

Abstract
Ca2+ mediates the functional coupling between L-type Ca2+ channel (LTCC) and sarcoplasmic reticulum (SR) Ca2+ release channel (ryanodine receptor, RyR), participating in key pathophysiological processes. This crosstalk manifests as the orthograde Ca2+-induced Ca2+-release (CICR) mechanism triggered by Ca2+ influx, but also as the retrograde Ca2+-dependent inactivation (CDI) of LTCC, which depends on both Ca2+ permeating through the LTCC itself and on SR Ca2+ release through the RyR. This latter effect has been suggested to rely on local rather than global Ca2+ signaling, which might parallel the nanodomain control of CDI carried out through calmodulin (CaM). Analyzing the CICR in catecholaminergic polymorphic ventricular tachycardia (CPVT) mice as a model of RyR-generated Ca2+ leak, we evidence here that increased occurrence of the discrete local SR Ca2+ releases through the RyRs (Ca2+ sparks) causea depolarizing shift in activation and a hyperpolarizing shift inisochronic inactivation of cardiac LTCC current resulting in the reduction of window current. Both increasing fast [Ca2+]i buffer capacity or depleting SR Ca2+ store blunted these changes, which could be reproduced in WT cells by RyRCa2+ leak induced with Ryanodol and CaM inhibition.Our results unveiled a new paradigm for CaM-dependent effect on LTCC gating and further the nanodomain Ca2+ control of LTCC, emphasizing the importance of spatio-temporal relationships between Ca2+ signals and CaM function.
Introduction
Dynamic modulation of cellular Ca2+ flows from either the extracellular space or the intracellular Ca2+ store into the cytoplasm participates in key pathophysiological processes, which depends on the ability of cells to properly sort ‘global’ and ‘local’ Ca2+ signals [1]. In this respect, the functional coupling of the sarcolemmal L-type Ca2+ channels (LTCC) and the sarcoplasmic reticulum (SR) Ca2+ release channels (ryanodine receptor, RyR), plays an important role in ventricular cardiomyocytes [2], [3]. Depolarizing stimuli open voltage-gated LTCC, leading to Ca2+ entry (ICa) and a subsequent rise in the cytoplasmic free Ca2+ concentration ([Ca2+]i). While such [Ca2+]i elevations are initiated by LTCC, they are also influenced by Ca2+ transporting organelles such as the mitochondria and the SR. Notably, in response to these increases in [Ca2+]i, Ca2+ binds to and activates RyRs thereby amplifying the initial Ca2+ signal through the locally controlled Ca2+-induced Ca2+-release (CICR) process to support the excitation-contraction coupling (ECC) and thus heart function [4]. On the other hand, the opening of LTCCs is tightly controlled to prevent intracellular Ca2+overload. A major intrinsic negative feedback mechanism is the Ca2+-dependent inactivation (CDI) of the widely distributed voltage-gated Ca2+ channels [5], [6], [7], [8], [9], [10]. From the pioneering descriptions [11], CDI manifests as the hallmark time-dependent current decay during prolonged depolarization but also determines the voltage-dependent availability of Ca2+ channel during double-pulse protocols. Early studies of CDI were mainly focused on Ca2+ entry, but SR Ca2+ release also contributes significantly to the CDI [5]. In cardiac myocytes, depletion of SR Ca2+ stores or abolition of SR Ca2+ release causes a reduction of CDI [3], [12], whereas increasing SR Ca2+ loading results in CDI enhancement [13]. CDI depends linearly on the rate and magnitude of SR Ca2+ release from the RyRs [12]. Thus, ∼70% of CDI that occurs during ECC in rat ventricular myocytes arises from SR Ca2+ released, which might reduce Ca2+ influx during action potential up to 50% [14], [15]. Furthermore, it has been shown that SR Ca2+ release dominates CDI initially, then, as [Ca2+]i decreases due to SR Ca2+ reuptake, the SR dependent contribution declines with participation from Ca2+ entry via ICa dominating [16]. Now, SR Ca2+ release from the RyRs results in discrete and localized rises of [Ca2+]i (Ca2+ sparks) triggered by I Ca [4]. The large local releases of Ca2+during CICR modulate in turn LTCC [3], [12], suggesting that discrete Ca2+ cross-signaling occurs in the microdomains of LTCC-RyRs [17].
Over the past decades there has been rapid progress toward understanding the molecular basis for CDI, cumulating with the identification of constitutively complexed calmodulin (CaM) with the Ca2+channel as the specific resident Ca2+ sensor [5], [6], [7], [8], [9], [10]. CaM relies on its N and C lobes to detect spatiotemporal aspects of the Ca2+ signal for regulation of Ca2+ channels [6]. In cardiac myocytes, for example, C lobe–mediated CDI of LTCCs appears to be an essential regulator of action potential duration [18], whereas in nerve terminals, N lobe–mediated CDI appears to underlie use-dependent short-term plasticity of synaptic transmitter release via Ca2+ channels [19]. More recently, a model has been proposed where the C lobe of CaM senses local [Ca2+]i as a result of Ca2+ influx from the LTCC, while the N-lobe can operate as a tunable detector of global or local [Ca2+]i arising from distant Ca2+ sources [8]. In addition, an N-terminal cytoplasmic region of LTCCs has been shown to restrict CaM to respond almost exclusively to local Ca2+ signals in the immediate vicinity of the channel pore [7]. These results gave rise to the concept of nanodomaincontrol of the CDI. This was at first focused on Ca2+ influx as source for modulation of the channel, formulated upon measurements from nonexcitable cells that lack CICR nanodomains. A pertinent question is, therefore, whether and how discrete and local SR Ca2+release participates to the Ca2+ control of LTCC?
We have chosen to further explore this question by comparing RyRR4496C catecholaminergic polymorphic ventricular tachycardia (CPVT) mutant mice to their wild type littermates (WT) by simultaneous recordings of ICa and the evoked [Ca2+]i transients and Ca2+ sparks. CPVT is a severe inherited cardiac disorder that manifests as malignant exercise-emotion-triggered arrhythmias leading to syncope and sudden death. Mutations in the cardiac RyR account for an autosomal-dominant form in approximately 50% of CPVT cases. In knock-in transgenic mouse model (CPVT mice), the R4496C mutation of the RyR increased the Ca2+ sensitivity of RyR, leading to diastolic Ca2+ leak and arrhythmogenic triggered activity [20]. Because the RyR Ca2+ leak is increased in this CPVT mouse model we investigated several key steps in the process of ECC that might provide insights into local [Ca2+]i control of LTCC.
Results
Increased CICR-gain at low voltage in CPVT cells
The ability of the SR to amplify the trigger Ca2+ influx, or CICR-gain, reflects not only the operation of the fundamental processes that underlie normal ECC, but also those involved in important pathological conditions of the heart, such as triggered arrhythmias produced by uncontrolled SR Ca2+ release [4], [21]. Figure 1A shows representative experiments of ICa traces (normalized to cell capacitance) and line scan images of the evoked [Ca2+]i transients (using the fluorescence Ca2+ indicator fluo-3) recorded in freshly isolated ventricular myocytes from wild type (WT, top) and CPVT (bottom) mouse hearts using simultaneous patch-clamp current recording and high resolution confocal Ca2+ imaging techniques [22]. The measurements were used to calculate the CICR-gain, defined as the ratio of the peak [Ca2+]i transient (F/F0) over the corresponding Ca2+ influx through the LTCC, calculated as the IC a integral, in response to voltage steps (Figure 1B) [22]. At −30 and −20 mV, the curve for CPVT cells bends upward and deviates significantly from that obtained from the WT cells, then the CICR-gain curve essentially overlaps with that of the WT. This happens even though the SR Ca2+ content was constant, as estimated by the integral of the caffeine-evoked inward current (in pC: 381.2±70.1 vs 326.0±92.6, in 13 WT vs 14CPVT cells, respectively, P>0.05) [23].
