

Abstract
Despite our rapidly growing knowledge about the human genome, we do not know all of the genes required for some of the most basic functions of life. To start to fill this gap we developed a high-throughput phenotypic screening platform combining potent gene silencing by RNA interference, time-lapse microscopy and computational image processing. We carried out a genome-wide phenotypic profiling of each of the ~21,000 human protein-coding genes by two-day live imaging of fluorescently labelled chromosomes. Phenotypes were scored quantitatively by computational image processing, which allowed us to identify hundreds of human genes involved in diverse biological functions including cell division, migration and survival. As part of the Mitocheck consortium, this study provides an in-depth analysis of cell division phenotypes and makes the entire high-content data set available as a resource to the community.
To target the ~21,000 protein-coding genes in the human genome, we used a chemically synthesized short interfering RNA (siRNA) library designed to uniquely target each gene with 2–3 independent sequences (Supplementary Methods). The siRNAs in this library were tested individually and reduced the messenger RNAs of targeted genes to below 30% of original levels (to an average of 13%) for 97% of more than 1,000 genes tested (Supplementary Table 1). To allow high-throughput phenotyping of each individual siRNA in triplicates by live-cell imaging, we used a previously established workflow for solid-phase transfection using siRNA microarrays coupled to automatic time-lapse microscopy1. As a high-content phenotypic assay we chose to monitor fluorescent chromosomes in a human cell line stably expressing core histone 2B tagged with green fluorescent protein (GFP)1. After seeding on the siRNA microarrays, on average 67 (±30) cells for each siRNA of the library were imaged in triplicates for 2 days, thus documenting many of their basic functions such as cell division, proliferation, survival and migration.
Image processing reveals mitotic hits
This resulted in a large data set of ~190,000 time-lapse movies providing time-resolved records of over 19 million cell divisions. To automatically score and annotate phenotypes in this large data set, we developed a computational pipeline2 (Fig. 1) extending previously established methods of morphology recognition by supervised machine learning3–6. In brief, after segmentation, about 200 quantitative features were extracted from each nucleus and used for classification into one of 16 morphological classes (Fig. 1 and Supplementary Movies 1–30) by a support vector machine classifier previously trained on a set of ~3,000 manually annotated nuclei (Supplementary Methods). This classifier automatically recognizes changes in nuclear morphology due to the cell cycle, cell death or other phenotypic changes with an overall accuracy of 87% (Supplementary Fig. 1) and allows us to convert each time-lapse movie into a phenotypic profile that quantifies the response to each siRNA (Fig. 1a). In addition, the position of each nucleus is tracked over time. Using stringent significance thresholds for each morphological class, nuclear mobility as well as proliferation rate, significant and reproducible (majority of three or more technical replicates) deviations caused by each siRNA are computed (Fig. 1 and Supplementary Methods).
Figure 1. Data analysis and hit detection.

a, All nuclei in the 187,226 movies (each consisting in 92 images) are classified into 1 out of 16 predefined morphological classes. The workflow is illustrated for a RAD23A RNAi experiment; for clarity, only four morphological classes are shown: mitotic delay/arrest (prometaphase plus metaphase alignment problems (MAP)), polylobed, grape and cell death. For each morphological class, the score is defined as the maximal difference over time between the relative cell count curve in one morphological class and the corresponding negative control curve, averaged over eight scrambled siRNA experiments on the same slide (shown for mitosis). b, 1,918,544,775 nuclei from all movies (controls removed) classified into 16 different nuclei morphology classes. Classes used for mitotic hit detection are underlined. c, Genome-wide score distribution for the four classes used to detect potential mitotic hits—mitotic delay/arrest (prometaphase plus MAP), binuclear, polylobed and grape—automatically computed for all 51,810 siRNAs. Each siRNA is considered as a potential mitotic hit if the median score of its replicates exceeds a manually defined threshold (dotted lines) in at least one of the four morphological classes.
