as cleavage of the peptide chain, among others. Expanding the scope of this sequence is an area of active investigation in our laboratory.
- 14.Corey EJ, Reichard GA. Tetrahedron Lett. 1993;34:6973–6976. [Google Scholar]
- 15.Since the identity of the deprotected material could not be confirmed by further structural characterization, we synthesized 39 independently by iterative solution phase couplings. Although the linear heptapeptide remained in solution during HPLC purification, the lyophilized material was similarly insoluble in aqueous and organic solvent.
- 16.Tickler AK, Clippingdale AB, Wade JD. Protein Pept Lett. 2004;11:377–384. doi: 10.2174/0929866043406986. [DOI] [PubMed] [Google Scholar]
- 17.A conceptually related approach that is thought to disrupt intermolecular forces by promoting “internal solubilization” using polyethylene glycol–based protecting groups has been articulated: Mutter M, Mutter H, Uhmann R, Bayer E. Biopolymers. 1976;15:917–927. doi: 10.1002/bip.1976.360150508.Zier A, Ryan D, Mutter M. Tetrahedron Lett. 1994;35:1039–1042.Zinieris N, Zikos C, Ferderigos N. Tetrahedron Lett. 2006;47:6861–6864.Kocsis L, Bruckdorfer T, Orosz G. Tetrahedron Lett. 2008;49:7015–7017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.