Figure 1. CICR gain is increased in CPVT mice due to reduced ICa at low voltages.

A. Representative examples of Ca2+ entry and release fluxes simultaneously recorded in WT (black traces) and CPVT (red traces) myocytes. Beneath the images is the corresponding profile of fluorescence, expressed as F/F0, where F is fluorescence and F0 is diastolic fluorescence, after background correction. B. Voltage-dependent Ca2+ induced-Ca2+ release gain (CICR-gain) decreased monotonically, giving rise to an L-shaped in CPVT (filled symbols, n = 14) and WT (open symbols, n = 16) cells. C & D. Voltage dependence of peak [Ca2+]i transients (C) and peak of ICa density (D) displayed bell-shaped, graded function with the membrane potential. * P<0.05 and ** P<0.005.
The enhanced CICR-gain at more negative voltages, despite maintained SR Ca2+ load, might reflect an increased efficiency of crosstalk between LTCCs and RyRs. Figures 1C and D compares the average voltage dependence of peak [Ca2+]i transient and ICa in WT and CPVT myocytes from experiments such as those shown in Figure 1A. Both ICa and [Ca2+]i transients displayed bell-shaped, graded function with the membrane potential. Whereas no difference on [Ca2+]i transient was observed between WT and CPVT cells at any potential (Figure 1C), the peak ICa-voltage relationships showed significant reductions at low voltages in CPVT cells, leaving unmodified the maximal ICa (Figure 1D).
These results indicated that the RyRR4496C mutation lowers ICa at negative voltages without global [Ca2+]i transient alteration, resulting in enhancement of CICR-gain, consistent with the increased Ca2+ sensitivity of RyRs [20]. This could be explained by a modified activity of Na+/Ca2+ exchanger (NCX), which might rapidly and reversibly alter the Ca2+ concentration in the vicinity of the LTCCs [24]. However, the NCX currents (normalized to cell size) showed similar values in WT and CPVT myocytes (peak current density normalized by peak caffeine-evoked [Ca2+]i transient, as evaluated by synchronous confocal images, in pA/pF: −0.88±0.09 vs −0.82±0.13, in 10 WT vs 12 CPVT cells, respectively, P>0.05).
Changes of voltage-dependent availability of Ca2+ channel reduce window current
A change in the time- and/or voltage-dependence of ICa kinetics could account for the observed alteration of ICa. Over the whole voltage range, neither activation (Figure 2A) nor inactivation (Figure 2B) kinetics of ICa were significantly different between WT and CPVT cells. In addition, the increase in current area upon repetitive stimuli (during trains of voltage pulses), or frequency-dependent facilitation, was not modified in CPVT cells (data not shown).
Figure 2. CPVT cells demonstrated rightward and leftward shifts in the voltage-dependent activation and inactivation of ICa, respectively.

A. Activation kinetics of ICa over the whole voltage range were not significantly different between WT (open symbols) and CPVT (filled symbols) cells. B. The time course of inactivation of ICa, which encompass slow and fast components, were similar in WT and CPVT cells at all voltages studied. C. Superimposed voltage-dependence of ICa activation and inactivation. ICa activationis shifted to more positive values in CPVT vs WT cells, whereas inactivation of ICa is shifted to more hyperpolarized potential in CPVT cells compared with WT cells. D. Voltage dependence of ICa window current (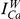 ) display a bell-shaped voltage-dependence, however, the peak of
) display a bell-shaped voltage-dependence, however, the peak of 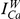 is reduced in CPVT cells (continuous line) compared to WT cells (dashed line). * P<0.05 and ** P<0.005.
is reduced in CPVT cells (continuous line) compared to WT cells (dashed line). * P<0.05 and ** P<0.005.
Difference in the availability of ICa as function of the voltage might underlie the reduced peak current density at low voltages. The activation-voltage relationships were constructed by converting the peak current values from each current-voltage relationship data set to the chord conductance using the equation: G = I/(V-Erev) and then the ratio G/Gmax were plotted against the membrane potential (Erev and Gmax as determined by the current-voltage fits [25]). In both CPVT and WT cells, the relations rise sigmoidally from 0 to 1 over the range −50 to +30 mV, but the voltage ranges for activation of ICa in CPVT cells were significantly more positive than for WT cells (Figure 2C). To determine the activation variable (d ∞), curves through the data points were fitted by the Boltzmann function d ∞ (V ) = 1/{1+exp[(V0.5-V )/k]}, where V0.5 is the potential at which the conductance is half maximally activated and k is the slope factor describing the steepness of the curve. Whereas k was unchanged, a significant rightward shift in V0.5 was observed in CPVT cells compared to WT (Table 1).
Table 1. Parameters of Boltzmann fittings of activation (d∞) and inactivation (f∞) curves (in mV).
| WT | CPVT | |||||||
| d∞ | f∞ | d∞ | f∞ | |||||
| Control | V0.5−12.6±1.4 | k6.3±0.5 | V0.5−22.8±1.1 | k6.1±0.5 | V0.5−8.9±1.2* | k5.3±0.2 | V0.5−29.8±1.6* | k5.9±0.5 |
| (n = 14) | (n = 8) | (n = 16) | (n = 8) | |||||
| Iso | −21.1±1.6# | 4.7±0.3# | −36.5±1.3# | 4.4±0.6# | −20.0±2.2† | 4.6±0.5† | −36.3±1.4† | 4.2±0.8† |
| (n = 6) | (n = 6) | (n = 7) | (n = 7) | |||||
| BAPTA | −16.2±0.9# | 3.6±0.3# | −24.5±0.4 | 5.3±0.2 | −15.4±1.0† | 4.0±0.2† | −24.0±0.4† | 5.8±0.2 |
| (n = 12) | (n = 12) | (n = 13) | (n = 11) | |||||
| Thapsi | −17.5±1.7# | 4.7±0.5# | −24.7±1.0 | 5.8±0.5 | −19.9±2.1† | 4.2±0.6† | −25.1±0.5† | 6.5±0.3 |
| (n = 11) | (n = 10) | (n = 8) | (n = 8) | |||||
| Ryanod | −7.6±0.7# | 5.2±0.1 | −25.7±0.8# | 5.2±0.1 | − | − | ||
| (n = 12) | (n = 10) | |||||||
| W7 | −9.2±0.5# | 5.9±0.2 | −31.7±0.6# | 5.5±0.1 | −8.3±0.4 | 5.9±0.2 | −31.9±0.4 | 5.5±0.2 |
| (n = 12) | (n = 11) | (n = 13) | (n = 12) | |||||
| CALP2 | −8.2±1.6# | 6.3±0.3 | −32.9±1.2# | 5.9±0.5 | −7.7±1.2 | 5.6±0.5 | −30.7±1.1 | 6.0±0.2 |
| (n = 8) | (n = 8) | (n = 11) | (n = 10) | |||||
*p<0.05 vs WT;
p<0.05 vs control WT;
p<0.05 vs control CPVT.