The key biological function that motivated this screen was mitosis, studied systematically within the Mitocheck consortium. Cell division phenotypes are rare and transient in human cell culture and are therefore typically missed in endpoint assays; however, they can be particularly well detected by time-lapse microscopy1,7. In addition, live imaging data reveal the primary defect and secondary consequences of the phenotype and thereby allow a more precise interpretation of the function of already identified genes. Despite genome-wide screening in a number of model organisms7–9, candidate genes for key mitotic processes such as the restructuring or segregation of mitotic chromosomes remain to be discovered. To score an initial set of potential mitotic genes identified reproducibly with at least one siRNA, 5 of our 16 morphological classes describing chromosome configurations were used (see Fig. 1 and Supplementary Methods). These classes included early mitotic chromosome configurations such as ‘prometaphase’ and ‘metaphase alignment problems’ (MAP) that will be enriched by delays or arrests in mitosis, and we therefore combined these classes to score ‘mitotic arrest/delay’ phenotypes (we did not find significant deviations in normal ‘metaphase’ or ‘anaphase’ classes and therefore did not use these for scoring mitotic hits) (Fig. 1b). Also included were morphological classes such as ‘polylobed’, exhibiting multilobed nuclei, ‘grape’, exhibiting many micronuclei, as well as ‘binuclear’, representing cells with two nuclei (Fig. 1b). These three classes specifically arise as a consequence of distinct problems during mitotic exit including premature nuclear assembly, chromosome segregation errors or cytokinesis failures. A total of 1,042 genes deviated significantly from controls in one or more of these four phenotypic groups (Fig. 1c). In addition, 207 genes below the stringent significance thresholds of automatic scoring were identified by manual annotation of the movies during training, quality control and threshold evaluation (see Supplementary Methods). The combined 1,249 genes (Supplementary Table 2) are thus the potential mitotic hits from this first pass genome-wide screen (Fig. 1c).
Validation of mitotic hits
Comparison of our potential hits with previously published RNA interference (RNAi) screens that scored cell division is not suitable for validation of our hit list, because the overlap between such screens tends to be relatively low (in our case ranging between 6–36%) due to poor comparability of the different screens (Supplementary Table 3). To minimize the risk of reporting false positives, we therefore carried out a second pass validation screen against 90% (1,128) of these genes with two additional independent siRNAs. Combined with the results from the first pass genome-wide screen, 46% (572 out of 1,249) of the potential hits showed consistent phenotypes with two or more siRNAs (Supplementary Table 4). This set of validated genes contained 61% (41 out of 67) of a manually curated human gene set for which a requirement for mitosis had already been established in low-throughput RNAi experiments in HeLa cells with comparably specific mitotic assays (Supplementary Table 5). In addition, we also carried out phenotypic complementation experiments for a subset of the potential mitotic hits. To this end, the genomic copy of the mouse orthologue was tagged with a combined localization and affinity tag at the last exon in a bacterial artificial chromosome10,11, and stably expressed under its endogenous promoter in the HeLa cell strain used for the screen. Because of the DNA sequence divergence between mouse and human, 89% of mouse genes are not targeted by our siRNAs against human genes. We created 21 cell lines with such RNAi-resistant BAC transgenes. In 12 (57%) of these lines the phenotype was fully complemented (Fig. 2 and Supplementary Table 6). These rescues were specific as the mouse transgenes did not suppress the knockdown of the endogenous human gene nor the phenotype of siRNAs targeting other genes (Supplementary Fig. 2). Suppression of the target genes was thus responsible for the phenotype. Phenotypes were partially complemented in three (14%) additional cell lines and not complemented in six (29%) lines (data not shown), indicating correct specificity of the siRNAs targeting 15 out of 21 (71%) genes. Unsuccessful complementation could be due to off-target effects of the siRNA, but can also be caused by incorrect expression regulation or malfunction of the mouse gene in human cells (for example, the previously validated mitotic gene NUSAP112 could not be complemented). Most of the hits that we complemented had scored with two or more siRNAs (11 out of 14); however, some hits that scored with only one siRNA could also be rescued (4 out of 7).
Figure 2. Rescue experiments.
