

Abstract
We have constructed a public gene expression data repository and online data access and analysis, WWW and FTP sites for serial analysis of gene expression (SAGE) data. The WWW and FTP components of this resource, SAGEmap, are located at http://www.ncbi.nlm.nih.gov/sage and ftp://ncbi.nlm.nih.gov/pub/sage, respectively. We herein describe SAGE data submission procedures, the construction and characteristics of SAGE tags to gene assignments, the derivation and use of a novel statistical test designed specifically for differential-type analyses of SAGE data, and the organization and use of this resource.
Gene expression quantifying techniques promise to shape our understanding of the distribution and regulation of the products of transcription in normal and abnormal cell types. cDNA microarray (DeRisi 1997), high-density oligo DNA array (Wodicka 1997) and serial analysis of gene expression (Velculescu 1995) techniques have all been developed to quickly and efficiently survey genome-wide transcript expression. However, each of these techniques has the potential to produce, in a single experiment, vast amounts of data which must be sifted and ordered for useful information to become apparent. Additional challenges are met when attempts are made to compare, merge and contrast data from experiments conducted under differing conditions and locales.
As a prototype for the handling, analysis and exchange of gene expression data in the public forum, we have undertaken the production of a public repository and resource for a particular set of gene expression data, i.e., serial analysis of gene expression (SAGE) data. This repository was designed initially to archive SAGE data produced through the Cancer Genome Anatomy Project (CGAP) (Strausberg 1997; http://www.ncbi.nlm.nih.gov/cgap) but is now capable of accepting submissions of SAGE sequence data from any source, without fee or restriction on dissemination or use. It is our goal to provide free and open access to raw SAGE sequence data, precomputed tag extractions, and several modest analysis tools.
This resource currently contains over two million tags from 47 SAGE libraries. We call this resource SAGEmap. Its two online components are available via the World Wide Web (http://www.ncbi.nlm.nih.gov/sage) and anonymous FTP (ftp://ncbi.nlm.nih.gov/pub/sage).
RESULTS AND METHODS
SAGE Library Construction
At this time, data from 47 human SAGE libraries have been submitted to SAGEmap. These libraries were made from mRNA extracts from both bulk tissue and cell lines (neoplastic and non-neoplastic). The tissue sources of these libraries currently include human brain, breast, colon, ovary, prostate, skin and vascular tissue. All of the SAGE libraries currently in SAGEmapwere constructed as previously described in great detail (Velculescu 1995; Zhang 1997). Information and data for species other than Homo sapiens as well as other tissue and tumor types will be posted as they become available.
Library Information and Sequence Data Submission and Storage
Information about the tissue and library treatment and preparation (including organ and tissue type, neoplastic state, any special treatment, tissue submitter, and library producer) is gathered, submitted, and stored in a Sybase relational database maintained at the National Center for Biotechnology Information (NCBI), National Institutes of Health. A unique library name is assigned by NCBI and is used to track submitted SAGE sequences as they are archived. This sequencing information is submitted using the same format and general guidelines governing the submission of expressed sequence tag (EST) sequence information to NCBI's EST data repository, dbEST (Boguski 1993; http://www.ncbi.nlm.nih.gov/dbEST). Information about SAGE library and sequencing data submission can be gained from the SAGEmap website or by sending email to sage@ncbi.nlm.nih.gov. All library and sequence information is made publicly available through the SAGEmap web and FTP sites within days of its successful submission.
SAGEmap Submission Tool
In order to facilitate submissions, a program called SAGEmap Submission Tool, or SST, has been written in the Java programming language. SST not only makes submissions from small laboratories easier by allowing direct submitter annotation of library and reference information, but performs the extraction procedure detailed below. SST has been successfully operated on Windows 95/98/NT and Sun Solaris (UNIX) operating systems running Sun Microsystems's Java Runtime Environment version 1.2.2 (http://java.sun.com/products/jdk/1.2/jre/). A beta version of SST is freely available from the SAGEmap web and FTP sites.
