

Abstract
We built whole-genome trees based on the presence or absence of particular molecular features, either orthologs or folds, in the genomes of a number of recently sequenced microorganisms. To put these genomic trees into perspective, we compared them to the traditional ribosomal phylogeny and also to trees based on the sequence similarity of individual orthologous proteins. We found that our genomic trees based on the overall occurrence of orthologs did not agree well with the traditional tree. This discrepancy, however, vanished when one restricted the tree to proteins involved in transcription and translation, not including problematic proteins involved in metabolism. Protein folds unite superficially unrelated sequence families and represent a most fundamental molecular unit described by genomes. We found that our genomic occurrence tree based on folds agreed fairly well with the traditional ribosomal phylogeny. Surprisingly, despite this overall agreement, certain classes of folds, particularly all-beta ones, had a somewhat different phylogenetic distribution. We also compared our occurrence trees to whole-genome clusters based on the composition of amino acids and di-nucleotides. Finally, we analyzed some technical aspects of genomic trees—e.g., comparing parsimony versus distance-based approaches and examining the effects of increasing numbers of organisms. Additional information (e.g. clickable trees) is available from http://bioinfo.mbb.yale.edu/genome/trees.
Introduction: Traditional Single-gene Phylogeny
The sequencing of whole genomes of microbial organisms allows us to reassess how we place organisms into groups and relate them to each other in phylogenetic trees. Traditionally, microorganisms have been grouped together into trees based on the sequence similarity of small subunit ribosomal RNA (SSU rRNA) (Woese et al. 1990; Woese 1987). This approach uses a single important and highly conserved gene, which has complex interactions with many other RNAs and proteins, as a basis of phylogeny. Despite its popularity, there are a number of long-standing difficulties with this approach—for example, long-branch attraction, unresolved tree differences, rate variation among sites, and mutational saturation (Lopez et al. 1999; Doolittle 1999; Lawrence 1999; Jain et al. 1999; Gogarten and Olendzenski 1999). Some researchers have even proposed that rRNA itself can be horizontally transferred between organisms (Nomura 1999; Yap 1999).
Many researchers have also tried building trees based on sequence similarity of individual protein families, such as the cytochromes, ATPases, elongation factors, aminoacyl tRNA synthases, beta-tubulins, or RNA polymerases (Makarova et al. 1999; Teichmann and Mitchison 1999a; Tumbula t al. 1999; Lake et al. 1999; Doolittle 1998; Rivera et al. 1998; Ibba et al. 1999, 1997; Edlind et al. 1996; Baldauf et al. 1996; Brown and Doolittle 1995; Andersson et al. 1998; Tomb et al. 1997; Bult et al. 1996; Lake 1994). These studies have often resulted in a wide range of implied phylogenies. The differences from the accepted ribosomal phylogeny are usually attributed to such factors as horizontal transfer or the existence of ambiguous paralogs. However, sometimes they have been used to argue that the rRNA tree is not representative of the true phylogeny. This has been particularly effective when the protein tree is based on a complex and fundamental protein such as RNA polymerase, or when the protein tree is backed up by extensive natural history evidence (Hirt et al. 1999; Edlind et al. 1996).
Whole-genome Trees and the Current Controversy
Because SSU rRNA and other individual gene families each correspond to only a tiny fraction of the genomic material in most microorganisms, focusing exclusively on them ignores the bulk of the genetic information in constructing phylogenetic trees. (In particular, the ∼1.8 kb of SSU rRNA makes up less than 0.2% of most microbial genomes, which are ≧1 Mb.) Now with the advent of completely sequenced genomes, it is possible to build trees that encompass much more of the genetic information in an organism. This has led to a profusion of new approaches toward phylogenetic estimation and a heated controversy about the structure of the fundamental tree of life, which has even been featured in the popular press (Pennisi 1998, 1999; Stevens 1999). On one extreme, some prefer traditional ribosomal trees. On the other extreme, some argue that trees are not really meaningful given the widespread evidence of horizontal transfer in microorganisms and that nets or more general graph structures should be used instead (Hilario and Gogarten 1993). In between, a third perspective maintains that most microorganisms can be arranged into meaningful trees; however, these trees might not always reflect the branching pattern suggested by rRNA.
To contribute to this debate, we build trees considering progressively more and more of the information in a genome and compare them with the traditionally proposed phylogeny. In particular, we are proposing a number of novel trees based on the occurrence of specific features, either folds or orthologs, throughout the whole genome. We call these genomic trees or whole-genome trees. Our approach toward genomic trees, which is schematized in Table 1, is similar to the practice in traditional phylogenetic analysis using the presence or absence of morphologic characteristics or heritable traits such as hair or vertebrae to group organisms(Hennig 1965; Maisey 1986).
Table 1.
Schematization of the Different Levels
| Scale | Levels | Description | |
|---|---|---|---|
| Single-gene | Traditional ribosomal | Nucleotide sequence | Based on the small and large subunit ribosomal rRNA |
| Individual orthologs | Protein sequence | Based on a variety of established orthologous proteins | |
| Partial-proteome (multi-gene) | Ortholog occurrence | Protein function | Based on the occurrence of specific orthologs in the proteome |
| Protein fold occurrence | Protein structure | Based on the occurrence of specific protein folds in the proteome | |
| Entire genome | Amino acid frequency | Proteome composition | Based on frequency of single amino acids in the entire genome |
| Dinucleotide frequency | Genome composition | Based on frequency of pairs of adjacent nucleotides in entire genome | |
This table delineates the three levels on which we compared the microbial genomes. On the single-gene level, both ribosomal RNA and orthologous proteins were used for phylogenetic analysis. These used sequence-based methods for tree construction. The second level uses the presence or absence of orthologous protein groups or protein folds. Because some parts of the genome are assigned neither to a protein fold nor to an ortholog, we call this scale “partial proteome.” This scale is based on both protein function and protein structure. The most encompassing scale uses dinucleotide and amino acid composition as a basis for genome comparison. The entire genome is considered.