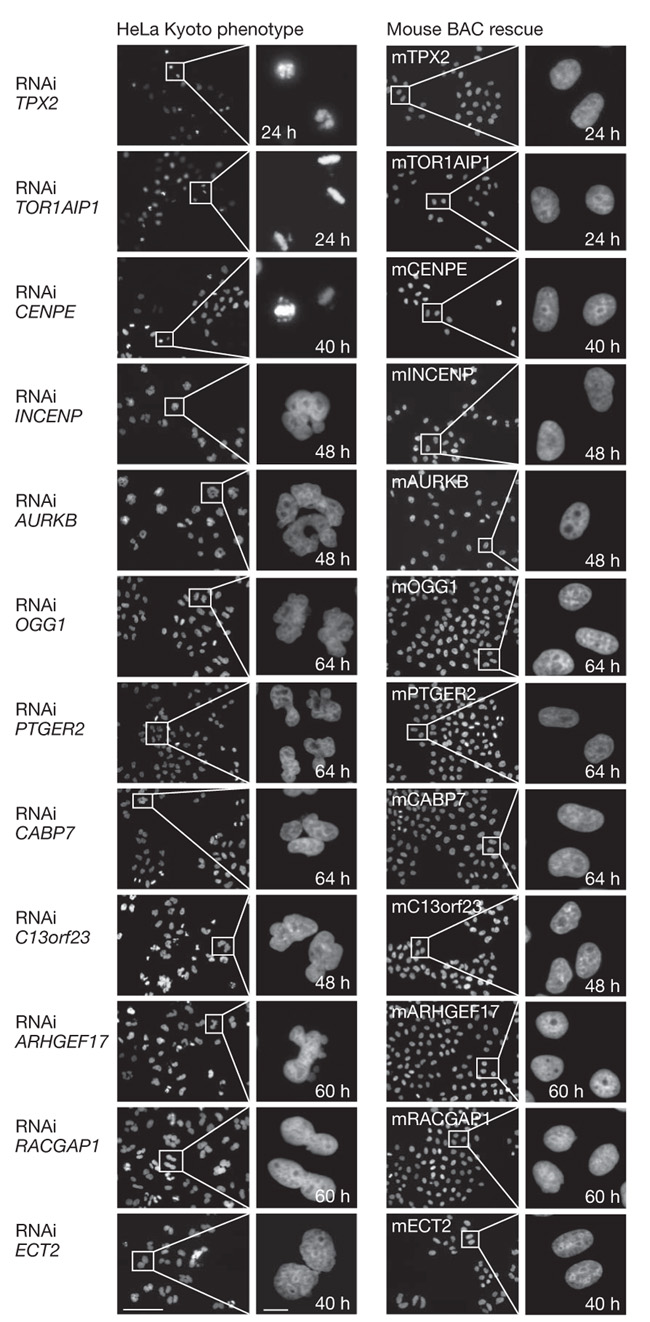
a, HeLa cells and HeLa cells stably expressing 12 different mouse BACs are stained with Hoechst after RNAi knockdowns. The specific mouse proteins could rescue the RNAi nuclear morphology phenotypes (left panel) in all rescues shown (right panel). Scale bars represent 100 μm (left) and 10 μm (right). Out of the 21 rescues performed, 12 (57%) were successful, 3 (14%) partial and 6 (29%) failed (Supplementary Table 6).
We focus this study on the detailed analysis of the 572 validated mitotic genes found with two or more siRNAs in the first pass genome-wide and validation screen. We note that another 677 genes scored with only one siRNA. Although a fraction of these is expected to contain true positives (see complementation results above), our current data set cannot provide further validation on these genes. Because we expect future experiments to provide further validation, we have made the results on genes identified with only one siRNA available online at http://www.mitocheck.org. Ultimately, only development of high-throughput phenotypic complementation assays in mammalian cells will allow validation without any doubt of all hits from RNAi screens.