Sequence Data Processing—Tag Extraction
The primary data product of the SAGE technique is the clone insert sequence, which represents the concatenated tags, in pairs (ditags), separated by four base punctuation signals (e.g., NlaIII sites). Processing of the sequence data from the SAGE libraries described in this paper was performed as previously described (Zhang 1997), using the SAGEnet SAGE extraction software (currently called SAGE300, and freely available at http://www.sagenet.org/), as well as by a number of highly customized UNIX operating system shell scripts and C programs at NCBI. All of the data represented by the SAGEmap resource was derived using the latter method. Briefly, the steps in the latter algorithm are as follows:
Locate the NlaIII sites (i.e., CATG “punctuation signals”) within the ditag concatemer,
extract ditags of length 20–26 bases which fall between these sites,
remove repeat occurrences of ditags, including repeat occurrences in the reverse-complemented orientation,
define tags as the end-most 10 bases of each ditag, reverse-complementing the right-handed tag,
remove tags corresponding to linker (e.g., TCCCCGTACA and TCCCTATTAA), as well as those with unspecified bases (i.e., bases other than A, C, G, or T), and
for each tag, count its number of occurrences.
The product of this processing is a list of tags with their corresponding count values, and thus is a digital representation of cellular gene expression.
Tag-to-Gene Assignments
The UniGene project (http://www.ncbi.nlm.nih.gov/UniGene) is an experimental system for automatically partitioning GenBank transcript-source sequences (e.g., proteins, well-characterized mRNA/cDNA sequences and ESTs) into a non-redundant set of gene-oriented clusters. Each UniGene cluster fundamentally contains a group of sequences which are sequence-similar, and therefore represents a unique trancript. The UniGene project thus provides a single identifier and gene description for each cluster of sequence-similar, transcript-source sequences. These identifiers are used in the construction of a SAGE tag to gene mapping. The construction process of the tag to UniGene cluster assignments (hereafter called tag-UniGene assignments or pairs) itself is a multistep, automated process which consists of a number of highly customized UNIX operating system shell scripts and C programs designed for system-dependent file manipulations. UniGene-represented EST sequences and well-characterized mRNA/cDNA sequences are treated similarly, but separately. The reasons for separately processing sequences of different types will be described later. The following steps detail the construction process for making tag-UniGene assignments based on the SAGE anchor enzyme NlaIII.
Separate out individual human sequences from GenBank submission records which are represented in UniGene,
assign sequence orientation through a combination of identification poly-adenylation signal (ATTAAA or AATAAA), poly-adenylation tail (minimum of eight A's), and orientation annotation (3′ or 5′),
extract a 10-base tag which is 3′-adjacent to the 3′-most NlaIII site (CATG),
assign a UniGene identifier to each human sequence with a SAGE tag, and
for each tag-UniGene pair, calculate two frequencies from the number of times this tag-UniGene pair has been seen divided by, separately, the number of sequences with this tag, and the number of sequences with tags in this UniGene cluster.
The result of this process is a “full” tag to gene mapping, with forward and reverse sequence frequency weights given to the edges. Full mappings for human, rat and mouse, based on NlaIII (CATG) or Sau3A (GATC) anchor enzymes, are available for download from the SAGEmap web and/or FTP sites.
Reliable Assigments
When extracting tags, five arbitrary sequence “classes” can be defined to describe the reliability of the tag-UniGene assignment (Table 1). Of highest reliability are tag-UniGene pairs derived from well-characterized mRNA/cDNA sequences from GenBank. Of less reliability are tags extracted from EST sequences, which can be further partitioned into four classes based upon sequence orientation identifiers and annotations. Among the EST sequences, sequences with a polyadenylation signal and/or polyadenylation tail and annotated as 3′ sequences are the most reliable. Next are sequences with a polyadenylation signal and/or polyadenylation tail, but without a 3′ or 5′ annotation. Of the third level of reliability are EST sequences with a polyadenylation signal and/or polyadenylation tail, but annotated as 5′ orientation. Of lowest reliability are EST sequences without a polyadenylation signal or tail but annotated as having a 3′ orientation.
Table 1.