Even if this shift, toward slightly more positive membrane potentials, might be involved in the reduction of ICa at low voltages, maintained ICa at more positive voltages might reflect alteration in ICa inactivation. We then determined isochronic inactivation with a double-pulse protocol, in which the relative amplitude of ICa during the test pulse (normalized to the maximum test current, Imax) is proportional to the fraction of available channels at that given time. As shown in Figure 2C, the relations are sigmoid over −50 to 0 mV voltage range, but at prepulse potentials more positive than 0 mV the extent of inactivation decreased resulting in an U-shaped inactivation curve, consistent with inactivation arising from voltage- and Ca2+-dependent processes (VDI and CDI, respectively) [11]. In contrast to activation, we observed that the voltage range where channels are experiencing inactivation is significantly more negative in CPVT than in WT cells. In addition, the turn up of inactivation curve at positive potentials is significantly reduced in CPVT. Data negative to 0 mV from individual cells were fit to the Boltzmann equation f∞(V) = (1-A)/{1+exp[(V-V0.5)/k]}+A; where V0.5 is the potential of half-maximal inactivation, k is the slope factor, A is the amplitude of the non-inactivating ICa. A significant leftward shift is observed for V0.5 in CPVT compared to WT cells (Table 1).
The overlap of activation and inactivation voltage curves delimits a window current region that allows the channels to be tonically active at these membrane potentials, as the channel population dynamically equilibrates among open, closed and inactivated states, resulting in a steady-state current [26]. Due to opposite shifts in activation and inactivation curves (Figure 2C), the inactivation-activation overlap “window area” for ICa was substantially reduced in CPVT cells compared with controls. The Ca2+ window current (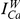 ) was estimated by the product of available channels (d∞.f∞) and the relative peak conductance using the classical Hodgkin and Huxley formulation: Gmax.d
∞.f
∞.(V-Erev). A marked reduction in
) was estimated by the product of available channels (d∞.f∞) and the relative peak conductance using the classical Hodgkin and Huxley formulation: Gmax.d
∞.f
∞.(V-Erev). A marked reduction in 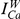 was observed in CPVT myocytes compared with WT myocytes (Figure 2D). Of note, under β-adrenergic stimulation, which induced ∼1.5 fold increase in Gmax (in pS/pF: 124.1±4.0 vs 173.9±13.1 in 6 WT cells and 134.1±7.5 vs 210.3±28.3 in 7 CPVT cells, before and after 1 µmol/L isoproterenol, respectively), no more differences in activation or inactivation parameters (Table 1) were observed between WT and CPVT myocytes.
was observed in CPVT myocytes compared with WT myocytes (Figure 2D). Of note, under β-adrenergic stimulation, which induced ∼1.5 fold increase in Gmax (in pS/pF: 124.1±4.0 vs 173.9±13.1 in 6 WT cells and 134.1±7.5 vs 210.3±28.3 in 7 CPVT cells, before and after 1 µmol/L isoproterenol, respectively), no more differences in activation or inactivation parameters (Table 1) were observed between WT and CPVT myocytes.
Ca2+ dependence of reduced window current
Our observations furthered investigation on the molecular mechanism by which the R4496C mutation in RyR alters the 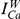 . In cardiomyocytes, due to the close functional association of LTCCs with RyRs in the dyadic space, the Ca2+ flux through either channel modifies the activity of the other channel. Thereby SR Ca2+ release influences at least partly ICa
[5]. Whereas [Ca2+]i transient is unaltered in CPVT cells (Figure 1C), CPVT myocyte show a marked increased in RyR Ca2+ leak, which might be visualized as Ca2+ sparks [4], [20]. We analyzed voltage-activated Ca2+ sparks in patch-clamped cells, applying small depolarizing steps from −48 to −42 mV for 300 ms, in 2 mV increments. At these voltages, the number of activated LTCCs is limited and it is possible to resolve the evoked Ca2+ sparks [22]. Figure 3A shows that the Ca2+ spark frequencies were significantly higher over the studied voltage range in CPVT compared to WT cells. As previously reported on spontaneous Ca2+ sparks [20], no other change in spatiotemporal spark properties were denoted in ICa-evoked Ca2+ sparks (data not shown). These results lead us to probe the plausible Ca2+ dependence of the cross-signaling between ICa and RyR in the reduced
. In cardiomyocytes, due to the close functional association of LTCCs with RyRs in the dyadic space, the Ca2+ flux through either channel modifies the activity of the other channel. Thereby SR Ca2+ release influences at least partly ICa
[5]. Whereas [Ca2+]i transient is unaltered in CPVT cells (Figure 1C), CPVT myocyte show a marked increased in RyR Ca2+ leak, which might be visualized as Ca2+ sparks [4], [20]. We analyzed voltage-activated Ca2+ sparks in patch-clamped cells, applying small depolarizing steps from −48 to −42 mV for 300 ms, in 2 mV increments. At these voltages, the number of activated LTCCs is limited and it is possible to resolve the evoked Ca2+ sparks [22]. Figure 3A shows that the Ca2+ spark frequencies were significantly higher over the studied voltage range in CPVT compared to WT cells. As previously reported on spontaneous Ca2+ sparks [20], no other change in spatiotemporal spark properties were denoted in ICa-evoked Ca2+ sparks (data not shown). These results lead us to probe the plausible Ca2+ dependence of the cross-signaling between ICa and RyR in the reduced 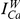 . To explore this possibility, further experiments were carried out by increasing the Ca2+-buffering capacity of the cytoplasm through the introduction of exogenous buffer inside the patch pipette. We used the fast Ca2+chelator 1,2-bis (2-ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 10 mmol/L). When compared to control conditions, increasing the cytoplasmic Ca2+-buffering capacity with BAPTA in CPVT cells completely blunted the shifts in d∞ activation and f∞ inactivation variables eliminating the difference with WT cells (Table 1). As a results,
. To explore this possibility, further experiments were carried out by increasing the Ca2+-buffering capacity of the cytoplasm through the introduction of exogenous buffer inside the patch pipette. We used the fast Ca2+chelator 1,2-bis (2-ethane-N,N,N′,N′-tetraacetic acid (BAPTA, 10 mmol/L). When compared to control conditions, increasing the cytoplasmic Ca2+-buffering capacity with BAPTA in CPVT cells completely blunted the shifts in d∞ activation and f∞ inactivation variables eliminating the difference with WT cells (Table 1). As a results, 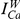 was merely identical to that observed in WT cells when [Ca2+]i is clamped with BAPTA (Figure 3B). This suggests that local [Ca2+]i increases lead to the reduced
was merely identical to that observed in WT cells when [Ca2+]i is clamped with BAPTA (Figure 3B). This suggests that local [Ca2+]i increases lead to the reduced 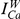 in CPVT mice. To test the contribution of the SR in the Ca2+-dependent effects in
in CPVT mice. To test the contribution of the SR in the Ca2+-dependent effects in 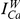 , we functionally disabled intracellular Ca2+ stores by application of the SR Ca2+ ATPase inhibitor (thapsigargin, 5 µmol/L) to block the ability of the cell to pump Ca2+ into the SR and to deplete the store by consecutive depolarizations to 0 mV. Consequently, SR Ca2+ release was blunted. In the same way as for buffering [Ca2+]i, abolishing SR Ca2+ release restored
, we functionally disabled intracellular Ca2+ stores by application of the SR Ca2+ ATPase inhibitor (thapsigargin, 5 µmol/L) to block the ability of the cell to pump Ca2+ into the SR and to deplete the store by consecutive depolarizations to 0 mV. Consequently, SR Ca2+ release was blunted. In the same way as for buffering [Ca2+]i, abolishing SR Ca2+ release restored 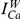 in CPVT cells to WT levels (Figure 3C) by preventing the shifts in d∞ and f∞ variables (Table 1). It is noteworthy that both BAPTA dialysates and thapsigargin-treatment induced in WT cells a significant shift in the hyperpolarizing direction and a steepening of the activation curves (Table 1), resulting in a slight increased peak
in CPVT cells to WT levels (Figure 3C) by preventing the shifts in d∞ and f∞ variables (Table 1). It is noteworthy that both BAPTA dialysates and thapsigargin-treatment induced in WT cells a significant shift in the hyperpolarizing direction and a steepening of the activation curves (Table 1), resulting in a slight increased peak 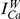 (compare Figures 1 to
3B and C). This emphasizes the physiological relevance of RyR Ca2+ leak-mediated
(compare Figures 1 to
3B and C). This emphasizes the physiological relevance of RyR Ca2+ leak-mediated 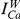 reduction.To confirm this interpretation, we exposed WT cells to 50-µmol/L Ryanodol, which has been shown to increase the overall Ca2+ spark rate without global change on SR Ca2+ load [27]. Similarly to CPVT cells, upon Ryanodol exposition, the activation of ICa in WT cells was shifted toward positive voltages while the inactivation was shifted toward negative voltages (Table 1, Ryanod) resulting in a decrease of
reduction.To confirm this interpretation, we exposed WT cells to 50-µmol/L Ryanodol, which has been shown to increase the overall Ca2+ spark rate without global change on SR Ca2+ load [27]. Similarly to CPVT cells, upon Ryanodol exposition, the activation of ICa in WT cells was shifted toward positive voltages while the inactivation was shifted toward negative voltages (Table 1, Ryanod) resulting in a decrease of 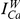 (Figure 3D).