Analysis of temporal order of mitotic phenotypes
Bioinformatic analysis of Gene Ontology annotations showed that less than one-half of the 572 validated mitotic genes we identified had previously been annotated with cellular processes consistent with a function in mitosis, whereas for the majority of the hit genes our data provide a novel functional link to cell division (Fig. 3a). To obtain a global picture of the different types of mitotic phenotypes caused by all mitotic genes, we analysed the temporal patterns of phenotypic deviations that occurred in the depleted cell populations by computing the relative order of phenotypic events for each gene (Supplementary Methods). This analysis allowed us to centre complex phenotypes in time on particularly interesting events such as mitotic delays or binucleation in event order maps (Fig. 3b and Supplementary Fig. 5). Manual inspection of the corresponding movies showed that event order maps mostly link phenotypes that occur consecutively in the same cell and thus allow interpretations about the causality of different events. The event order map of mitotic delays revealed that they typically occur as the first phenotypic deviation and are transient and therefore result in secondary and tertiary phenotypes (Fig. 3b). Interestingly, similar mitotic delays differed markedly in their consequences (Fig. 3b). The most common consequence of a mitotic delay was cell death either in direct succession to mitosis (MFSD3) or via an intermediate aberrant chromosome segregation phenotype. The second most common consequence of a mitotic delay was polylobed or grape-shaped nuclei indicative of mitotic exit with aberrant chromosome segregation (HAUS3) that did not cause cell death. Only few mitotic delays occurred as the only detectable phenotype, indicating that most early mitotic perturbations trigger long-lasting and frequently detrimental cellular responses. The event order map for binuclear cells indicative of cytokinesis defects was markedly different (Fig. 3b). Most frequently, binucleation was the only detected phenotype, indicating that in contrast to problems in spindle assembly or chromosome segregation, a failure of cell body separation is most of the time well tolerated by the cell and most of the genes in this pathway have no additional essential function in the cell division process (Fig. 3b). If binucleation was coupled with other phenotypes, it was often the first phenotypic event followed by polylobed phenotypes as a secondary consequence (RGMA). Only a very small group of genes showing a binucleation phenotype was essential for survival (RAB24). Event order maps of polylobed or grape-shaped nuclei revealed that these morphologies indicative of chromosome segregation problems occur mostly or, in case of grape-shaped nuclei, almost exclusively as a consequence of earlier problems in mitosis (Fig. 3b).
Figure 3. Gene Ontology (GO) terms distribution and event order maps.

a, GO analysis. Biological process annotations of the 572 validated mitotic hits. To deal with multiple annotations for each gene, categories were arbitrarily ordered by relevance to mitosis and each gene was assigned to the first term from this list that it had been annotated with. The same analysis was performed for the whole set of potential hit genes (1,249) in Supplementary Fig. 3. b, Event order maps. To each gene of the validated mitotic hit list (found by at least two siRNAs), a representative order of phenotypic events and a normalized penetrance score in each of the morphological classes is assigned (Supplementary Methods). For each gene, the event order can be visualized by a sequence of coloured fields where different colours correspond to different phenotypic classes, the colour intensity to the corresponding penetrance and the colour order to the phenotypic event order. The event orders are then centred on the phenotypic classes mitotic delay, polylobed, binuclear and grape. Genes with similar event orders are grouped together, resulting in centred event order maps, where the rows correspond to genes and the columns to the event order relative to the main phenotypic event. On the left, the locations of four genes (MFSD3, HAUS3, RMA, RAB24) in the maps are indicated by grey arrows; the corresponding order of phenotypic events are illustrated on a single cell level (numbers indicate time after transfection (h:min)). The corresponding event order maps for the whole set of potential hit genes (1,249) are shown in Supplementary Fig. 4. Single gene resolution event order maps are shown in Supplementary Fig. 5.
Clustering of mitotic genes by phenotype kinetics
Event order maps provide an excellent overview of the global patterns of temporal coupling between different mitotic perturbations. Our next goal was to use the full information of our phenotypic profiles to order genes by phenotypic similarity. We therefore performed hierarchical clustering of genes by their phenoprints in all morphological classes relevant for mitosis, taking both the temporal change as well as the severity of the phenotype into account. This resulted in a phenoprint heat map for all mitotic hits (Fig. 4a and Supplementary Fig. 7). Globally, there are two main first-order clusters, one for early mitotic defects and their consequences and one representing problems in cytokinesis. The first can roughly be subdivided into three second-order clusters, one with strong mitotic delays, usually coupled with cell death, polylobed and/or grape-shaped nuclei (Fig. 4a, mitosis 1), and two with mild mitotic delays with subtle consequences (Fig. 4a, mitosis 2) or followed by formation of polylobed nuclei (Fig. 4a, mitosis 3). Within the mitotic clusters, we identified two relatively small third-order clusters where large or dynamic nuclei occur in combination with mild mitotic arrest and polylobed nuclei. Manual inspection of the corresponding movies indicated that the non-mitotic classes ‘large’ and ‘dynamic’ occur mostly in distinct sets of cells in the knocked-down population, indicating multiple gene functions during the cell cycle. Cytokinesis defects are rarely preceded by mitotic delays or arrests, but they can lead to severe segregation defects if the cells undergo mitosis again.
Figure 4. Time-resolved heat map.