Sequence Classes
| Class | Sequence type | PolyA signal and/or tail | Annotation |
|---|---|---|---|
| 1 | well-characterized mRNA/cDNA | n/a | n/a |
| 2 | EST | yes | 3′ |
| 3 | EST | yes | none |
| 4 | EST | yes | 5′ |
| 5 | EST | no | 3′ |
Well-characterized mRNA/cDNA sequences are assumed to have no sequencing errors. However, since EST sequences are sequenced in a single pass, a certain measurable error rate must be assumed. Previous studies of EST sequences have calculated this error rate to be approximately 1 out of 100 bases, or 1%, on average (Hillier 1996). Assuming an overall error rate of 1%, the resultant chance for one or more sequence errors over 10 bases is 1.00–(0.99)10 = 0.096, or approximately 10%. Therefore, we expect 10% of the tag-UniGene pairs in the full tag-to-gene mapping to be due entirely to sequencing error. Since the most infrequent tag-to-gene assignments are most likely due to error, the most suspect group of tag-UniGene assignments consists of the 10% most infrequent tag-UniGene assignments. Conservative removal of these most infrequently seen tag-UniGene assignments from the “full” tag-to-gene mapping produces “reliable” tag-to-gene mapping. The reliable mappings for human, mouse, and rat, based on NlaIII (CATG) or Sau3A (GATC) anchor enzymes, are also available for download from the SAGEmap web and FTP sites.
Virtual Northern Tool
In order to support mRNA sequence-based queries of the SAGE data in the repository, we have constructed a “virtual Northern” tool which extracts possible SAGE tags and orientation signals (i.e., polyA signal and tail) from any entered mRNA/cDNA sequence. The most likely tag for this sequence may then be selected based on the orientation. These tags are hotlinked to the tag display tool (described below) which, among other things, displays absolute and relative tag abudance in currently held experimental SAGE data (hence the name virtual Northern). In order for this tool to extract, display and link to the correct tag, the submitted sequence must contain the 3′ end of an mRNA sequence. Because it is possible for the same SAGE tag to be present in the transcriptome ambiguously (i.e., the same tag is present in more than one dissimilar transcript), results from the use of this tool should be confirmed with independent methods.
Tag and Gene Display Tools
The tag and gene display tools are used to query multiple data sets. The tag display tool shows the tag's relative and absolute representation in the currently held SAGE libraries, as well as its most reliable gene assignment(s) (via the “reliable” tag-to-gene mapping) and its representation in all of the sequences represented in UniGene (via the “full” tag-to-gene mapping).
Conversely, the gene display tool uses an index based upon the set of valid UniGene cluster ids, and displays the gene's reliable tag assignments (via the “reliable” gene-to-tag mapping) and their absolute and relative representations in the currently held SAGE data, as well as all possible tags extracted from the sequences representing that UniGene cluster (via the “full” gene-to-tag mapping).
Differential SAGE Data Analysis Statistics
Several statistical approaches can be used to test for differential expression in SAGE data. Assume that within two types of cells Y and Z, a particular mRNA species has unknown respective concentrations y and z. A total of A tags are sequenced from cell type Y, and B tags from cell type Z, and among these, a and b tags, respectively, correspond to the mRNA of interest. What may be inferred about the relative size of the actual concentrations, y and z?
Audic & Claverie (1997) have described a classical statistical approach. They consider the null hypothesis H0 that y=z, and the alternative that y≠′z, and derive formulas based upon the observed data for rejecting H0 with various degrees of confidence. If a/A and b/B differ significantly, one rejects H0, concluding that the expression levels y and z are unequal.
An alternative Bayesian approach was described by Chen et al. (1998). They consider the quantity x = y/(y +z), and assume it has a prior probability density function f(x) over the interval [0,1]. If the total number of tags sequenced for each cell type is equal, i.e., if A = B, the posterior probability density for x is, up to a normalizing constant, g(x) = f(x)xa(1-x)b. The concentration y exceeds z by a factor of at least F when x ≥ L, where L = F/(F +1). The posterior probability P of this being the case is given by
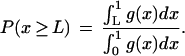 |
The application of this equation is illustrated in Figure 1.
Figure 1.