Our first set of genomic trees is based on the occurrence of orthologous proteins. This work builds upon recent work done by other researchers clustering genomes based on the occurrence of protein families (Tekaia et al. 1999; Snel et al. 1999; Teichmann and Mitchison 1999a). In particular, Tekaia et al. (1999) developed a methodology for comparing the whole proteome content of one genome against another and using the loss or acquisition of genes to build trees. We add to this work by dividing the proteome into various classes, building trees based on these, and looking at their consistency.
Our second set of genomic trees is based on protein folds. Folds group together a number of protein families that may not share sequence similarity but do share the same essential molecular shape. As each protein fold represents a unique three-dimensional shape used by an organism, folds are ideal characteristics for building phylogenetic trees. To build our fold trees, we used a similar approach to that for the ortholog trees, in this case using the presence or absence of folds in particular genomes for tree construction. Our fold tree work is an extension of our previous investigations (Gerstein 1998a). Related work comparing genomes in terms of the occurrence of protein folds has been done by Wolf et al. (1999), who used somewhat different definitions to cluster organisms based on folds.
In a strict sense, our fold occurrence and ortholog occurrence trees are partial proteomes rather than whole-genome trees, because they are based on considering simultaneously a large, but not complete, portion of the protein-coding regions of genomes. The entire genome cannot be used, because not all of the proteins can be classified as part of orthologous groups or can be assigned to fold families. In the last part of our analysis, we look at trees that are based on information from the entire genome, using amino acid and dinucleotide composition. Although these trees represent an unbiased consideration of all the information in the genome, they condense everything to a simple composition vector, discarding much important detail. Our composition tree analysis follows up on our previous work (Gerstein, 1998b) and extensive work done by Karlin and co-workers (Karlin and Burge 1995; Karlin and Mrazek 1997; Campbell et al. 1999). Finally, it is worth pointing out that a number of additional approaches toward whole-genome trees have been advanced beyond those discussed here. In particular, Gupta (1998) used insertions and deletions along with cell-membrane structures for tree reconstruction.
Note that, whereas the occurrence and composition trees have the advantage over the single-gene trees in that they incorporate more genome information, they are not as clearly associated with an evolutionary mechanism. That is, underlying the ribosomal tree there is a specific biologic mechanism, internal to each organism, generating sequence diversity: the mutation of single base pairs, which happens at a rate roughly proportional to time. However, the way a single organism expands or contracts its repertoire of folds or orthologs cannot be explained simply in terms of individual molecular events. If one explains the acquisition of folds with horizontal transfer, the tree is no longer based on ancestral characteristics evolving internally but on the interaction with other organisms. Therefore, as opposed to true evolutionary phylogenies, whole-genome trees would then more appropriately be described as clusterings.
RESULTS AND DISCUSSION
Genomes Analyzed and Tree Techniques Used
We focus on the first eight microbial genomes to be sequenced, which include representatives from all three domains of life (Table 2). All our trees were built with the standard programs using both maximum-parsimony and distance-based methods (Felsenstein 1993, 1996; Swofford 1998). In general, we found that distance-based methods gave more reasonable results, probably because of the great divergence of the taxa, which has been remarked upon in other contexts (Swofford et al. 1996). Additional information (clickable trees, plots, etc.) is available on the Internet at http://bioinfo.mbb.yale.edu/genome/trees.
Table 2.
The Eight Completely Sequenced Organisms Used in this Study
| Abbrev. | Organism | Phylogeny | Size (Mb) | Proteins | Reference | ||
|---|---|---|---|---|---|---|---|
| Ecol | Escherichia coli | Bacteria | Proteobacteria | gamma subdivision | 4.653 | 4283 | Blattner et al. 1997 |
| Hinf | Haemophilus influenzae | Bacteria | Proteobacteria | gamma subdivision | 1.830 | 1703 | Fleischmann et al. 1995 |
| Hpyl | Helicobacter pylori | Bacteria | Proteobacteria | epsilon subdivision | 1.667 | 1566 | Tomb et al. 1997 |
| Mjan | Methanococcus jannaschii | Archaea | Euryarchaeota | Methanococcales | 1.739 | 1736 | Bult et al. 1996 |
| Mgen | Mycoplasma genitalium | Bacteria | Firmicutes | Bacillus/Clostridium | .58 | 468 | Fraser et al. 1995 |
| Mpne | Mycoplasma pneumoniae | Bacteria | Firmicutes | Bacillus/Clostridium | .816 | 677 | Himmelreich et al. 1996 |
| Scer | Saccharomyces cerevisiae | Eukaryota | Fungi | Ascomycota | 12.068 | 5932 | Goffeau et al. 1997 |
| Syne | Synechocystis sp. | Bacteria | Cyanobacteria | Chroococcales | 3.573 | 3168 | Kaneko et al. 1996 |
This table lists the currently published microbial genomes, discussed in the text, from the TIGR web site (http://www.tigr.org/tdb/mdb/mdb.html). The first column lists the abbreviations that are used for the figures in this paper, corresponding to the genome name in the second column. Column three shows the top three levels of the phylogenetic lineage of the organisms as shown in the NCBI Taxonomy Browser (http://www.ncbi.nlm.nih.gov/Taxonomy/tax.html), which is sufficient to locate the taxa on the traditional tree. The size of the complete genome of the particular organism is shown in the fourth column with the total number of proteins in the organism listed on the next column. The final column lists the original publication citation for each of the genomes.
Ortholog and Fold Assignments
Orthologs were selected from the COGs (clusters of orthologous groups) database (Tatusov et al. 1997; Koonin et al. 1998). This database lists groups of ortholog sequences in eight of the first genomes sequenced based on whether or not they form a mutually consistent sequence of best matches between genomes. This database is in wide use and is accepted as a valid source of functional annotation. It groups orthologs into a hierarchy of functional classes–(e.g. class J, transcription, is part of the “Information Storage and Processing” superclass). The presence or absence of specific proteins for all the COGs for different genomes can be derived from the web site data files.
