

Human retinal pigment epithelial cells are extremely resistant to apoptosis. Blocking the interaction between p53 and Mdm2 sensitizes these cells to apoptosis, suggesting a novel therapeutic target in the treatment of proliferative and degenerative retinopathies.
Abstract
Purpose.
Because recent studies indicate that blocking the interaction between p53 and Mdm2 results in the nongenotoxic activation of p53, the authors sought to investigate whether the inhibition of p53-Mdm2 binding activates p53 and sensitizes human retinal epithelial cells to apoptosis.
Methods.
Apoptosis was evaluated by the activation of caspases and DNA fragmentation assays. The Mdm2 antagonist Nutlin-3 was used to dissociate p53 from Mdm2 and, thus, to increase p53 activity. Knockdown of p53 expression was accomplished by using p53 siRNA.
Results.
ARPE-19 and primary RPE cells expressed high levels of the antiapoptotic proteins Bcl-2 and Bcl-xL. Exposure of these cells to camptothecin (CPT) or TNF-α/ cycloheximide (CHX) failed to induce apoptosis. In contrast, treatment with the Mdm2 antagonist Nutlin-3 in the absence of CPT or TNF-α/CHX increased apoptosis. Activation of p53 in response to Nutlin-3 also increased levels of Noxa, p53-upregulated modulator of apoptosis (PUMA), and Siva-1, decreased expression of Bcl-2 and Bcl-xL, and simultaneously increased caspases-9 and -3 activities and DNA fragmentation. Knockdown of p53 decreased the basal expression of p21Cip1 and Bcl-2, inhibited the Nutlin-3–induced upregulation of Siva-1 and PUMA expression, and consequently inhibited caspase-3 activation.
Conclusions.
These results indicate that the normally available pool of intracellular p53 is predominantly engaged in the regulation of cell cycle checkpoints by p21Cip1 and does not trigger apoptosis in response to DNA-damaging agents. However, the blockage of p53 binding to Mdm2 frees a pool of p53 that is sufficient, even in the absence of DNA-damaging agents, to increase the expression of proapoptotic targets and to override the resistance of RPE cells to apoptosis.
The retinal pigment epithelium is essential for maintaining retinal homeostasis and visual function by supplying metabolic support for photoreceptors, constituting a blood-retinal barrier, and providing other major support functions.1,2 RPE (retinal pigment epithelial) cells are normally nonmitotic and survive for a person's lifetime, but disease, trauma, and aging can lead to alterations in the retinal pigment epithelium that can have profound consequences on vision. In response to severe retinal injury or ocular trauma, RPE cells can detach, dedifferentiate, rapidly proliferate, and infiltrate the retina and vitreous cavity,3 leading to proliferative vitreoretinopathy (PVR), which causes significant visual impairment. Dedifferentiated RPE cultures have been used to study the early phases of PVR in vitro and in vivo.4,5 Age-related macular degeneration (AMD), a leading cause of blindness in patients older than 60, may be caused by (or result in) a loss of RPE cells through apoptosis.6,7 Mechanisms that support RPE cell proliferation in PVR and cell loss in AMD (geographic atrophy) are as yet poorly understood. To prevent or better treat these blinding retinal diseases, it is necessary to understand the cellular and molecular events that regulate the survival and death of RPE cells.
The tumor suppressor and transcription factor p53 is arguably the most important regulator of cellular longevity.8 Activation of p53 triggers apoptosis, cell cycle arrest, or cellular senescence. Posttranslational modifications, localization, and degradation tightly regulate the activity of p53.9,10 The murine double minute (Mdm2), a product of a p53-inducible gene, binds directly with the transactivation domain of p53 and assists in ubiquitin-proteasomal degradation of p53, thereby acting as a negative regulator.11 Radiation and chemotherapeutic drugs that cause DNA damage activate the p53-signaling pathway and have been previously used for the treatment of PVR and various forms of cancer.12,13 Another promising approach for activating p53 in the absence of DNA-damaging agents is to dissociate p53 from Mdm2. Nutlin-3, a small-molecule Mdm2 inhibitor,14 preferentially binds to the p53-binding pocket of Mdm2, disrupts p53-Mdm2 association, and effectively activates p53, thereby representing a potential drug target. Nutlin-3 has antitumor activity in various forms of cancers, which express wild-type p53, including osteosarcoma,15 neuroblastoma,16 leukemia,17 and retinoblastoma.18 It is unknown whether destabilization of the p53-Mdm2 interaction induces cell cycle arrest or reverses the resistance to apoptosis in RPE cells.
Relative to many other cell types, RPE cells are resistant to apoptosis. Ionizing radiation,19 blue light,20 4-hydroxynonenal,21 and diphenyleneiodonium22 are known to induce p53-dependent apoptosis in RPE cells. Other inducers such as TNF-α23 and oxidative stress,24 which trigger apoptosis in a wide range of cells, are generally ineffective in this cell type. Relatively little is known about the mechanisms involved in the inherent resistance of normal RPE cells to apoptosis. There are studies that demonstrated the presence of carotenoid pigments (antioxidants) in the macula,25 synthesis of docosahexaenoic acid (DHA) bioderivative neuoprotectin D1 (known to inactivate proapoptotic and inflammatory signaling26), expression of antiapoptotic protein Bcl-xL,27 and decreased levels of caspase-8.28 However, there is limited information about the mechanism of p53 activation and the identification of its downstream signals, including p53-upregulated modulator of apoptosis (PUMA), Bax, Noxa, Siva-1, and the expression of Bcl-2 family proteins in RPE cells.