a, The quantitative time-resolved phenoprints for all validated mitotic hit genes (572) can be used for phenotypic clustering, taking into account both the penetrance values and the joint temporal evolution in several phenotypic classes illustrated at the top of each column. Colour code at the right: reference genes (Supplementary Table 5) with known function are marked in blue (cytokinesis) or green (early mitotic phenotype). On the right, interesting clusters are highlighted. On the left, the dendrogram corresponding to the hierarchical clustering is shown. The same analysis has been performed for the whole set of potential mitotic genes (1,249) in Supplementary Fig. 6. The single gene resolution heat map is available as Supplementary Fig. 7. b, Early mitotic phenotypes (magnified view of the red rectangle at the top of panel a); the rescued gene TOR1AIP1 is highlighted in red. c, Binuclear phenotypes (magnified view corresponding to the red rectangle at the bottom of panel a); the rescued gene CABP7 is highlighted in red. In b and c, the numbers in parentheses represent the identifiers of the siRNAs that produced the phenotypic profile illustrated in the heat map.
We hypothesized that phenotypic similarities should allow us to make predictions about the mitotic functions of new genes. To test this, we used genes for which a mitotic loss of function phenotype in human cells has been reported previously, including well known genes with functions in spindle assembly and cytokinesis (Fig. 4a and Supplementary Table 5). For example, one of the two large first-order phenoprint clusters consisted of genes that exhibited early mitotic delay followed by polylobed nuclei and finally cell death, a phenotypic signature consistent with a severe defect in spindle assembly and consequently chromosome segregation (Fig. 4b). Indeed, this cluster contained several well known mitotic spindle genes such as PLK1 (ref. 13), TPX2 (ref. 14), CKAP5 (also called ch-TOG)15 and KIF11 (ref. 16). In addition, this cluster contained several completely uncharacterized genes including LSM14A (the orthologue of which has a similar phenotype in Caenorhabditis elegans) but also well characterized genes believed to have non-mitotic functions such as the inner nuclear membrane protein TOR1AIP1 (ref. 17). A second phenotypic cluster is formed by genes exhibiting binucleated cells, sometimes followed by polylobed nuclei, indicative of a severe cytokinesis defect which allows the cells to continue nuclear divisions (Fig. 4a). This cluster contained many genes whose loss was previously demonstrated to affect cytokinesis, such as RACGAP1 (ref. 18), ANLN (ref. 18), ECT2 (ref. 18), PRC1 (ref. 18) and KIF23 (ref. 18) (Supplementary Fig. 7). Many uncharacterized genes, such as C14orf54, and genes believed to have non-mitotic functions, such as the calcium binding protein CABP7 (Fig. 4c), had a similar, albeit weaker, phenotypic profile indicative of a function in the same process.
Functional analysis of predicted mitotic spindle phenotypes
To validate the predictions that CABP7 is required for cytokinesis and that TOR1AIP1 is required for spindle assembly, we decided to carry out a functional analysis of these genes. Both genes represent true positives, as their RNAi phenotypes could be complemented by RNAi-resistant transgenes (Fig. 2). To test directly the phenotypic clustering predictions that the binucleation phenotype of CABP7 arose from a cytokinesis defect and that the chromosome alignment phenotype of TOR1AIP1 arose from spindle assembly problems, we performed high-resolution four-dimensional confocal microscopy with microtubule and chromosome markers. Indeed, CABP7 suppression caused cytokinesis failure after normal chromosome segregation, resulting in a single cell containing four centrosomes and two nuclei (Fig. 5). Knockdown of TOR1AIP1 caused spindle formation to fail in prometaphase, because centrosomes could form only weak mitotic asters and failed to establish a bipolar spindle or align chromosomes, leading to aberrant mitotic exit and cell death (Fig. 5). Thus, CABP7 and TOR1AIP1 are bona fide cytokinesis and spindle assembly genes, respectively, revealing a novel connection between calcium binding and cytokinesis on the one hand and nuclear membrane proteins and the assembly of the mitotic microtubule spindle on the other hand.
Figure 5. Functional analysis of spindle phenotypes.