(Figure 3D).
Figure 3. Increased Ca2+ spark occurrence limits ICa window current (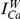 ).
).

A. Analyze of the patch clamped-Ca2+ sparks revealed that the average of Ca2+ sparks frequencies in CPVT cells (filled circles, n = 10) were significantly higher compared to WT cells (open circles, n = 3). * P<0.05. Right insets. Representative examples of line-scan images of Ca2+ sparks elicited by depolarizing step to −48 mV. B & C. BAPTA dialysates (B) and thapsigargin-treatment (C) eliminates the difference in 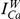 between CPVT and WT cells. D. In presence of Ryanodol in the perfusion solution,
between CPVT and WT cells. D. In presence of Ryanodol in the perfusion solution, 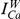 is reduced in WT cells compared to control condition, shown as light gray line.
is reduced in WT cells compared to control condition, shown as light gray line.
CaM antagonists promoted window current reduction
Collectively, our findings suggest that increased RyR Ca2+ leak limits 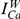 . But what could the essential mechanistic ingredient of this effect be? The ubiquitous Ca2+ binding protein calmodulin (CaM) has a central role in determining LTCC Ca2+ sensitivity [5], [6], [7], . We therefore proceeded to probe the plausible involvement of CaM in reduction of
. But what could the essential mechanistic ingredient of this effect be? The ubiquitous Ca2+ binding protein calmodulin (CaM) has a central role in determining LTCC Ca2+ sensitivity [5], [6], [7], . We therefore proceeded to probe the plausible involvement of CaM in reduction of 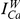 . As a first approximation of events that may be CaM dependent, we used [N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide] (W7, 1 µmol/L), a water-soluble, cell membrane–permeant competitive antagonist of Ca2+/CaM [28]. Surprisingly, 30-min W7 cell-treatment did not affect either availability variables (Table 1) or
. As a first approximation of events that may be CaM dependent, we used [N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide] (W7, 1 µmol/L), a water-soluble, cell membrane–permeant competitive antagonist of Ca2+/CaM [28]. Surprisingly, 30-min W7 cell-treatment did not affect either availability variables (Table 1) or  in CPVT cells (Figure 4A), whereas it shifted both activation curve to more positive voltages and inactivation curve to more negative voltages (Table 1) in WT cells compared to control conditions. Consequently, W7-treated WT cells behave such as CPVT cells, showing a reduced
in CPVT cells (Figure 4A), whereas it shifted both activation curve to more positive voltages and inactivation curve to more negative voltages (Table 1) in WT cells compared to control conditions. Consequently, W7-treated WT cells behave such as CPVT cells, showing a reduced 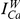 (Figure 4A). Because the results obtained with W7 should not be taken as absolute proof of a CaM-dependent process, we repeated the experiments with a cell-permeable CaM-inhibitory peptide, CALP2, designed to bind to EF-hand four-amino acid sequence of CaM [29]. Similarly to W7-treatment, a 60-min incubation with 100-µmol/L CALP2 did not affect either
(Figure 4A). Because the results obtained with W7 should not be taken as absolute proof of a CaM-dependent process, we repeated the experiments with a cell-permeable CaM-inhibitory peptide, CALP2, designed to bind to EF-hand four-amino acid sequence of CaM [29]. Similarly to W7-treatment, a 60-min incubation with 100-µmol/L CALP2 did not affect either 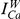 (Figure 4B) or availability variables (Table 1) in CPVT cells. However, in WT cells CALP2 significantly shifted the activation curve rightward and the inactivation curve leftward (Table 1) leading to a reduction of
(Figure 4B) or availability variables (Table 1) in CPVT cells. However, in WT cells CALP2 significantly shifted the activation curve rightward and the inactivation curve leftward (Table 1) leading to a reduction of 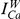 towards the level corresponding to that of CPVT cardiomyocytes (Figures 4B). As an important control, neither in CPVT nor in WT CaM antagonists altered the diastolic RyR activity in intact cells, visualized in situ by confocal microscopy as spontaneous Ca2+ sparks frequencies (Figure 4C). As well, the amount of soluble (Figure 4D) and membrane bound CaM (Figure 4E) are not altered in CPVT mice hearts compared to control WT, consistent with unaltered ICa decay kinetics (Figure 2B).
towards the level corresponding to that of CPVT cardiomyocytes (Figures 4B). As an important control, neither in CPVT nor in WT CaM antagonists altered the diastolic RyR activity in intact cells, visualized in situ by confocal microscopy as spontaneous Ca2+ sparks frequencies (Figure 4C). As well, the amount of soluble (Figure 4D) and membrane bound CaM (Figure 4E) are not altered in CPVT mice hearts compared to control WT, consistent with unaltered ICa decay kinetics (Figure 2B).
Figure 4. Calmodulin inhibition reduced 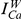 in WT cells.
in WT cells.

A & B. CaM antagonists, W7 (A) or CALP2 (B) reduced 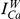 in WT cells (dashed line) without affecting it in CPVT cells (continuous line). C. Comparison of the Ca2+ sparks occurrence at rest in myocytes from WT (open bars) and CPVT (closed bars) cells in control conditions and after incubation with W7 (left hatched bars) or CALP2 (right hatched bars). * P<0.05 vs WT. D & E. Representative immunoblots and quantification of CaM protein levels in cardiac heart lysates (D, normalized to the corresponding actin level and normalized to respective controls) and detected in the membrane fraction (E, normalized to the corresponding Cav1.2 level and normalized to respective controls) from WT (open bars, n = 4) and CPVT (closed bars, n = 4) mice.
in WT cells (dashed line) without affecting it in CPVT cells (continuous line). C. Comparison of the Ca2+ sparks occurrence at rest in myocytes from WT (open bars) and CPVT (closed bars) cells in control conditions and after incubation with W7 (left hatched bars) or CALP2 (right hatched bars). * P<0.05 vs WT. D & E. Representative immunoblots and quantification of CaM protein levels in cardiac heart lysates (D, normalized to the corresponding actin level and normalized to respective controls) and detected in the membrane fraction (E, normalized to the corresponding Cav1.2 level and normalized to respective controls) from WT (open bars, n = 4) and CPVT (closed bars, n = 4) mice.