Folds were assigned to the genome sequences based on a previously described approach (Gerstein 1997, 1998b; Teichmann and Mitchison 1999b; Hegyi and Gerstein 1999). We compared the structure databank (the PDB) against the genome sequences by using both pairwise and multiple-sequence methods and standard thresholds (FASTA and PSI-BLAST, Lipman and Pearson 1985; Pearson 1996; Altschul et al. 1997). We used the SCOP classification to group the domain-level structure matches into different fold families (Murzin et al. 1995). The SCOP (structural classification of proteins) classification is assembled based on expert manual judgement, and we have augmented it with our automatically derived protein-structural alignments (Gerstein and Levitt 1998). Like the COG scheme, the SCOP classification is in wide use and accepted as a reliable classification of a protein's fold.
Single-gene Trees, a Reference Point
Traditional Ribosomal RNA Trees
As a reference point, we started our survey by constructing a traditional phylogenetic tree based on the small subunit ribosomal RNA. This established a basis of comparison for the trees generated in this study. The ribosomal tree in Figure 1A resulted in three general clusters corresponding to Archaea, Bacteria, and Eukaryota. The construction of a tree based on the large subunit ribosomal RNA in Figure 1B showed some variation in the topology of the tree.
Figure 1.

Representative single-gene trees. (A) The traditional small subunit ribosomal phylogenetic tree. This is a tree of eight completely sequenced representative organisms constructed with the SSU rRNA. Trees could be constructed using data from two different sources: the Ribosomal Database Project (RDP, http://www.cme.msu.edu/RDP, Maidak et al. 1999) and the rRNA WWW Server (http://www-rrna.uia.ac.be, Van de Peer et al. 1999). Although a tree can be abstracted from the RDP, the tree cannot contain both prokaryotes and eukaryotes. Instead, we took sequences from the RDP and the rRNA WWW server and aligned them with Clustal (Thompson et al. 1997). Phylip and PAUP were used to construct trees from the aligned sequences using distance and parsimony methods. There was little variation in the resulting trees displayed using TreeView (Page 1996), which was used to show all the trees used in this survey. The PAUP distance-based tree is shown. (B) The large subunit ribosomal tree. Another common method of building phylogenetic trees is the use of the large subunit rRNA (De Rijk et al. 1999). Because of the lack of large subunit rRNA information from the RDP, the sequences were downloaded from the rRNA WWW Server. The same method of tree construction was used as in A. The tree shown in B is the PAUP distance-based tree. Because of the large divergence of the species, the topology of the tree varied slightly when compared to the SSU ribosomal tree in A. The placement of Synechocystis was slightly different, as it is placed closer to the eukaryote and Archeae in the large subunit tree. This was relatively less significant when considering the branch lengths of the tree in A . (C , D) Representative trees based on sequence similarity of orthologs. The sequences of proteins for the different organisms were obtained from the COGs web site (http://www.ncbi.nlm.nih.gov/COG, Tatusov et al. 1999). Clusters of orthologous groups were chosen that had one protein for each organism in the group. There were eight such COGs with representatives from four different classes. Distance-based trees and parsimony trees were both constructed for each of the orthologous groups. There was great variation in the resulting trees. The tree, which had the highest similarity to the traditional ribosomal tree, is shown in C. In fact, the distance-based tree based on the 30S ribosomal protein S3 (COG92, Class J) in C is exactly the same in topology to the traditional tree. This is not surprising because we expect a ribosomal protein tree to be similar to ribosomal rRNA trees because of their interaction and conservation. For the bootstrap values, all bootstrap replicates grouped E. coli with H. influenzaee, S. cerevisiae with M. jannaschii, and M. genitalium with M. pneumoniae. In all, the conserved topology coupled with high bootstrap values shows that phylogenetic trees with even a single protein can exhibit very high fidelity to the traditional ribosomal tree. Besides trees with high similarity to the traditional tree as in C, there were trees that varied significantly from the traditional ribosomal tree. Part D shows a distance-based tree based on the metabolic enzyme triosephosphate isomerase (TIM). In general, there are a lot of differences between this tree and the traditional tree. M. jannaschii is grouped with M. genitalium and M. pneumoniae; M. jannaschii is not grouped with S. cerevisiae at all. The connectivity of S. cerevisiae and H. pylori is also different from the traditional tree. The low bootstrap values of 59% and 40% suggest that within the sequence there is great variation and the tree is generated with lower certainty. In general, there were a wide variety of trees produced using sequence similarity of orthologous proteins.
Single-gene Ortholog Trees
Next we examined the trees based on individual orthologous proteins chosen from the COGs database. Again this was to establish a reference for comparison and also to see the variation within single-gene trees. We focused on orthologous groups for which each of our eight organisms had only one representative, to minimize the possible effects of unrecognized paralogy (Teichmann and Mitchison 1999b). As shown in Figure 1, as has been remarked on in previous studies, the clusterings exhibit great variation depending on the protein chosen. Furthermore, many of the trees have only marginal bootstrap values if we consider 95% to be the cutoff for reasonable confidence (Efron et al. 1996). In Figure 1C, we show a representative tree that agrees well with the traditional ribosomal phylogeny. It is based on the 30S ribosomal protein S3 (COG 92, Class J). It has relatively good bootstrap values compared to other single-gene trees that we constructed and compared to the findings of others (Teichmann and Mitchison 1999a), but not all of the values are above 95%. Figure 1D shows an example of a tree that differs significantly from the traditional phylogeny, that of triosephosphate isomerase (TIM, COG 149, Class C). Perhaps predictably, we found that trees that agreed well with the traditional ribosomal phylogeny tended to be based on proteins involved in transcription and translation, especially those with extensive RNA interactions. [This latter observation is also true for some proteins, such as the signal recognition particle (SRP) GTPase, which are not involved in transcription or translation.] In contrast, soluble enzymes, such as TIM, tended to produce trees with greater variation. These discrepancies provide evidence that trees built on sequence-similarity of individual genes would result in different phylogenies, because different genes have different mutational rates and some are horizontally transferred.