The solid curve represents an assumed prior probability density for x, given within a normalizing constant by f(x) = x3(1-x)3. The prior probability that the mRNA concentration y is at least 4 times z is represented by the area beneath this curve and to the right of the vertical line at x=0.8, or about 3.3%. Assume that 25 tags corresponding to the mRNA in question are then sequenced from the Y cells, and two tags from the Z cells, in an experiment in which the same total number of tags are sequenced from each of the two cell types. The posterior probability density for x is then proportional to g(x) = x28(1-x)5, represented by the dotted curve. The posterior probability that x ≥0.8 is approximately 70.0%.
This approach may be generalized to the case where A ≠′ B (Lal et al. 1999). Some simple calculus, omitted here, shows that the posterior probability density function is given, up to a normalizing constant, by the equation
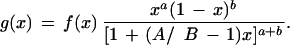 |
When A = B, this reduces to the formula for g(x) given previously (Chen et al. 1998).
The Bayesian approach has both advantages and disadvantages. Rather than simply rejecting the hypothesis that the concentrations y and z are equal, one can estimate the probability that they differ by any desired factor. However, this ability depends upon the assumption of a prior probability density function f(x). It is clearly desirable that f(x) be peaked at and symmetric about 0.5. For mathematical convenience, f(x) may be chosen to be a beta function with both parameters equal (Chen 1998). Ignoring the normalizing constant as usual, this function has the form f(x) = xc(1-x)c. The larger the value specified for c, the more peaked the prior distribution is about 0.5, and the more evidence needed to infer a statistically significant departure from near-equal expression levels.
An appropriate value for the c parameter depends upon the distribution of mRNA expression level changes between cells under various experimental conditions. Chen et al. (1998) proposed choosing c=1, but an analysis of data from other studies (Polyak 1997; Zhang 1997) suggests that a higher and therefore more conservative value, closer to c=3, might be appropriate (Lal 1999).
The value of c is relevant mainly for mRNA species for which little data is available. When the numbers of observed copies a and b of a given tag are large, the data overwhelm prior assumptions. If one is interested mainly in ordering mRNA species for further study, the absolute probabilities estimated for various tags are less important than their relative size. The choice of c is then largely irrelevant, and the classical and Bayesian approaches also are likely to yield very similar orderings by significance.
Online Comparison Tool
We have constructed a differential display-type library comparison tool using the statistical method described above. Counts for a particular tag from individual libraries placed in the same group may be filtered first for homogeneity by using a cutoff value for relative coefficient of variance. The coefficient of variance is a scalar value and is calculated in the usual way as the standard deviation divided by the arithmetic mean, but the frequency of tag expression within the library or group is used in this calculation, rather than absolute tag counts. This cutoff is initially disabled with a value of 0%, but may be altered by the user. As an example, if this cutoff were set to a value of 30%, the standard deviation of the expression frequency of a particular tag within a group differs from the mean by no more than 30%. Once a tag passes this filter, the counts are summed within the group. This filter may be used to reduce the effects of outliers, and thereby imposes a certain degree of homogeneity within each group.
Following the summation of tag count values within each of the groups, the expression levels of the tags within each of the groups are compared using the statistical test described above. The factor used for inter-group comparison is initially set at a 2.0-fold difference, but may be altered by the user. A statistic equivalent to the probability that the two levels differ by at least the given factor is used to order the results.
In order to allow browsing of the results, without requiring a large “download investment” up front (since results may include many tens of thousands of tags), the first 100 tags with their associated gene assignments, tag counts and statistics are displayed in the browser window (in linked HTML), in an abbreviated format. If complete results are desired, a tab-delimited text file of the results may be downloaded from this results webpage, to the user's local computer, where it can be manipulated using spreadsheet and database computer programs.
DISCUSSION
SAGE is a technique designed to take advantage of high-throughput sequencing technology to obtain a quantitative profile of cellular gene expression. Essentially, the SAGE technique measures not the expression level of a gene, but quantifies a “tag” which represents a gene transcript. A tag, for the purposes of SAGE, is a nucleotide sequence of a defined length, directly 3′-adjacent to the 3′-most restriction site for a particular restriction enzyme. As originally described (Velculescu 1995), the length of the tag was nine bases, and the restriction enzyme was NlaIII. Current SAGE protocols produce a 10- to 11-base tag (Zhang 1997), and, although NlaIII remains the most widely used restriction enzyme, enzyme substitutions (e.g., Sau3A) are possible.