Discussion
The purpose of our study was to examine whether discrete and local increase of SR Ca2+ release has any effect on ICa with respect to nanodomain control of LTCCs. Using CPVT mice as a model with high spontaneous RyR-generated Ca2+ leak, our results pinpoint that the increase in RyRCa2+ leak caused opposite shifts in activation and in activation voltage curves of the cardiac LTCC. The resulting reduction of window current is prevented by manoeuvres that minimize variations in [Ca2+]i due to SR Ca2+ release. Surprisingly, application of CaM antagonists did not have any effect on the cells from the CPVT mouse but altered the inactivation and activation curves in the WT mouse to make these more like the CPVT mouse, revealing a new paradigm for CaM-dependent effect on LTCC gating. These effects might represent an adaptive mechanism to cytotoxic SR Ca2+ leak and Ca2+ overload.
RyRCa2+ leak limits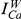
Analysis of skeletal myotubes derived from RyR-knockout (dyspedic) mice has revealed that in addition to the orthograde signal of ECC, there is also a retrograde signal whereby RyR promotes the Ca2+ conducting activity of the skeletal LTCC [30]. But this bi-directional coupling is thought to result from direct physical protein-protein interaction [30]. Nevertheless, it has been recently suggested that this feedback mechanism, which results in reduction of 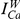 , might also depend on local SR Ca2+ signaling [2]. Despite some controversial results, no such structural interaction has been found in cardiac cells. However, the close physical association between LTCCs and RyRs that in cardiac muscle takes place at the narrow site of the tubulo-reticular junction where each LTCC is closely associated with a group of RyRs [4], creates a bi-directional Ca2+ signaling microdomain such that Ca2+ flux through one channel modifies the functional behavior of the other channel. In this study, we show that either buffering intracellular Ca2+ or eliminating SR Ca2+ release erased the difference in
, might also depend on local SR Ca2+ signaling [2]. Despite some controversial results, no such structural interaction has been found in cardiac cells. However, the close physical association between LTCCs and RyRs that in cardiac muscle takes place at the narrow site of the tubulo-reticular junction where each LTCC is closely associated with a group of RyRs [4], creates a bi-directional Ca2+ signaling microdomain such that Ca2+ flux through one channel modifies the functional behavior of the other channel. In this study, we show that either buffering intracellular Ca2+ or eliminating SR Ca2+ release erased the difference in 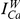 between CPVT and WT mice. Thus, we conclude that the mechanism underlying the reduced
between CPVT and WT mice. Thus, we conclude that the mechanism underlying the reduced 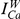 in CPVT mice is SR Ca2+ dependent. In cardiac cells, SR Ca2+ release is indeed the major component determining CDI [3], [12], [13], [14], [15], [16]. However, these studies as well as the majority of CDI analyses, relied almost exclusively on the accelerated current inactivation during depolarizing voltage steps whereas the CDI manifestation on current availability [11] has not been yet the subject of in depth investigation. Nonetheless, when compared to neonatal cardiomyocytes, where there is poor T-tubule development, adult cardiomyocytes show a reduced
in CPVT mice is SR Ca2+ dependent. In cardiac cells, SR Ca2+ release is indeed the major component determining CDI [3], [12], [13], [14], [15], [16]. However, these studies as well as the majority of CDI analyses, relied almost exclusively on the accelerated current inactivation during depolarizing voltage steps whereas the CDI manifestation on current availability [11] has not been yet the subject of in depth investigation. Nonetheless, when compared to neonatal cardiomyocytes, where there is poor T-tubule development, adult cardiomyocytes show a reduced 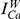 [31] and a slight reduction in ICa availability has been observed with static elevation of [Ca2+]i
[32].
[31] and a slight reduction in ICa availability has been observed with static elevation of [Ca2+]i
[32].
Although a global increase in [Ca2+]i can participate to CDI, there is evidence suggesting that local Ca2+ signaling between RyRs and LTCCs might mediate a Ca2+ functional crosstalk between LTCCs and RyRs [3], [16], [17]. Changes in local subsarcolemmal Ca2+ caused by the alteration of normal Ca2+ extrusion via NCX could be involved. But we observed no difference in NCX activity between WT and CPVT cells. Such an alteration in Ca2+ driving force, as well as in surface charges, will be difficult to reconcile with the absence of modification in peak ICa density, apparent reversal potential and opposite shifts of activation and inactivation voltage ranges. Long term changes are known to complicate the mechanistic attribution of cause and effect in genetically modified animals. One might thus suggest that the observed enhancement of inactivation of LTCCs could reflect a survival adaptation to increased [Ca2+]i, rather than a mechanistic insight into the functional coupling between LTCCs and RyRs. However, acute intervention buffering Ca2+ (Figure 3B) or depleting the SR Ca2+ store (Figure 3C) restores 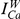 in CPVT cells to WT levels by preventing the shifts in d∞ and f∞ variables. These considerations let us suggest a tonic effect rather than long term adaptation. The alteration in Ca2+ signaling observed in CPVT cells is the increase in RyR Ca2+ leak. Indeed, the ICa evoked [Ca2+]i transients (Figure 1) were not altered. This is consistent with our previous observations and the normal cardiac function described under basal conditions in the CPVT mice, whereas a rate-dependent defect exists [20]. The increased RyR Ca2+ leak seems then to be insufficient to precipitate by itself changes in [Ca2+]i transient under basal stimulation condition, effect which might reflect a sufficient time interval to maintain the physiological SR Ca2+ load, as we observed by caffeine application. Then, we conclude that the tonic reduction in
in CPVT cells to WT levels by preventing the shifts in d∞ and f∞ variables. These considerations let us suggest a tonic effect rather than long term adaptation. The alteration in Ca2+ signaling observed in CPVT cells is the increase in RyR Ca2+ leak. Indeed, the ICa evoked [Ca2+]i transients (Figure 1) were not altered. This is consistent with our previous observations and the normal cardiac function described under basal conditions in the CPVT mice, whereas a rate-dependent defect exists [20]. The increased RyR Ca2+ leak seems then to be insufficient to precipitate by itself changes in [Ca2+]i transient under basal stimulation condition, effect which might reflect a sufficient time interval to maintain the physiological SR Ca2+ load, as we observed by caffeine application. Then, we conclude that the tonic reduction in 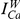 is mediated through local changes of [Ca2+]i in the restricted space where LTCCs and RyRs are located in the dyadic junctions, a mechanism paralleling the local and global [Ca2+]i sensing for the CDI of LTCCs [6], [7], [8]. This interpretation is further supported by the use of Ryanodol, which mimics RyR Ca2+ leak [27], although we cannot exclude a direct effect of this ryanoid on LTCCs.
is mediated through local changes of [Ca2+]i in the restricted space where LTCCs and RyRs are located in the dyadic junctions, a mechanism paralleling the local and global [Ca2+]i sensing for the CDI of LTCCs [6], [7], [8]. This interpretation is further supported by the use of Ryanodol, which mimics RyR Ca2+ leak [27], although we cannot exclude a direct effect of this ryanoid on LTCCs.