Genomic Occurrence Trees
Now that we have described the single-gene perspective as a reference, we can progress to the focus of our analysis, constructing genomic trees that consider more than the variation of individual genes. These trees were defined in terms of presence or absence of shared characteristics throughout the whole genome. Broadly, these characteristics potentially could be orthologs, homologs, or folds. We focused on orthologs and folds. We used both distance-based and parsimony methods for tree construction. For the distance-based methods, we defined the distance between two organisms with a normalized Hamming distance, which was expressed as the fraction of unshared characters divided by the total number of characters in the genomes—that is (A + B−2S)/(A + B), where A and B are the characters in the first and second genomes, respectively, and S is the number of shared characters between A and B.
We had hoped that parsimony would produce reasonable trees, because this method would automatically propose ancestral organisms that had the intermediate configurations of orthologs or folds. However, we found that, in general, distance-based methods resulted in trees closer to the traditional phylogeny. This may be because of the great divergence of the organisms studied. We give an example of the superiority of distance-based methods in Figure 3, which compares fold trees based on distances and parsimony. Also, because of the divergence of the organisms, a number of our trees may show some evidence of long-branch attraction. This arises when there are differing rates of variation among different sites in a gene, resulting in the clustering of organisms with higher rates of sequence change (Felsenstein 1996). However, we feel long-branch attraction affects the ribosomal tree as much as, if not more than, the whole-genome trees, as evident in its longer relative branch lengths and more star-shaped appearance (i.e., with less well-resolved branching). Furthermore, it is known that distance-based methods are less sensitive to long-branch attraction than parsimony, perhaps suggesting why we found the distance-based trees more reasonable.
Figure 3.

Genomic trees based on the occurrence of folds. (A) Genomic tree based on the overall occurrence of folds in the genomes, generated by a distance-based method. For each of the microbial organisms the presence or absence of folds was marked with 1 or 0, respectively. The folds that were not present in any of the genomes were excluded, because this does not provide any distinguishing information. Similar to the ortholog occurrence, a distance matrix was generated with the Hamming distance. (B) Genomic tree based on the overall occurrence of folds in the genomes, generated by parsimony. Instead of generating a distance matrix, parsimony can be used for tree construction. For this task, PAUP was used and the resulting tree is mostly similar to the distance-based tree. However, the locations of S. cerevisiae and H. pylori are switched. Also, in contrast to the traditional ribosomal tree, S. cerevisiae is placed closer to M. jannaschii, whereas H. pylori is placed with the other bacteria. Therefore, the distance-based method as described in A seems to be better. For all the trees presented here, both distance-based and parsimony trees were generated; in general, as observed in this instance, the distance-based tree is closer to the ribosomal tree. The star shown in the bootstrap value represents a node where the bootstrap consensus tree results in a star decomposition and cannot be resolved. (C, D, E, and F) Distance-based genomic trees based on occurrence of folds in particular fold classes. In this analysis, instead of dividing the COGs into functional classes, the folds are fractionated into classes: all-alpha, all-beta, alpha+beta, and alpha/beta. As seen in the pie chart, the distribution of folds among the different classes is rather equal; each has approximately one quarter of the total. Of the four divisions of folds, the alpha+beta group is most similar to the overall tree, having the exact same topology. It also has the largest number of folds (81, 29% of the total). The all-alpha fold group has 27% (75) of the total folds and has almost the exact topology of the overall tree, except that H. pylori and Synechocystis are grouped together instead of just being close to each other. The alpha/beta group has 24% (68) of the total folds and is also very similar to the overall fold tree. The most surprising tree is that of the all-beta group. This is based on the smallest number of folds, which is 20% (55) of the total folds.
Genomic Tree Derived from Occurrence of Orthologs
Figure 2 shows the trees built in terms of the overall occurrence of ortholog families in the eight genomes. We used the COGs database to determine whether a genome had a particular orthologous group. The overall clustering based on all the orthologs in the COGs database is notably different from the traditional ribosomal tree. The Mycoplasma pneumoniae and Mycoplasma genitalium cluster is placed with the Methanococcus jannaschii, and the generally conserved grouping of Escherichia coli and Haemophilus influenzae does not occur.
Figure 2.