The SAGE technique, as was noted earlier, produces a digital output. This is not to imply that no loss of fidelity occurs from the conversion of an actual transcript and its expression level to a tag and its count value. Accuracy in both the assignment of tags to genes as well as the ability to quantify a gene's expression level are sacrificed in order to increase throughput, and therefore increase the speed and lower the cost of analysis. A ten-base tag is by no means a perfect representation of a gene's entire transcript. There will be instances in which two or more genes share the same tag (i.e., the tag-to-gene assignment is ambiguous), and instances in which one gene has more than one tag (i.e., through alternate termination in an individual, or polymorphism in a population, the gene-to-tag assignment is not specific). However, some of the tag-to-gene ambiguities may be resolved using the 11th and 12th bases of the SAGE tag, where present, or independent transcript quantitation methods (e.g., Northern blot analysis).
Sequencing Error Effects on SAGE Data
Single-pass sequencing error plays an important role in tag production. An error, if it occurs in the generation of a SAGE tag datum, will, of course, lower the correct tag count by one, but will also either increase the tag count of an already established tag by one, or will establish and count a tag which does not, in reality, exist. The former effect is not of great concern when drawing conclusions from tags with relatively high counts, since raising or lowering a tag count by one or two should, overall, have no great effect. The former and latter effects, on the other hand, do much to increase suspicion of the tags with low counts, particularly those with a count of 1. The only compensation for counting errors of this type to date has been to remove tags counted only once from the data. This empirical approach has been used in the past for libraries in which roughly 250,000 total tags have been sequenced. For libraries or pools of libraries with, for example, over one million total tags sequenced, it might be necessary to exclude tags with counts of less than 2, 3 or more. This method of compensation for counting errors due to sequencing error may not be an optimal approach, and investigations are currently underway to determine whether a better approach exists.
One such possible approach is to calculate, and make use of, the expected number (or percentage) of nearest neighbors (i.e., one base substitution, insertion or deletion) with a count of one, two, etc., given the number of total tags sequenced, and the actual tags in the data set. Another possible approach is to make use of automated sequencer trace quality scores, such as those generated by the phred algorithm (Ewing 1998). For the data represented on the SAGEmap website, we chose not to attempt these more sophisticated sequencing error compensation steps until we have an opportunity to study this further.
Tag-to-Gene Assignments
It would be preferable if specific and unambiguous gene assignments could be made for every experiment-derived tag, but this is definitely not the case. The difficulties are several, and begin with the set of sequences from which tags are derived. The sets of transcripts from Homo sapiens, Mus musculus, and Rattus norvegicus have yet to be sequenced, let alone characterized. Until they are, there is only an incomplete set of sequences from which to derive tags. Next, considering the nature of the roughly 1.3 million transcript-source human sequences used for the mapping detailed here, only about 19,000 (0.1%) are well-characterized mRNA/cDNA sequences, while the vast majority are expressed sequence tag (EST) sequences. The problem with using EST sequences for the derivation of 10-base tags is that they are, as SAGE sequences are, usually only single-pass sequenced, and therefore have a 1% error rate on average (Hillier 1996). This means that there is roughly a 10% chance that a 10-base tag will include one or more errors. Considering that tag-to-UniGene assignments are based upon the sequence from these tags, it stands to reason that roughly 10% of the assignments that are extracted from these sequences will be incorrect. This compounds the “naturally” unspecific and ambiguous tag-to-gene assignments which are already expected without considering sequencing error.
The naturally occurring unspecific and ambiguous tag-to-gene assignments for human might be reasonably approximated by extracting SAGE tags from the 19,000 or so, nearly errorless, well-characterized mRNA/cDNA sequences in GenBank, and matching those tags to some set of defined gene-units. As noted above, since the set of transcripts from Homo sapiens has not yet been sequenced or characterized, an artificial method for defining gene-units must be chosen. This may be as simple as taking the title of the GenBank sequence entry, or as complicated as using a set of gene contigs, or using gene-based sequence clusters, such as the UniGene gene set (http://www.ncbi.nlm. nih.gov/UniGene). The latter approach was the one adopted for SAGEmap. When tags are extracted from these nearly errorless sequences, two distributions of the tag-to-gene assignments give an idea of baseline specificity and ambiguity before higher sequencing error rates are considered (Fig. 2A,B). Tags can be derived from error-prone EST sequences, but some consideration should be made to how sequencing error might affect tag-to-gene assignments.