A new paradigm of CaM effect on LTCC gating
Our results indicate that RyR Ca2+ leak influence gating properties of the LTCCs. One intriguing but remarkable aspect of our findings is that the changes in the voltage-dependence of LTCC activation and inactivation were not paralleled by changes in the kinetic properties of ICa (Figure 2A & B), similar to that observed by others [33]. Whereas this might indicate that recovery from the inactivated state was impeded, this interpretation will be difficult to reconcile with the maintenance of ICa peak density at more depolarized voltages (Figure 1D). This emphases the complex nature of ICa inactivation mechanisms, and thus might not reflect the same process. The absence of influences on ICa decay suggested that discrete Ca2+ control is unlikely on LTTC open state, whereas shifted steady-state inactivation suggested a stronger effect on inactivated state than on rested state. However, the conformational distinct Ca2+ channel populations (resting, open and inactivated states) are closely interrelated. We reasoned on the possible crosstalk between the CDI and VDI gating of the LTCCs, such that a change in the stability of the open state is predicted to affect both VDI and CDI gating [34]. In this way, we suspected that the RyR Ca2+ leak induced enhancement of inactivation might induce the rightward shift in the activation. In fact, conformational changes caused by Ca2+ binding to the resident CaM, the primary initiatory event for CDI [5], [6], [7], [8], may allosterically reduce activation gating [10] and functionally link the VDI machinery [9], [35]. We observed that CaM antagonists had no effect on CPVT but reduced 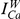 in WT. These results echo the shift in ICa availability curve to more negative potential with calmidozolium, another CaM antagonist [36] and are consistent with previous results in recombinant systems. Similarly to our results, splice variants or mutants of the LTCC IQ motif for CaM binding show a rightward shift in activation and a leftward shift in inactivation [37], [38], whereas CaM overexpression shifts the activation to more negative potential [39]. This prompt us to suggest that the RyR Ca2+ leak-dependent reduced
in WT. These results echo the shift in ICa availability curve to more negative potential with calmidozolium, another CaM antagonist [36] and are consistent with previous results in recombinant systems. Similarly to our results, splice variants or mutants of the LTCC IQ motif for CaM binding show a rightward shift in activation and a leftward shift in inactivation [37], [38], whereas CaM overexpression shifts the activation to more negative potential [39]. This prompt us to suggest that the RyR Ca2+ leak-dependent reduced 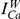 inversely related to CaM is due to an intrinsic effect on LTCC. Besides its role as a signal transduction molecule, CaM also functions as a ubiquitous endogenous fast Ca2+ buffer, an effect frequently overlooked [40]. We therefore propose that Ca2+ buffer capacity of CaM normally prevents Ca2+ to access to other regulator sites, since free CaM is locally enriched in the vicinity of the channels [41]. CaM antagonists in WT cells or increased RyR Ca2+ leak in CPVT cells outdo this CaM effect, allowing Ca2+ feedback to other Ca2+ sensor domains involved in CDI [7], [24], [35].
inversely related to CaM is due to an intrinsic effect on LTCC. Besides its role as a signal transduction molecule, CaM also functions as a ubiquitous endogenous fast Ca2+ buffer, an effect frequently overlooked [40]. We therefore propose that Ca2+ buffer capacity of CaM normally prevents Ca2+ to access to other regulator sites, since free CaM is locally enriched in the vicinity of the channels [41]. CaM antagonists in WT cells or increased RyR Ca2+ leak in CPVT cells outdo this CaM effect, allowing Ca2+ feedback to other Ca2+ sensor domains involved in CDI [7], [24], [35].
Pathophysiological perspectives
Taken together, our study identifies a new Ca2+ regulatory mechanism acting as a powerful switch that determines LTCC gating by interfering with CaM modulation. This effect might serve as a compensatory response to counteract excessive SR Ca2+ release and SR store overload [42], and thus participates to the SR Ca2+ load decrease observed at high stimulation frequencies [20]. One might speculate on a yin-yang effect of the diastolic Ca2+ leakage on trigger activities for fatal acquired or genetic cardiac arrhythmias and sudden death. Elevation of diastolic [Ca2+]i through increased Ca2+ spark frequency is regarded as an arrhythmogenic mechanism: Ca2+ sparks are believed to participate as crucial events in the initiation and propagation of Ca2+ waves in cardiomyocytes and the elimination of cytosolic Ca2+ via the NCX generates a depolarizing current, which can give rise to delayed after depolarizations (DADs) [4]. Conversely, the 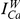 has a central role in arrhythmogenesis in the setting of action potential (AP) prolongation because it can reverse repolarization to create fluctuation of membrane voltage during the repolarization phase of the AP, or early after depolarizations (EADs) [26]. EADs may give rise to salvos of premature APs leading to after contractions through a subsequent secondary SR Ca2+ release, triggered activity and promote reentrant arrhythmias such as Torsades de pointes [43]. Thus, diastolic Ca2+ leak would promote DADs [21], while would be protective from EADs by reducing
has a central role in arrhythmogenesis in the setting of action potential (AP) prolongation because it can reverse repolarization to create fluctuation of membrane voltage during the repolarization phase of the AP, or early after depolarizations (EADs) [26]. EADs may give rise to salvos of premature APs leading to after contractions through a subsequent secondary SR Ca2+ release, triggered activity and promote reentrant arrhythmias such as Torsades de pointes [43]. Thus, diastolic Ca2+ leak would promote DADs [21], while would be protective from EADs by reducing 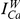 . Our results indeed echo a prevention of EAD induced Torsades de pointes by W7 through a decrease of
. Our results indeed echo a prevention of EAD induced Torsades de pointes by W7 through a decrease of 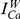 [33], [44].
[33], [44].
Methods
Experiments were performed on male and female heterozygous RyR2R4497C mice (CPVT) and their WT littermates (F3 to F5 generation), as previously described [20], [22], in accordance to the ethical principles laid down by the French (Ministry of Agriculture) and ECC directive 96/609/EEC and was approved by the Comité Régional d' Ethique sur l'expérimentation animale of Languedoc-Roussillon on the Use and Care of Animals. All persons who participated in the experiments had the training and authorization to do so (authorization B34-172-16 for animal facility manager).
Mice
The generation of RyRR4496C knock-in mice was previously described [45]. Mice were age-matched littermates (between 4 to 6 months old and weighing 22–25 g) maintained on a C57BL/6 background after >3 backcrosses to C57BL/6.
Cell isolation
Isolated ventricular myocytes from knock-in mouse-model carrier of the RyR R4496C mutation (CPVT) and their gender-matched littermates wild-type (WT) were prepared using an enzymatic perfusion method. Animals were treated with heparin (1000 units kg-1) and anaesthetized with Na+ pentobarbital (50 mg/kg) administered intraperitoneally. The heart was excised rapidly via a thoracotomy and the pericardium was removed. The heart was placed in ice-cold (0 °C) oxygenated Tyrode solution containing (mmol/L): NaCl 130, NaH2PO4 0.4, NaHCO3 5.8, MgCl2 0.5, KCl 5.4, glucose 22, Hepes 25 and insulin 10−3 (titrated to pH 7.4 with NaOH). The aorta was cannulated above the aortic valve and was perfused by gravity (70 cm column height) with warm (37 °C), preoxygenated Tyrode solution supplemented with 0.1 mmol/L EGTA for 2 min. Enzyme solution containing 1 g/L collagenase Type II (Worthington) in Tyrode solution supplemented with 0.1 mmol/L CaCl2 was then perfused until the aortic valve was digested (attested by the increased outflow of perfusate). The heart was transferred to a Petri dish containing enzyme solution supplemented with 2 g/L bovine serum albumin (BSA) and gently shaken for 2–3 min at 37 °C to disperse individual myocytes. The resulting cell suspension was filtered through a 250 µm nylon mesh and centrifuged for 3 min at 20 g. The cell pellet was suspended in Tyrode solution supplemented with 0.5 mmol/L CaCl2 and 2 g/L BSA and was centrifuged again at the same speed. Finally, the cell pellet was suspended in storage solution comprising Tyrode solution supplemented with 1 mmol/L CaCl2 and 2 g/L BSA.