Genomic trees based on the occurrence of orthologs. (A) Distance-based genomic tree based on the overall occurrence of orthologous proteins in the complete genome. One of the alternative methods we used for phylogenetic analysis involved building trees based on the presence or absence of orthologs in the complete genome, using the information from the original COGs web site with eight genomes (http://www.ncbi.nlm.nih.gov/COG, Tatusov et al. 1999). For each of the microbial organisms, the occurrence of proteins in each of the clusters of orthologous groups was tabulated with 1 for present and 0 for absent. With the parsed data, a distance matrix was then calculated using the normalized Hamming distance, as described in the text. The trees were subsequently constructed using the kitsch program in the PHYLIP package, which allowed for easy automation. For the bootstrap values, we used PAUP. The resulting tree shown is a distance-based tree using the information of the occurrence of all the COGs in the genomes. As expected, the M. pneumoniae and M. genitalium are grouped with bootstrap values of 100%. However, interestingly, E. coli and Synechocystis are also clustered with this bootstrap value—a grouping that is not in the traditional tree. Also, M. jannaschii is clustered with M. pneumoniae and M. genitalium with a bootstrap value of 81%. Furthermore, the eukaryote, S. cerevisiae, is placed among the bacteria. (B, C, and D) Ortholog occurrence genomic trees based on a three-way partition of the whole ortholog set. As described in the text, the total COGs were divided into three large subsets, the information, cellular, and metabolic subsets. The pie chart in Figure 3 shows the number of COGs in each group as percentages. The metabolic subset dominated the total group with 362 COGs, approximately half of all the COGs. The information subset has 190 COGs, just above one quarter, while the cellular subset has 132 COGs, just less than a quarter. For each of the subsets, distance-based trees were generated using the same methods described in A. Because of the smaller sizes of these subsets, the bootstrap values were often ill-defined. The largest subset was the metabolic partition shown in B. There was a high correlation between the trees in A and B. Aside from the different placement of H. pylori and S. cerevisiae, the trees are nearly identical, even having similar branch lengths. The second largest partition was the information subset shown in C. Surprisingly, this subset produced a tree almost identical to the traditional ribosomal tree. The only difference is the switch in the placement of H. pylori and Synechocystis. This shows that although using the entire group of COGs may produce trees much different from the traditional tree, using a smaller subset may in fact produce a tree that is closer to the traditional topology. Part D shows the smallest partition, the cellular subset. (E and F) Representative genomic trees of ortholog occurrence based on specific functional classes J and H. Using the functional classes obtained from the COGs web site, the metabolic, information, and cellular partitions were subdivided further, into specific functional classes. For each of the different functional classes, there was a range of trees produced. Two representatives were chosen to show this variety. Class J (translation, ribosomal structure, and biogenesis), which has 108 clusters of orthologous proteins, is a further subdivision of the information subset. It has a tree very similar to the traditional ribosomal tree in Figure 1A. Class H (Coenzyme metabolism), which has 77 clusters of orthologous proteins, is a further subdivision of the metabolism subset. It produced a tree that did not correspond well to the traditional phylogeny.
Subdivision by Functional Class
The COGs database contains three main functional subdivisions: metabolism, information storage and processing, and cellular processes. For convenience, we will refer to these as the metabolic, information, and cellular subdivisions. More than half of the total ortholog groups and half of the signal in the overall tree come from proteins in the metabolic subset. If we remove the metabolic subset, we get a tree much more consistent with the traditional phylogeny. Figure 2C shows a genomic tree based on the occurrence of orthologs just in the information set. Its topology is almost identical to the traditional tree, the only difference being the placement of Synechocystis and Helicobacter pylori, which are reversed. This difference is minor as the divergence between these two organisms is very small even in the traditional tree. One sees, furthermore, that the traditional topology is preserved even when one selects a subset of the information set, namely the orthologs involved in transcription (class J) in Figure 2F.
In contrast to the information-subset tree, the metabolic-subset tree in Figure 2B corresponds closely to the topology of the overall ortholog-occurrence tree, dominating it and giving rise to its nontraditional topology. This unusual tree topology is accentuated even more when we look at a subset of the metabolic proteins corresponding to less essential functions that are less evenly maintained across organisms. This is shown in Figure 2E, which shows a tree with 65 COGs involved in coenzyme metabolism (class H).
The topology of the metabolic subset, in fact, seems skewed by the number of orthologous proteins that each genome in this subset has. The two genomes with the largest number of orthologous proteins in the subset, E. coli (350) and Synechocystis (330), were grouped together. Haemophilus influenzae (264), having the third highest number of clusters of orthologous metabolic proteins, branches off from this cluster, followed by S. cerevisiae (262), H. pylori (226), M. jannaschii (215), and the mycoplasmas (81 and 75). We tried a variety of alternative distance measures to correct for this effect (e.g., by dividing the number of shared orthologous groups over the number of groups only present in one) but were unsuccessful in getting the traditional topology from the metabolic subset.
Thus, our results suggest that the occurrence of proteins associated with transcription and translation is closer to the traditional rRNA phylogeny than that associated with metabolism. These outcomes are reasonable and in consonance with the results that show that trees based on individual proteins involved in metabolism are usually farther from the established phylogeny than those based on proteins involved with transcription (see references above).
Fold Trees
In Figure 3 we show trees based on the occurrence of protein folds throughout the genome. Folds unite protein families that share the same basic architecture but which might not have any appreciable sequence similarity. They provide an ideal type of character to use in the construction of occurrence trees, since it is believed that there are only a very limited number of protein folds (Chothia 1992). The occurrence of a particular fold within the genome represents the organism selecting a particular three-dimensional shape from the overall master parts list found in nature. Fold trees have the advantage over ortholog trees in that the assignment of a particular ORF to a fold can be done fairly automatically and objectively, whereas the assignment of ORFs to various orthologous groups is often more ambiguous and requires considerable manual intervention.
Our overall fold tree is shown in Figure 3A. It has a remarkably similar topology to the traditional ribosomal tree, especially when one considers how radically different the criteria are for building the trees. The tree shown in Figure 3A is built with distance-based methods, using a normalized Hamming distance (the number of different folds between genomes as a fraction of their total). As a contrast, in Figure 3B we show a fold tree built on the basis of parsimony. The parsimony tree clearly differs from the distance-based tree. Although these two trees were still similar, the parsimony tree alternates the position of H. pylori and S. cerevisiae, placing the latter much closer to the E. coli, H. influenzaee, Synechocystis bacterial cluster than to the Archaea, M. jannaschii.
Subdivision by Fold Class
As with the orthologous protein trees, we analyzed the composition of the total tree through various subdivisions. We can subdivide folds into four major structural classes, all-alpha, all-beta, alpha/beta, and alpha+beta (Levitt and Chothia 1976). One might not expect much variation between the classes, since unlike the orthologous proteins whose subsets had definite functional and evolutionary implications, the fold class subdivisions are not clearly related to any of these aspects. However, the trees are, in fact, different among the fold structural class subdivisions, as seen in Figure 3.