Left panel: phenotypic time curves (colour scheme as in Fig. 1) for nine corresponding RNAi experiments from the primary screen and one negative control experiment (scrambled; top panel). Right panels: confocal still images from movies of HeLa cells stably expressing GFP–tubulin (green) and H2B–mCherry (red) after RNAi knockdown show mitotic phenotypes in the first (b, d, g) cell cycle or mitotic consequences in the following one (a, c, e, f, h–j). First phenotype appearance is indicted by arrows. Mitotic phenotypes are shown in b–f, whereas in the CENPE knockdown (e) the consequence of the non-aligned chromosomes is indicated with an arrowhead. g, Binuclear cells after cytokinesis failure. h–j, Cytokinesis failure followed by the formation of a multipolar spindle during the second cell cycle resulting in multinucleated cells (arrows indicate the centrosomes). Scale bar, 10 μm.
Secondary four-dimensional confocal imaging assays are currently not high-throughput methods. We therefore focused our four-dimensional confocal spindle assembly assays on knockdown experiments of genes with successful rescue experiments (Fig. 5 and Supplementary Movies 31–40). Consistent with the predictions of gene function from the primary screen, prometaphase delays were explained by spindle assembly defects (AURKB, INCENP, TOR1AIP1), whereas binucleated cells resulted from chromosome alignment and/or segregation problems that subsequently prevented cytokinesis because chromatin persisted in the area of the cleavage furrow19,20 (CENPE, OGG1); in other cases chromosome segregation was normal but cytokinesis appeared to be specifically affected and cells contained two nuclei and four centrosomes (PTGER2, ECT2, CABP7, C13orf23). Binucleated cells either remained viable and ceased dividing (PTGER2) or formed multipolar spindles that resulted in aberrant chromosome segregation in the next cycle which, when coupled to another failed cytokinesis, resulted in large polylobed nuclei (ECT2, CABP7, C13orf23). Together this high-resolution assay for spindle formation, chromosome alignment and segregation demonstrates that the phenotypic predictions derived from the automatic mining of the primary genome-wide screen are valid. In addition, the analysis of phenotype development with high temporal resolution in single cells directly shows the causal relationship of different phenotypic classes. Thus, the detailed phenoprints of the primary RNAi screen provide mechanistic hypotheses for the observed phenotypes that can now be pursued in targeted biochemical and cell biological experiments for each gene. As exemplified by our imaging of the spindle microtubules, such future experiments should ideally complement the chromosome visualization assay of the primary screen with information about other key elements of the mitotic machinery, such as centrosomes, spindle microtubules and kinetochores.
Scoring of cell survival and cell migration phenotypes
The power of time-lapse microscopy makes our quantitative phenotypic profiles recorded for siRNAs targeting the whole genome informative about many other cellular functions that cannot be scored in endpoint assays, such as the rate of cell proliferation, cell migration and dynamic changes in nuclear structure. To provide scores for siRNAs belonging to these and additional phenotypic categories, we determined significance thresholds for specific dynamic and morphological changes that report on these functions (Supplementary Figs 8–11 and Supplementary Table 7). Because we have currently not carried out second-pass validation screens for these additional phenotypic categories, typically less than 5% of these genes are currently validated with at least two siRNAs. However, it is reasonable to expect a similar validation rate for these genes as for the mitotic genes. As a starting point for future experiments we have therefore made the results for genes identified reproducibly with at least one siRNA available online (http://www.mitocheck.org) and provide brief examples of these results here (for details of other phenotypic categories see Supplementary Figs 8–10).
Cell death was scored on the basis of significant deviations in chromosome fragmentation morphologies indicative of cell death (Supplementary Methods). This identified 783 potential cell survival genes. Notably, only 22% (124 out of 572) of our validated mitotic hits exhibited cell death phenotypes, showing that cell survival in human cell culture is not a good indicator of mitotic defects. Furthermore, the cell model that we chose, HeLa cells, has little to no p53 DNA damage response and therefore probably represents a sensitized background for certain cell death phenotypes. Nevertheless, HeLa cells have been used successfully to study many basic cellular processes and are probably the most widely used human cell line. Increased mobility was scored by a significant increase in either speed or distance covered by the nucleus (for details see Supplementary Methods). This identified 360 potential cell migration genes. Except for two genes (BARD1 and MYH9), mitosis and cell migration seem to be largely independent phenotypes. Further validation and analysis of the mobility genes should be of interest to the cell migration community in the future.