Patch-clamp and fluorescence measurements
Whole-cell currents were monitored with an Axopatch 200A patch-clamp amplifier. Capacitance compensation was optimized and series resistance was compensated by 40–80%. Ca2+ images were simultaneously acquired with a confocal microscope (MetaZeiss LSM510, objective oil immersion x40, numerical aperture 1.2) in line-scan mode (1.5 msec/line). ICa and fluorescence signals were simultaneously digitized (Digidata 1200, Axon Instruments) and acquired at sampling rate of 100 µsec using pClamp 8.1. During experiments, cells were superfused with an external solution containing (in mmol/L) 140 NaCl, 0.5 MgCl2, 5 CsCl, 1.8 CaCl2, 5.5 glucose, 5 Hepes (pH 7.4), while the patch pipette was filled with a solution containing (in mmol/L) 130 CsCl, 1 MgCl2, 1 NaH2PO4, 3.6 Na2 phosphocreatine, 5 MgATP, 10 HEPES and 0.05 Fluo-3 pentapotassium salt (Molecular Probes); pH 7.2. ICa and SR Ca2+ releases were elicited by 100 msec voltage steps in 10 mV increments from −50 to +60 mV, every 10 seconds. Prior to it, to allow steady-state SR load a voltage protocol including 4 voltage steps (150 msec) from −80 to 0 mV was apply and voltage-gated Na+ channels were inactivated by a 500-msec ramp from −80 to −42 mV [22].
The activation kinetic of ICa was measured for every depolarizing step as the time from the onset of the voltage step to the peak of current.
The time course of inactivation of ICa was determined by analysis of the decay phase of current traces in response to voltage steps. Best fits were obtained with an equation including a sum of two exponentials plus a constant expressed as A fastexp(-t/τfast)+A slowexp(-t/τslow)+A 0, where τ and A are the time constant and the initial amplitude of the two components subscripted fast and slow, respectively, and A 0 is the amplitude of the time-independent component.
Isochronic inactivation were performed with a double pulse protocol, in which a conditioning prepulse of variable amplitude (in the −50 to +60 mV range, from −80 mV) and 250 msec in duration (long enough to produce complete inactivation at each potential) was followed by a test pulse to 0 mV (selected based on the voltage at which peak ICa was maximum) and 100 msec in duration.
Fluo-3 fluorescence was excited with the 488-nm line of an argon ion laser. Emitted fluorescence was measured at wavelengths over 515 nm. Image acquisition was made in the line-scan mode. A single myocyte was scanned repetitively along a line parallel to the longitudinal cell axis. Image processing and analysis were performed using IDL software (Research Systems), as previously described [22], [23]. Briefly, each image was background-subtracted. The fluorescence transient was obtained by averaging the fluorescence values in a 1.4-µm frame over time. Amplitude was measured as the maximum value of F/F0, where F is the fluorescence signal and F0 is the basal fluorescence (measured as the average of the 50 lowest values on the fluorescence transient).
For SR Ca2+ load estimation, myocytes were previously stimulated (4 voltage 150 msec steps from −80 to 0 mV) for 1 min, then 10 mmol/L caffeine was added, the fluorescence image was recorded by confocal microscopy and the associated NCX current was recorded by patch clampand integrated to estimate the amount of Ca2+ released by the SR [23].
Spontaneous Ca2+ sparks (Figure 3A) were imaged on intact Fluo-3AM loaded cells superfused with Tyrode solution (in mmol/L: NaCl 140, MgCl2 1.1, CaCl2 1.8, KCl 4, glucose 10 and Hepes 10; pH 7.4).
Drugs
BAPTA and W7 were purchased from Calbiochem; CALP2 and Ryanodine were from Tocris Bioscience; and other chemical products were from Sigma. Ryanodol was generated as previously described [27].
Western blotting
Mouse heart homogenates were prepared with homogenization buffer (in mmol/L: sucrose 300, sodium-fluoride 20, HEPES 20, Aprotinin 5.2 10−4, Benzamidine 10−2, Leupeptin, 12 10∼3; PMSF 0.1, 0.5% sodium desoxicholate, 0.1% SDS, pH 7.2) using a Potter-Elvehjem and spun at 2,000 g for 10 min. Membrane fractions were isolated by ultracentrifugation at 40 000 g for 30 minutes at 4°C [21]. Protein concentration was assessed by the Bradford method. 25 µg of protein were fractionated on gradient (8 to 20%) SDS-PAGE gels, transferred onto PVDF membranes (30 min in the semidry transfer chamber at 15 V using the 0.45 µm Amersham Hybond-P membrane, GE Healthcare Biosciences, Waukesha, WI, USA) and probed with anti-calmodulin monoclonal antibody (1∶500, Thermo Scientific, Waltham, MA, USA) or ant-CaV1.2 policlonal antibody (1∶1000, Millipore Co, Billerica, MA, USA), in TBS buffer (in mmol/L: Tris-HCl 50, NaCl 150, +0.1% v/v Tween 20, pH 7.4) with 1% w/v non-fat dried milk powder. Membranes were blocked overnight with 5% w/v non-fat dried milk powder in TBS buffer before primary antibody addition. Membranes were incubated 1 h with corresponding secondary peroxidase-conjugate goat antiserum (diluted 1∶5,000 in TBS, EMD Chemicals Inc, Gibbstown, NJ, USA). Signals were developed by chemiluminiscence (Supersignal West Pico Chemiluminescent substrate, Thermo Scientific, Waltham, MA, USA). The relative amount of protein on the blots was determined by densitometry using KodakID Software (v. 3.635, Molecular Imaging Systems, New Haven CT, USA). Actin signals were detected in the same blots with anti-actin serum (1∶20000, Sigma-Aldrich, Inc. México) and used as loading controls.
Statistical Analysis
Data are presented as means ± SEM and compared using t test or an unequal variance t statistic (Welch test), when appropriated, with the PAST program (http://folk.uio.no/ohammer/past/). Differences with values of P<0.05 were considered significant.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This research program was funded by Inserm, a grant from the Agence National de la Recherche (ANR-09-GENO-034) and a grant from the European Union (FPG, Life Science Genomics and Biotechnology for Health, contract CT 2005 N°018802, CONTICA). MF-V was funded by Ministerio de Educacion y Ciencia of Spain, AR by Conacyt (Project N°80960), PN by Fondation de la Recherche Medicale, and Telethon grants N°GGP04066 and GGP06007 and FIRB RBNE01XMP4_006, RBLA035A4X_002 from the Ministero dell' Università e della Ricerca Scientifica e Tecnologica (to SGP and CN). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 2.Andronache Z, Hamilton SL, Dirksen RT, Melzer W. A retrograde signal from RyR1 alters DHP receptor inactivation and limits window Ca2+ release in muscle fibers of Y522S RyR1 knock-in mice. Proc Natl Acad Sci U S A. 2009;106:4531–4536. doi: 10.1073/pnas.0812661106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sham JS, Cleemann L, Morad M. Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci U S A. 1995;92:121–125. doi: 10.1073/pnas.92.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
- 5.Budde T, Meuth S, Pape HC. Calcium-dependent inactivation of neuronal calcium channels. Nat Rev Neurosci. 2002;3:873–883. doi: 10.1038/nrn959. [DOI] [PubMed] [Google Scholar]
- 6.Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, et al. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- 7.Dick IE, Tadross MR, Liang H, Tay LH, Yang W, et al. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–834. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tadross MR, Dick IE, Yue DT. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell. 2008;133:1228–1240. doi: 10.1016/j.cell.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Findeisen F, Minor DL., Jr Disruption of the IS6-AID linker affects voltage-gated calcium channel inactivation and facilitation. J Gen Physiol. 2009;133:327–343. doi: 10.1085/jgp.200810143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tadross MR, Ben Johny M, Yue DT. Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels. J Gen Physiol. 2010;135:197–215. doi: 10.1085/jgp.200910308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brehm P, Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 1978;202:1203–1206. doi: 10.1126/science.103199. [DOI] [PubMed] [Google Scholar]