While the alpha+beta and all-alpha subdivisions look most similar to the overall fold tree and the traditional phylogeny, the all-beta class has an unusual clustering. Specifically, E. coli is not placed with H. influenzae, and S. cerevisiae is placed deep within the bacterial cluster. Perhaps this reflects the less even distribution of all-beta proteins and their selective proliferation in various taxa. It has been suggested before, based on bulk structure prediction, that there seems to be a different distribution of all-beta proteins in eukaryotes than prokaryotes (Gerstein 1997).
Genome Composition Trees
Because we cannot assign every single ORF in a genome to a known fold or ortholog family, the genomic occurrence trees based on these characters are in a strict sense partial proteome trees. Consequently, they suffer from potential biases because the orthologs or folds that are selected may not represent a truly random sampling of proteins in the genomes. One simple way of building a tree that takes into account all the information within the genome in unbiased fashion is using the overall nucleotide composition. To complete our analysis, we built trees based on dinucleotide and amino acid (essentially trinucleotide) composition. We constructed vectors representing the normalized composition of the genome, denoted by f, and then took the difference of these vectors to define our tree. We thus used the following relation for the distance between genomes i and j:
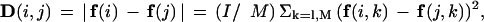 |
where f(i,k) represents the composition and the sum over k runs from 1 to M=16 or M=20, depending on whether the tree is for dinucleotide composition or amino acid. Clearly, in the computation of composition, while we are broadly considering the entire genome, we are discarding much information by reducing the entire genome into a single composition vector.
Dinucleotide Composition
Dinucleotide composition results in a tree that is very different from the traditional tree. As seen in Figure 4A, the clustering did not show any of the patterns observed in most of the trees in this paper. Even M. pneumoniae and M. genitalium were not clustered together.
Figure 4.

Trees based on overall composition. (A) Dinucleotide composition tree. We counted the relative frequency of the dinucleotides for the complete genomes of the eight organisms. Distance between two species of dinucleotides is the distance between the 16-dimensional vectors, with each axis representing a dinucleotide pair. PAUP then generated trees using the distance matrix. Figure 4A shows the resulting dinucleotide composition tree, which has almost no resemblance to the traditional ribosomal tree in Figure 1A. Even the M. genitalium and M. pneumoniae clustering, which is conserved throughout the survey, does not appear. This suggests that the dinucleotide method is not very accurate in the production of phylogenetic trees. Although it encompasses entire genomes, it reduces them to a 16-dimensional vector, losing much information. (B) Amino acid composition tree. This shows the tree generated from amino acid composition. Again, the relative frequencies of the amino acids were counted and a similar distance measure as in A was used. The distances between the genomes are calculated using 20-dimensional vectors, one for each amino acid. The resulting distance matrices were used to generate trees using PAUP. Interestingly, although this tree is still significantly different from the traditional tree in Figure 1A, it indeed is a great improvement upon the dinucleotide composition tree. Relatively, the organisms are closer in position to the traditional tree.
Amino Acid Composition
The amino acid composition tree contained great similarity to other trees presented in this survey. The mycoplasmas are clustered together, as are E. coli, Synechocystis, and H. influenzae; M. jannaschii is far from the main clusters. As can be seen in Figure 4B, simple amino acid composition, for even the entire sequence, cannot generate anything like the traditional tree; it is noteworthy that by changing from two nucleotides in the dinucleotide analysis to three in the amino acid analysis (and taking into consideration reading frames) much more information is revealed.
CONCLUSION
We built trees grouping organisms based on the overall occurrence of molecular features throughout their genomes. Most broadly, these characteristics could be orthologs, homologs, or folds. We focused on orthologs and folds. For folds we found that the overall genomic tree agreed surprisingly well with the traditional ribosomal tree. However, the distribution of all-beta folds was somewhat different. For orthologs we found that an occurrence tree based just on proteins involved in transcription and translation also agreed quite well with the traditional phylogeny. However, one built based on metabolic proteins had a rather skewed topology, and as the metabolic subset comprised most of orthologs, the overall ortholog tree also shared this appearance. This implies that adding more features does not necessarily increase the accuracy of the tree, an observation that, of course, has been made numerous times in relation to traditional phylogeny (Swofford et al. 1996). We compared our occurrence trees with many other possible trees, ranging from single-gene trees based on sequence similarity of individual orthologous proteins to entire-genome composition trees. We found that many of these alternate trees had rather unusual topologies, providing a good context for appreciating how remarkable was the agreement between whole-genome occurrence trees, particularly the fold tree, and the traditional tree.
Prospects: Trees Based on More Genomes
The number of genomes being sequenced is increasing at a fast rate and obviously the eight-genome scale of analysis presented here will be soon out of date. However, the real question is whether our approach of comparing genomes in terms of the occurrence of orthologs or folds will scale with more and more organisms. We believe it will. Recently, we have built whole-genome occurrence trees based on more than eight genomes and found that they had quite a reasonable topology. As an illustration, in Figure 5, we show a fold tree based on 20 genomes.
Figure 5.

Prospects for the future. The figure shows a 20-genome tree based on the occurrence of folds. This is similar to Figure 3A. The unit in the SCOP classification that was used was the structural superfamily rather than the fold. For eight genome occurrence trees there is no difference between one made at the fold or superfamily level. However, for the 20-genome tree this distinction matters. The additional species names in the 20-genome tree are: Aaeo (Aquifex aeolicus), Aful (Archaeoglobus fulgidus), Bsub (Bacillus subtilis), Bbur (Borrelia burgdorferi), Cpne (Chlamydia pneumoniae), Ctra (Chlamydia trachomatis), Ecol (Escherichia coli), Hinf (Haemophilus influenzae), Hpyl (Helicobacter pylori), Mthe (Methanobacterium thermoautotrophicum), Mjan (Methanococcus jannaschii), Mtub (Mycobacterium tuberculosis), Mgen (Mycoplasma genitalium), Mpne (Mycoplasma pneumoniae), Phor (Pyrococcus horikoshii), Rpro (Rickettsia prowazekii), Scer (Saccharomyces cerevisiae), Syne (Synechocystis sp.), and Tpal (Treponema pallidum).