Discussion
This study provides time-resolved profiles of RNAi-induced loss-of-function phenotypes resulting from siRNAs targeting the entire human genome. Our detailed analysis of mitotic phenotypes and mining for additional basic cell functions, including survival and migration, demonstrate the richness of this large data set, which we expect to be exploited further and to provide the starting point for many focused validation and detailed mechanistic studies. We therefore provide the entire data set as a scientific resource at http://www.mitocheck.org. This database is organized in a gene-centric view using the ENSEMBL human genome database as a reference and provides all the data in an intuitive and easily searchable manner. It also provides the ~190,000 phenotypic movies and the siRNA sequences of all gene silencing experiments. This latter aspect is crucial because gene definitions and transcripts in the human genome are still dynamic and RNAi-based phenotypes quickly become unassignable to genes unless the sequence of the silencing reagent is known and mapped to the latest transcript data. In addition, http://www.mitocheck.org provides the quantitative time-resolved phenoprints that we extracted through our computational pipeline for each of the 2,953 human genes that showed significant deviations with one or more siRNA. Users can easily search this data from any gene entry point and quickly obtain new genes with phenotypes relevant to their biological questions of interest.
The future challenges for functional genomics projects like Mitocheck lie in developing the methods for high-throughput molecular characterization of the proteins encoded by the genes identified in genome phenotyping experiments. The first step in this pipeline, to create RNAi-resistant functionally tagged transgenes, has been developed by the Mitocheck consortium11. It will also be important to develop high-throughput methods for phenotypic complementation, as shown for 21 genes here, which could become a future standard in RNAi screens—currently this is the only experiment to demonstrate without doubt the identity of the gene responsible for the RNAi phenotype. Furthermore, high-throughput methods to use functionally tagged transgenes in human cells to analyse protein–protein interactions, post-translational modifications, their subcellular localization and dynamics are needed to go from phenotypic profiling of the human genome to a systems-level understanding of the functional principles of the encoded protein machinery of human cells.
METHODS SUMMARY.
For the genome-wide RNAi screen, a genome-wide library of siRNAs (Ambion) based on ENSEMBL version 27 was used. Transfected cell microarrays were produced as previously described21 with minor modifications. Live imaging of HeLa cells expressing histone 2B–GFP for 48 h at a time-lapse of 30 min was performed with automated epifluorescence microscopes (IX-81; Olympus Europe) using an in-house modified version of ScanR and an image-based auto-focus routine22. For quality control, the positive and negative controls of each microarray were inspected both manually and automatically using an in-house developed database. Each passed time-lapse image sequence was processed fully automatically via an in-house developed chromosome morphology recognition pipeline, based on Python, the C++ image processing library VIGRA, the classification library libSVM and R for plotting and statistical analysis. The same methodological framework was used for the validation screen. For the mitotic spindle assay, HeLa cells stably expressing GFP–tubulin and histone 2B–mCherry were seeded on siRNA-coated 8-well chambered cover glasses and imaged on a Leica SP5 confocal microscope using the Matrix Screening Application (MSA) co-developed with Leica.
Supplementary Material
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Acknowledgements
We thank J. Gagneur for suggestions on data processing; S. Berthoumieux for assistance in computation; Y. Sun for coordination support in Mitocheck; U. Ringeisen for help in preparing the figures; S. Winkler and L. Burger and EMBL’s electronic and mechanical workshops for support in microscope development; Olympus Soft Imaging Solutions (OSIS) and Olympus Europe for collaboration; Leica Microsystems for collaboration; Applied Biosystems for providing unpublished validation data of the siRNA library; and all our colleagues in the Mitocheck consortium for collaboration. This project was funded by grants to J.E. (within the Mitocheck consortium by the European Commission (LSHG-CT-2004-503464) as well as by the Federal Ministry of Education and Research (BMBF) in the framework of the National Genome Research Network (NGFN) (NGFN-2 SMP-RNAi, FKZ01GR0403)), to R.P. (BMBF NGFN2 SMP-Cell, FKZ01GR0423) as well as to J.E. and R.P. by the Landesstiftung Baden Wuerttemberg in the framework of the research programme ‘RNS/RNAi’. R.D. is supported by the Wellcome Trust.