Figure 2.

(A,B) The percent distributions of the tag-to-gene and gene-to-tag mappings, respectively, from October 1999, for well-characterized mRNA/cDNA sequences only (i.e., no EST sequences). (C,D) The distributions for the “full” mappings (including data from tags paired to UniGene clusters). (E,F) The distributions for the “reliable” mappings (see text).
Perceived sequence errors are not removed from or corrected in GenBank, UniGene or this SAGE tag mapping, for the very reason that it is difficult—perhaps impossible—to separate these errors from tags resulting from alternative termination, and sequence polymorphisms. However, in this mapping, we can use certain assumptions about the error rates given above to “recommend” certain connections between tags and genes, and not others. We have constructed “reliable” mapping by accepting a certain fraction of the most frequently occurring tag-to-gene assignments, and have based this fraction on an estimation of the sequencing error rate (i.e., a 10% chance of one or more sequencing errors in 10 bases; see Results and Methods section). Two sets of distributions of the tag-to-gene assignments in this mapping give an idea of the effect of adding in error-prone EST sequences before (Fig. 2C,D) and after (Fig. 2E,F) the sequencing error correction is performed.
In Figure 2, A and B represent the best possible outcome of our sequencing error correction algorithm (being the distribution of tags to genes for well-characterized sequences), and C and D, the worst possible outcome (being the distribution of the uncorrected, full mapping). Realistically, we expect our correction algorithm to result in mappings somewhere in between these two extremes, which it does. However, in both the tag-to-gene and the gene-to-tag mappings, we would prefer our correction to remove the erroneous tags, and thereby reduce the number of tag-UniGene and UniGene-tag pairs, but leave the number of unique tags and the number of unique genes represented in the reliable mappings unchanged.
This correction works reasonably well on the tag-to-gene mapping (Fig. 2A,C, and E). For example, 96% of tags derived from well-characterized sequences map to one, and only one, UniGene cluster (Fig. 2A), and 85% of tags derived from ESTs and well-characterized sequences (full mapping) map to only one cluster (Fig. 2C). After the correction is imposed on the full mapping, 87% of tags map to only one cluster (Fig. 2E). In this correction process, the total number of tag-UniGene pairs is reduced by 9.9% from 158,332 to 143,700 while the number of unique tags is reduced a mere 0.065% from 123,978 to 123,898.
This correction also works reasonably well on the gene-to-tag mapping (Fig. 2B, D, and F). For example, 0.52% of UniGene clusters derived from well-characterized sequences map to more than four tags (Fig. 2B), with the mean being 1.8 tags/cluster, and 11% of clusters in the full mapping map to more than four tags (Fig. 2D), with the mean being 2.5 tags/cluster. After the correction is imposed on the full mapping, 11% of clusters map to greater than four tags (Fig. 2F), with the mean being reduced to about 2.3 tags/cluster. In this correction process, the total number of UniGene-tag pairs is reduced by 5.7% from 158,332 to 149,368 while the number of unique genes remains unchanged at 63,776.
While the mean tag/cluster and cluster/tag ratios were lowered, it is somewhat surprising that the correction algorithm did not appreciably change the distribution patterns of the reliable tag-to-gene and gene-to-tag mappings from those of the full mappings towards a distribution which more closely resembled that derived from the well-characterized sequences. In this sense, the estimate of an EST error rate of 10% over 10 bases could be thought to be too conservative. However, it is also possible that the well-characterized sequences represent a biased sample, because they have not been randomly chosen, and so comparing mappings derived from them with the more randomly-derived EST sequences is not entirely appropriate.