Acknowledgments
We thank H. Hegyi for help with the fold assigments and G. Naylor for discussions on phylogeny. M.G. thanks the NIH and the Donaghue Foundation for support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL Mark.Gerstein@yale.edu; FAX (203) 432-5175.
REFERENCES
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, Naslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- Baldauf SL, Palmer JD, Doolittle WF. The root of the universal tree and the origin of eukaryotes based on elongation factor phylogeny. Proc Natl Acad Sci USA. 1996;93:7749–7754. doi: 10.1073/pnas.93.15.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Brown JR, Doolittle WF. Root of the universal tree of life based on ancient aminoacyl-tRNA synthetase gene duplications. Proc Natl Acad Sci USA. 1995;92:2441–2445. doi: 10.1073/pnas.92.7.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- Campbell A, Mrazek J, Karlin S. Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc Natl Acad Sci USA. 1999;96:9184–9189. doi: 10.1073/pnas.96.16.9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C. Proteins—1000 families for the molecular biologist. Nature. 1992;357:543–544. doi: 10.1038/357543a0. [DOI] [PubMed] [Google Scholar]
- De Rijk P, Robbrecht E, de Hoog S, Caers A, Van de Peer Y, De Wachter R. Database on the structure of large subunit ribosomal RNA. Nucleic Acids Res. 1999;27:174–178. doi: 10.1093/nar/27.1.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle RF. Microbial genomes opened up. Nature. 1998;392:339–342. doi: 10.1038/32789. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree [see comments] Science. 1999;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Edlind TD, Li J, Visvesvara GS, Vodkin MH, McLaughlin GL, Katiyar SK. Phylogenetic analysis of beta-tubulin sequences from amitochondrial protozoa. Mol Phylogenet Evol. 1996;5:359–367. doi: 10.1006/mpev.1996.0031. [DOI] [PubMed] [Google Scholar]
- Efron B, Halloran E, Holmes S. Bootstrap confidence levels for phylogenetic trees. Proc Natl Acad Sci USA. 1996;96:13,429–13,434. [Google Scholar]
- Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package) version 3.5c. Distributed by the author. Department of Genetics, University of Washington, Seattle.
- ————— Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Meth Enzymol. 1996;266:418–427. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick J, et al. Whole-genome random sequencing and assembly of haemophilus influenzae rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Gerstein M, Levitt M. Comprehensive assessment of automatic structural alignment against a manual standard, the Scop classification of proteins. Protein Sci. 1998;7:445–456. doi: 10.1002/pro.5560070226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M. A structural census of genomes: Comparing eukaryotic, bacterial and Archaeal genomes in terms of protein structure. J Mol Biol. 1997;274:562–576. doi: 10.1006/jmbi.1997.1412. [DOI] [PubMed] [Google Scholar]
- Gerstein M. How representative are the known structures of the proteins in a complete genome? A comprehensive structural census. Fold Des. 1998a;3:497–512. doi: 10.1016/S1359-0278(98)00066-2. [DOI] [PubMed] [Google Scholar]
- Gerstein M. Patterns of protein-fold usage in eight microbial genomes: A comprehensive structural census. Proteins. 1998b;33:518–534. doi: 10.1002/(sici)1097-0134(19981201)33:4<518::aid-prot5>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Aert R, Agostini-Carbone ML, Ahmed A, et al. The yeast genome directory. Nature. 1997;387(Supp):5–105. [Google Scholar]
- Gogarten JP, Olendzenski L. Orthologs, paralogs and genome comparisons. Curr Opin Genet Dev. 1999;9:630–636. doi: 10.1016/s0959-437x(99)00029-5. [DOI] [PubMed] [Google Scholar]
- Gupta RS. Protein phylogenies and signature sequences: A reappraisal of evolutionary relationships among archaebacteria, eubacteria, and eukaryotes. Microbiol Mol Biol Rev. 1998;62:1435–1491. doi: 10.1128/mmbr.62.4.1435-1491.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi H, Gerstein M. The relationship between protein structure and function: A comprehensive survey with application to the yeast genome. J Mol Biol. 1999;288:147–164. doi: 10.1006/jmbi.1999.2661. [DOI] [PubMed] [Google Scholar]
- Hennig W. Phylogenetic systematics. Ann Rev Entomol. 1965;10:97–116. [Google Scholar]
- Hilario E, Gogarten JP. Horizontal transfer of ATPase genes—the tree of life becomes a net of life. Biosystems. 1993;31:111–119. doi: 10.1016/0303-2647(93)90038-e. [DOI] [PubMed] [Google Scholar]
- Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt RP, Logsdon JM, Jr, Healy B, Dorey MW, Doolittle WF, Embley TM. Microsporidia are related to fungi: Evidence from the largest subunit of RNA polymerase II and other proteins. Proc Natl Acad Sci USA. 1999;96:580–585. doi: 10.1073/pnas.96.2.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Losey HC, Kawarabayasi Y, Kikuchi H, Bunjun S, Soll D. Substrate recognition by class I lysyl-tRNA synthetases: A molecular basis for gene displacement. Proc Natl Acad Sci USA. 1999;96:418–423. doi: 10.1073/pnas.96.2.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Morgan S, Curnow AW, Pridmore DR, Vothknecht UC, Gardner W, Lin W, Woese CR, Soll D. A euryarchaeal lysyl-tRNA synthetase: Resemblance to class I synthetases. Science. 1997;278:1119–1122. doi: 10.1126/science.278.5340.1119. [DOI] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: The complexity hypothesis. Proc Natl Acad Sci USA. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S, et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions (supplement) DNA Res. 1996;3:185–209. doi: 10.1093/dnares/3.3.185. [DOI] [PubMed] [Google Scholar]