- 12.Adachi-Akahane S, Cleemann L, Morad M. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. J Gen Physiol. 1996;108:435–454. doi: 10.1085/jgp.108.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sipido KR, Callewaert G, Carmeliet E. Inhibition and rapid recovery of Ca2+ current during Ca2+ release from sarcoplasmic reticulum in guinea pig ventricular myocytes. Circ Res. 1995;76:102–109. doi: 10.1161/01.res.76.1.102. [DOI] [PubMed] [Google Scholar]
- 14.Grantham CJ, Cannell MB. Ca2+ influx during the cardiac action potential in guinea pig ventricular myocytes. Circ Res. 1996;79:194–200. doi: 10.1161/01.res.79.2.194. [DOI] [PubMed] [Google Scholar]
- 15.Puglisi JL, Yuan W, Bassani JW, Bers DM. Ca(2+) influx through Ca(2+) channels in rabbit ventricular myocytes during action potential clamp: influence of temperature. Circ Res. 1999;85:e7–e16. doi: 10.1161/01.res.85.6.e7. [DOI] [PubMed] [Google Scholar]
- 16.Sun H, Leblanc N, Nattel S. Mechanisms of inactivation of L-type calcium channels in human atrial myocytes. Am J Physiol. 1997;272:H1625–1635. doi: 10.1152/ajpheart.1997.272.4.H1625. [DOI] [PubMed] [Google Scholar]
- 17.Sham JS. Ca2+ release-induced inactivation of Ca2+ current in rat ventricular myocytes: evidence for local Ca2+ signalling. J Physiol. 1997;500(Pt 2):285–295. doi: 10.1113/jphysiol.1997.sp022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci U S A. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Velasco M, Rueda A, Rizzi N, Benitah JP, Colombi B, et al. Increased Ca2+ sensitivity of the ryanodine receptor mutant RyR2R4496C underlies catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2009;104:201–209, 212. doi: 10.1161/CIRCRESAHA.108.177493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez AM, Rueda A, Sainte-Marie Y, Pereira L, Zissimopoulos S, et al. Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation. 2009;119:2179–2187. doi: 10.1161/CIRCULATIONAHA.108.805804. [DOI] [PubMed] [Google Scholar]
- 22.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 23.Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, et al. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006;55:608–615. doi: 10.2337/diabetes.55.03.06.db05-1284. [DOI] [PubMed] [Google Scholar]
- 24.Isaev D, Solt K, Gurtovaya O, Reeves JP, Shirokov R. Modulation of the voltage sensor of L-type Ca2+ channels by intracellular Ca2+. J Gen Physiol. 2004;123:555–571. doi: 10.1085/jgp.200308876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benitah JP, Vassort G. Aldosterone upregulates Ca(2+) current in adult rat cardiomyocytes. Circ Res. 1999;85:1139–1145. doi: 10.1161/01.res.85.12.1139. [DOI] [PubMed] [Google Scholar]
- 26.Hirano Y, Moscucci A, January CT. Direct measurement of L-type Ca2+ window current in heart cells. Circ Res. 1992;70:445–455. doi: 10.1161/01.res.70.3.445. [DOI] [PubMed] [Google Scholar]
- 27.Ramos-Franco J, Gomez AM, Nani A, Liu Y, Copello JA, et al. Ryanodol action on calcium sparks in ventricular myocytes. Pflugers Arch. 2010;460:767–776. doi: 10.1007/s00424-010-0839-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hidaka H, Kobayashi R. Pharmacology of protein kinase inhibitors. Annu Rev Pharmacol Toxicol. 1992;32:377–397. doi: 10.1146/annurev.pa.32.040192.002113. [DOI] [PubMed] [Google Scholar]
- 29.Villain M, Jackson PL, Manion MK, Dong WJ, Su Z, et al. De novo design of peptides targeted to the EF hands of calmodulin. J Biol Chem. 2000;275:2676–2685. doi: 10.1074/jbc.275.4.2676. [DOI] [PubMed] [Google Scholar]
- 30.Dirksen RT. Bi-directional coupling between dihydropyridine receptors and ryanodine receptors. Front Biosci. 2002;7:d659–670. doi: 10.2741/A802. [DOI] [PubMed] [Google Scholar]
- 31.Cohen NM, Lederer WJ. Changes in the calcium current of rat heart ventricular myocytes during development. J Physiol. 1988;406:115–146. doi: 10.1113/jphysiol.1988.sp017372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altamirano J, Bers DM. Effect of intracellular Ca2+ and action potential duration on L-type Ca2+ channel inactivation and recovery from inactivation in rabbit cardiac myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H563–573. doi: 10.1152/ajpheart.00469.2006. [DOI] [PubMed] [Google Scholar]
- 33.Qi X, Yeh YH, Chartier D, Xiao L, Tsuji Y, et al. The calcium/calmodulin/kinase system and arrhythmogenic afterdepolarizations in bradycardia-related acquired long-QT syndrome. Circ Arrhythm Electrophysiol. 2009;2:295–304. doi: 10.1161/CIRCEP.108.815654. [DOI] [PubMed] [Google Scholar]
- 34.Yue DT, Backx PH, Imredy JP. Calcium-sensitive inactivation in the gating of single calcium channels. Science. 1990;250:1735–1738. doi: 10.1126/science.2176745. [DOI] [PubMed] [Google Scholar]
- 35.Peterson BZ, Lee JS, Mulle JG, Wang Y, de Leon M, et al. Critical determinants of Ca(2+)-dependent inactivation within an EF-hand motif of L-type Ca(2+) channels. Biophys J. 2000;78:1906–1920. doi: 10.1016/S0006-3495(00)76739-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klockner U, Isenberg G. Calmodulin antagonists depress calcium and potassium currents in ventricular and vascular myocytes. Am J Physiol. 1987;253:H1601–1611. doi: 10.1152/ajpheart.1987.253.6.H1601. [DOI] [PubMed] [Google Scholar]
- 37.Soldatov NM, Oz M, O'Brien KA, Abernethy DR, Morad M. Molecular determinants of L-type Ca2+ channel inactivation. Segment exchange analysis of the carboxyl-terminal cytoplasmic motif encoded by exons 40-42 of the human alpha1C subunit gene. J Biol Chem. 1998;273:957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- 38.Zuhlke RD, Reuter H. Ca2+-sensitive inactivation of L-type Ca2+ channels depends on multiple cytoplasmic amino acid sequences of the alpha1C subunit. Proc Natl Acad Sci U S A. 1998;95:3287–3294. doi: 10.1073/pnas.95.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravindran A, Lao QZ, Harry JB, Abrahimi P, Kobrinsky E, et al. Calmodulin-dependent gating of Ca(v)1.2 calcium channels in the absence of Ca(v)beta subunits. Proc Natl Acad Sci U S A. 2008;105:8154–8159. doi: 10.1073/pnas.0711624105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kubota Y, Putkey JA, Shouval HZ, Waxham MN. IQ-motif proteins influence intracellular free Ca2+ in hippocampal neurons through their interactions with calmodulin. J Neurophysiol. 2008;99:264–276. doi: 10.1152/jn.00876.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mori MX, Erickson MG, Yue DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004;304:432–435. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
- 42.Fabiato A. Simulated calcium current can both cause calcium loading in and trigger calcium release from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:291–320. doi: 10.1085/jgp.85.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 44.Mazur A, Roden DM, Anderson ME. Systemic administration of calmodulin antagonist W-7 or protein kinase A inhibitor H-8 prevents torsade de pointes in rabbits. Circulation. 1999;100:2437–2442. doi: 10.1161/01.cir.100.24.2437. [DOI] [PubMed] [Google Scholar]
- 45.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, et al. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]