Since these mappings are based upon the UniGene sequence clustering algorithm, any artificial “lumping” of sequences belonging to disparate gene units into the same cluster, and “splitting” of sequences belonging to the same gene unit into different clusters will have an effect on the mappings which will not be corrected through sequencing error estimation and correction schemes, such as that which we employed here. If it occurs at an appreciable level, lumping would have the effect of increasing the mean tag/cluster ratio, and skew the gene-to-tag mapping distribution away from 1 tag/cluster. Likewise, if splitting occurs at an appreciable level, the mean cluster/tag ratio would be increased, and the tag-to-gene mapping distribution would be skewed away from 1 cluster/tag. Unlike sequencing errors, these lumping and splitting anomolies, if present at an appreciable level, would be very difficult to correct or compensate for at the mapping level.
An STS-based estimate of lumping and splitting in UniGene suggests that both rates are below 5% (Wagner, unpubl.). This is likely to be an underestimate, because STSs like SAGE tags may be repeated in several members of a gene family, though not necessarily in exactly the same way.
A third source of discrepancies between identically SAGE-tagged sets of sequences and Unigene clusters is 3′ ESTs terminating elsewhere than the terminus of the single longest consensus sequence. There are several causes for such ESTs. Biologically relevant are the familiar phenomena of alternative splicing (though only alternately spliced terminal exons would affect SAGE tagging) and termination at multiple cleavage sites on a single transcript (these seem to occur in 25% of Unigene clusters). In addition, there are artifacts of library preparation: chimeric sequences, internally primed sequences, reversed inserts, and clones terminating at a restriction binding site internal to the transcript (Hillier 1996; Aaronson 1996). The combined effect of these phenomena is estimated to be 5%–10% (both from an inspection of sequences in UniGene and from the studies cited above).
Virtual Northern Tool
We envisioned the virtual Northern tool as a sequence-based query mechanism into the experimentally-derived SAGE data and the UniGene-based tag-to-gene and gene-to-tag, mappings. This tool allows the user to use any mRNA/cDNA sequence (even one which is not represented in the public repositories) with an intact 3′ end to link to a SAGE tag, and then to both relative and absolute abundance and tag-to-gene mapping information. An example of the use of this tool is shown graphically in Figure 3.
Figure 3.

Pasting text sequence information into the text box and clicking on the “Submit” button (left) retrieves the tag and orientation information given below the text box. Tags are given in four possible sequence orientations, the top two orientations being the most common. The correct tag and orientation can be chosen based on the poly-adenylation signal and tail location/presence information. Clicking on the tag (left) will retrieve tag-to-gene mapping information (right) including reliable gene assignment; relative and absolute tag abundance in the SAGE libraries currently in the NCBI SAGE repository; and every UniGene identifier, type, orientation, frequency, and GenBank Accession numbers for each GenBank and dbEST sequence which contained that tag. The information in the right panel can also be retrieved by typing in the 10-base tag sequence in the search box at the top of the page. This same information for UniGene clusters can be retrieved with a similar search interface (not shown) which is located at http://www.ncbi.nlm.nih.gov/SAGE/SAGEcid.cgi.
Online Comparisons
One of the major uses of performing differential analyses of gene expression between a tumor and its corresponding normal tissue is to identify candidate tumor suppressor genes and oncogenes, which can then be studied in depth. Of course, since tumor cells and normal cells have different growth and cellular differentiation characteristics, one always runs the risk of identifying instead abnormally expressed growth factors or cell cycle constituents, or factors expressed only in normal, terminally differentiated cells. We can nevertheless gain some insight into the expression differences of cancer-related genes between examples of changes in neoplastic state (i.e., normal cells into tumor cells). Perhaps we can also draw some conclusions about the likelihood that any given SAGE tag from an uncharacterized transcript which shows differing expression in these two cellular states might also represent a heretofore uncharacterized transcript important in tumorigenesis. Examples of differential comparisons of this sort are highlighted on the SAGEmap website, and one such example of the comparison tool is shown graphically in Figure 4. Example comparisons of this sort using this SAGEmap resource and detailed conclusions of such analyses are described elsewhere (Lal 1999).
Figure 4.