Footnotes
Author Information Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Neumann B, et al. High-throughput RNAi screening by time-lapse imaging of live human cells. Nature Methods. 2006;3:385–390. doi: 10.1038/nmeth876. [DOI] [PubMed] [Google Scholar]
- 2.Walter T, et al. Automatic identification and clustering of chromosome phenotypes in a genome wide RNAi screen by time-lapse imaging. J. Struct. Biol. doi: 10.1016/j.jsb.2009.10.004. (in the press) [DOI] [PubMed] [Google Scholar]
- 3.Boland MV, Markey MK, Murphy RF. Automated recognition of patterns characteristic of subcellular structures in fluorescence microscopy images. Cytometry. 1998;33:366–375. [PubMed] [Google Scholar]
- 4.Conrad C, et al. Automatic identification of subcellular phenotypes on human cell arrays. Genome Res. 2004;14:1130–1136. doi: 10.1101/gr.2383804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glory E, Murphy RF. Automated subcellular location determination and high-throughput microscopy. Dev. Cell. 2007;12:7–16. doi: 10.1016/j.devcel.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Wang M, et al. Novel cell segmentation and online SVM for cell cycle phase identification in automated microscopy. Bioinformatics. 2008;24:94–101. doi: 10.1093/bioinformatics/btm530. [DOI] [PubMed] [Google Scholar]
- 7.Sönnichsen B, et al. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature. 2005;434:462–469. doi: 10.1038/nature03353. [DOI] [PubMed] [Google Scholar]
- 8.Björklund M, et al. Identification of pathways regulating cell size and cell-cycle progression by RNAi. Nature. 2006;439:1009–1013. doi: 10.1038/nature04469. [DOI] [PubMed] [Google Scholar]
- 9.Kittler R, et al. Genome-scale RNAi profiling of cell division in human tissue culture cells. Nature Cell Biol. 2007;9:1401–1412. doi: 10.1038/ncb1659. [DOI] [PubMed] [Google Scholar]
- 10.Kittler R, et al. RNA interference rescue by bacterial artificial chromosome transgenesis in mammalian tissue culture cells. Proc. Natl Acad. Sci. USA. 2005;102:2396–2401. doi: 10.1073/pnas.0409861102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poser I, et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nature Methods. 2008;5:409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raemaekers T, et al. NuSAP, a novel microtubule-associated protein involved in mitotic spindle organization. J. Cell Biol. 2003;162:1017–1029. doi: 10.1083/jcb.200302129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sumara I, et al. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr. Biol. 2004;14:1712–1722. doi: 10.1016/j.cub.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 14.Gruss OJ, et al. Chromosome-induced microtubule assembly mediated by TPX2 is required for spindle formation in HeLa cells. Nature Cell Biol. 2002;4:871–879. doi: 10.1038/ncb870. [DOI] [PubMed] [Google Scholar]
- 15.Cassimeris L, Morabito J. TOGp, the human homolog of XMAP215/Dis1, is required for centrosome integrity, spindle pole organization, and bipolar spindle assembly. Mol. Biol. Cell. 2004;15:1580–1590. doi: 10.1091/mbc.E03-07-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu C, et al. Functional analysis of human microtubule-based motor proteins, the kinesins and dyneins, in mitosis/cytokinesis using RNA interference. Mol. Biol. Cell. 2005;16:3187–3199. doi: 10.1091/mbc.E05-02-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaudouin J, Gerlich D, Daigle N, Eils R, Ellenberg J. Nuclear envelope break down proceeds by microtubule-induced tearing of the lamina. Cell. 2002;108:83–96. doi: 10.1016/s0092-8674(01)00627-4. [DOI] [PubMed] [Google Scholar]
- 18.Glotzer M. The molecular requirements for cytokinesis. Science. 2005;307:1735–1739. doi: 10.1126/science.1096896. [DOI] [PubMed] [Google Scholar]
- 19.Steigemann P, et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 20.Mora-Bermúdez F, Gerlich D, Ellenberg J. Maximal chromosome compaction occurs by axial shortening in anaphase and depends on Aurora kinase. Nature Cell Biol. 2007;9:822–831. doi: 10.1038/ncb1606. [DOI] [PubMed] [Google Scholar]
- 21.Erfle H, et al. Reverse transfection on cell arrays for high content screening microscopy. Nature Protocols. 2007;2:392–399. doi: 10.1038/nprot.2006.483. [DOI] [PubMed] [Google Scholar]
- 22.Liebel U, et al. A microscope-based screening platform for large-scale functional protein analysis in intact cells. FEBS Lett. 2003;554:394–398. doi: 10.1016/s0014-5793(03)01197-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.