- Karlin S, Burge C. Dinucleotide relative abundance extremes: A genomic signature. [Review] Trends Genetics. 1995;11:283–290. doi: 10.1016/s0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- Karlin S, Mrazek J. Compositional differences within and between eukaryotic genomes. Proc Natl Acad Sci USA. 1997;94:10,227–10,232. doi: 10.1073/pnas.94.19.10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Tatusov RL, Galperin MY. Beyond complete genomes: From sequence to structure and function. Curr Opin Struct Biol. 1998;8:355–363. doi: 10.1016/s0959-440x(98)80070-5. [DOI] [PubMed] [Google Scholar]
- Lake JA. Reconstructing evolutionary trees from DNA and protein sequences: Paralinear distances. Proc Natl Acad Sci USA. 1994;91:1455–1459. doi: 10.1073/pnas.91.4.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake JA, Jain R, Rivera MC. Mix and match in the tree of life. Science. 1999;283:2027–2028. doi: 10.1126/science.283.5410.2027. [DOI] [PubMed] [Google Scholar]
- Lawrence JG. Gene transfer, speciation, and the evolution of bacterial genomes. Curr Opin Microbiol. 1999;2:519–523. doi: 10.1016/s1369-5274(99)00010-7. [DOI] [PubMed] [Google Scholar]
- Levitt M, Chothia C. Structural patterns in globular proteins. Nature. 1976;261:552–558. doi: 10.1038/261552a0. [DOI] [PubMed] [Google Scholar]
- Lipman DJ, Pearson WR. Rapid and sensitive protein similarity searches. Science. 1985;227:1435–1441. doi: 10.1126/science.2983426. [DOI] [PubMed] [Google Scholar]
- Lopez P, Forterre P, Philippe H. The root of the tree of life in the light of the covarion model. J Mol Evol. 1999;49:496–508. doi: 10.1007/pl00006572. [DOI] [PubMed] [Google Scholar]
- Maidak BL, Cole JR, Parker CT, Jr, Garrity GM, Larsen N, Li B, Lilburn TG, McCaughey MJ, Olsen GJ, Overbeek, R. R, et al. A new version of the RDP (Ribosomal Database Project) Nucleic Acids Res. 1999;27:171–173. doi: 10.1093/nar/27.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisey JG. Heads and tails: A chordate phylogeny. Cladistics. 1986;2:201–256. doi: 10.1111/j.1096-0031.1986.tb00462.x. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Galperin MY, Grishin NV, Tatusov RL, Wolf YI, Koonin EV. Comparative genomics of the Archaea (Euryarchaeota): Evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 1999;9:608–628. [PubMed] [Google Scholar]
- Murzin A, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Nomura M. Engineering of bacterial ribosomes: Replacement of all seven Escherichia coli rRNA operons by a single plasmid-encoded operon [see comments] Proc Natl Acad Sci USA. 1999;96:1820–1822. doi: 10.1073/pnas.96.5.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page R D. TreeView: An application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Pearson WR. Using the FASTA program to search protein and DNA sequence databases. Meth Mol Biol. 1994;24:307–331. doi: 10.1385/0-89603-246-9:307. [DOI] [PubMed] [Google Scholar]
- ————— Effective protein sequence comparison. Meth Enzymol. 1996;266:227–258. doi: 10.1016/s0076-6879(96)66017-0. [DOI] [PubMed] [Google Scholar]
- Pennisi E. Genome data shake tree of life [news] Science. 1998;280:672–674. doi: 10.1126/science.280.5364.672. [DOI] [PubMed] [Google Scholar]
- ————— Is it time to uproot the tree of life? [news] Science. 1999;284:1305–1307. doi: 10.1126/science.284.5418.1305. [DOI] [PubMed] [Google Scholar]
- Rivera MC, Jain R, Moore JE, Lake JA. Genomic evidence for two functionally distinct gene classes. Genetics. 1998;11:6239–6244. doi: 10.1073/pnas.95.11.6239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat Genet. 1999;21:108–110. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- Stevens, W.K. 1999. Rearranging the branches on a new tree of life. The New York Times: Aug. 31 1999: F1.
- Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*;and Other Methods). Version 4. Sunderland, MA: Sinauer Associates; 1998. [Google Scholar]
- Swofford D, Olsen GJ, Waddell PJ, Hillis DM. Molecular Systematics. Sunderland, MA: Sinauer Associates; 1996. Phylogenetic Inference; pp. 407–514. [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Teichmann SA, Mitchison G. Is there a phylogenetic signal in prokaryote proteins? J Mol Evol. 1999a;49:98–107. doi: 10.1007/pl00006538. [DOI] [PubMed] [Google Scholar]
- ————— Making family trees from gene families. Nat Genet. 1999b;21:66–67. doi: 10.1038/5001. [DOI] [PubMed] [Google Scholar]
- Tekaia F, Lazcano A, Dujon B. The genomic tree as revealed from whole proteome comparisons. Genome Res. 1999;9:550–557. [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomb J-F, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Tumbula D, Vothknecht UC, Kim H, Ibba M, Min B, Li T, Pelaschier J, Stathopoulos C, Becker H, Soll D. Archaeal aminoacyl-tRNA synthesis. Diversity replaces dogma [In Process Citation] Genetics. 1999;152:1269–1276. doi: 10.1093/genetics/152.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Peer Y, Robbrecht E, de Hoog S, Caers A, De Rijk P, De Wachter R. Database on the structure of small subunit ribosomal RNA. Nucleic Acids Res. 1999;27:179–183. doi: 10.1093/nar/27.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Brenner SE, Bash PA, Koonin EV. Distribution of protein folds in the three superkingdoms of life. Genome Res. 1999;9:17–26. [PubMed] [Google Scholar]
- Yap WH, Zhang Z, Wang Y. Distinct types of rRNA operons exist in the genome of the actinomycete Thermomonospora chromogena and evidence for horizontal transfer of an entire rRNA operon. J Bacteriol. 1999;181:5201–5209. doi: 10.1128/jb.181.17.5201-5209.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]