This is an example comparison of colon carcinoma libraries and normal colon libraries. (Left) Names can be chosen for groups A and B, if desired, and the factor parameter upon which the comparison statistic is based; the coefficient of variance cutoffs are set (see text); libraries which are to be put into groups A and B are selected, and the “Calculate” button is pressed. Results are returned in a new window (right), which gives a synopsis of the comparison at the top, lists the names and descriptions of libraries in groups A and B, and results of the statistical comparison (with tag gene assignments) appearing in the lower part of the browser window (see text). In addition, the results of the comparison may be downloaded to a local computer for further analysis.
Gene expression technologies allow us to confront large amounts of data with which we endeavor to glimpse the inner workings of the cell (transcriptome analyses), or differentiate one cellular state from another (differential expression analyses). However, such vast amounts of data greatly complicate attempts to analyze the data produced and compare the data to what has already been produced. We have attempted to construct a public, user-friendly expression data resource which goes beyond being merely a gene expression repository, by providing efficient, online analysis tools to users world-wide. The NCBI SAGE repository and the SAGEmap website are some of the first forays into the world of publicly accessible gene expression data and analysis tools, and we believe they provide a glimpse of what may be accomplished in the field of genomics.
Acknowledgments
The SAGEmap resource discussed in this paper operates on SAGE data which were produced by the NCI's Cancer Genome Anatomy Project or submitted directly to NCBI. The authors express their appreciation to C. Marcelo Aldaz (M.D. Anderson Cancer Research Center), Kenneth Kinzler (Johns Hopkins University), Anita Lal (Duke University), Patrice Morin (National Institute of Aging, NIH), Nickolas Papadopoulos (Columbia University), Kornelia Polyak (Dana-Farber Cancer Institute), Victor Velculescu (JHU), Bert Vogelstein (JHU), and Lin Zhang (JHU) for their work in producing SAGE libraries and data represented on this site, as well as those investigators who will have submitted data before and after this article is published. In addition, we thank the able staff of the I.M.A.G.E Consortium, Lawrence Livermore National Laboratory, Washington University Sequencing Center, and the NIH Intramural Sequencing Center for the clone arraying and/or sequencing of the CGAP-originated SAGE libraries. In addition, the authors express their appreciation to David Lipman of the National Center for Biotechnology Information for providing computational support, general review and helpful suggestions during the development of this public resource.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL alash@ncbi.nlm.nih.gov; FAX (301) 480-9241.
REFERENCES
- Aaronson JS, Eckman B, Blevins RA, Borkowski JA, Myerson J, Imran S, Elliston KO. Toward the development of a gene index to the human genome: an assessment of the nature of high-throughput EST sequence data. Genome Res. 1996;6:829–845. doi: 10.1101/gr.6.9.829. [DOI] [PubMed] [Google Scholar]
- Audic S, Claverie J-M. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Boguski MS, Lowe TM, Tolstoshev CM. dbEST—database for “expressed sequence tags.”. Nat Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- Chen H, Centola M, Altschul SF, Metzger H. Characterization of gene expression in resting and activated mast cells. J Exp Med. 1998;188:1657–1668. doi: 10.1084/jem.188.9.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Hillier LD, Lennon G, Becker M, Bonaldo MF, Chiapelli B, Chissoe S, Dietrich N, DuBuque T, Favello A, Gish W, et al. Generation and analysis of 280,000 human expressed sequence tags. Genome Res. 1996;6:807–828. doi: 10.1101/gr.6.9.807. [DOI] [PubMed] [Google Scholar]
- Lal A, Lash AE, Altschul SF, Zhang L, McLendon RE, Marra MA, Prange C, Morin PJ, Papadopoulos N, Strausberg RL, et al. A public database for quantitative gene expression analysis in human cancers. Cancer Res. 1999;59:5403–5407. [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweler JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Strausberg RL, Dahl CA, Klausner RD. New opportunities for uncovering the molecular basis of cancer. Nat Genet. 1997;15 Spec No:415–416. doi: 10.1038/ng0497supp-415. [DOI] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Wodicka L, Dong H, Mittmann M, Ho M-H, Lockhart DJ. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotech. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, Vogelstein B, Kinzler KW. Gene expression profiles in normal and cancer cells. Science. 1997;276:1268–1272. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]