We have used both primary human RPE cells and the commonly used RPE cell line (ARPE-19) to examine whether disruption of the interaction between p53 and Mdm2 is sufficient for the induction of apoptosis. For the first time, we show that the Mdm2 antagonist, Nutlin-3, triggers the p53-mediated expression of proapoptotic targets, inhibits antiapoptotic mediators (Bcl-2 and Bcl-xL), and ultimately causes apoptosis of primary RPE cultures and ARPE-19 cells. Understanding the interactions of p53 and Mdm2 and their roles in the survival and apoptosis of RPE cells could provide an important framework for the development of therapeutic strategies for PVR and AMD.
Materials and Methods
Reagents
Disposable cell culture ware was purchased from Corning Glass Works (Corning, NY) and Zellkulture Flaschen (Europe, Switzerland). Cell culture medium and fetal bovine serum (FBS) were obtained from Mediatech, Inc. (Herndon, VA) and Invitrogen (Grand Island, NY). Insulin and dialyzed FBS (dFBS) was purchased from Sigma (St. Louis, MO). The cell lines IEC-6 and ARPE-19 were obtained from American Type Culture Collection (Rockville, MD). Test results for mycoplasma were always negative. Recombinant TNF-α was obtained from BD PharMingen International (San Diego, CA). The enhanced chemiluminescence (ECL) Western blot detection system was purchased from Perkin Elmer (Boston, MA). Phospho-p53 Ser15, phospho-Mdm2 Ser166, total-p53, Bcl-xL, cleaved active caspase-3 (Asp 175), phospho-Akt Ser473, total-Akt, phospho-ERK1/2, total-ERK1/2, phospho-JNK1/2, total-JNK1/2, phospho-Bcl-2 Ser70, and total Bcl-2 antibodies were purchased from Cell Signaling (Beverly, MA). p21Cip1 antibodies were obtained from BD Biosciences (San Diego, CA). Nutlin-3, camptothecin (CPT), and cycloheximide (CHX) were purchased from Sigma. p53 siRNA–, Mdm2 (SMP14)–, and Bax (Δ21)–specific antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). A photometric enzyme immunoassay (Cell Death Detection ELISA Plus) kit was purchased from Roche Diagnostics Corp. (Indianapolis, IN). All chemicals were of the highest purity commercially available.
Cell Culture
Primary human RPE cells were isolated from postmortem donor eyes (age 29 years) provided by the Midsouth Eye Bank using procedures previously described.29 Primary cultures within the first five passages were used. The ARPE-19 cell line (ATCC CRL-2302) was derived from spontaneously arising retinal pigment epithelia of a healthy person, as described by Dunn et al.30 ARPE-19 and primary RPE stock cells were grown in T-75 flasks in a humidified, 37°C incubator in an atmosphere of 5% CO2. Cell culture medium consisted of Dulbecco's modified Eagle's medium (DMEM) and Ham's F10 medium (1:1 ratio) containing l-glutamine and 10% heat-inactivated FBS. For experimental purposes, 0.75 × 106 cells were seeded per 60-mm plate so that cells were 90% confluent by day 3.
The IEC-6 cell line (ATCC CRL 1592) was derived from normal rat intestine and was developed and characterized by Quaroni et al.31 IEC-6 cells originate from intestinal crypt cells, as judged by morphologic and immunologic criteria. They are nontumorigenic and retain the undifferentiated character of epithelial stem cells. IEC-6 cell stocks were maintained in T-150 flasks in a humidified, 37°C incubator in an atmosphere of 10% CO2, and passages 15 to 22 were used. The medium consisted of DMEM with 5% heat-inactivated FBS, 10 μg insulin, and 50 μg gentamicin sulfate per milliliter. Stock flasks for each cell line were passaged weekly and fed three times per week. For experiments, the cells were trypsinized with 0.05% trypsin and 0.53 mM EDTA and were counted by a dual threshold analyzer (Z1 Coulter Counter; Beckman Coulter, Brea, CA). IEC-6, ARPE-19, and primary RPE cells were grown for 3 days in control medium and serum starved for 24 hours (to allow synchronization in the same phase of the cell cycle); this was followed by the induction of apoptosis.
Treatment
Confluent serum-starved IEC-6, ARPE-19, and primary RPE monolayers were treated with a combination of TNF-α (20 ng/mL) and CHX (25 μg/mL) or 20 μM CPT for 4 hours for the induction of apoptosis. For cell proliferation assays, proliferating ARPE-19 cells were treated with 5 μM Nutlin-3 or CPT for 24 hours in serum-containing medium. For dose-response studies using Nutlin-3, both confluent serum-starved ARPE-19 and primary RPE monolayers were treated with 20, 40, and 60 μM Nutlin-3 for 2 hours. To investigate Nutlin-3–induced p53 signaling, primary RPE cells were treated with 60 μM Nutlin-3 for 4 hours, and ARPE-19 cells were treated with 60 μM Nutlin-3 for 2 and 4 hours. Protein synthesis was blocked by pretreatment of cells with CHX (25 μg/mL) in the culture medium for 1 hour, followed by treatment with 20 μM CPT for 3 hours or 60 μM Nutlin-3 for 2 hours. To investigate the role of Src and PI3-K/Akt in ARPE-19 survival, confluent ARPE-19 cells were pretreated with Src kinase inhibitor (PP2, 10 μM) or PI3-K inhibitor (LY294002, 10 μM) for 1 hour, followed by treatment with or without 60 μM Nutlin-3 for 3 hours.
Apoptosis
The quantitative DNA fragmentation assay was carried out using a cell death detection ELISA kit as described earlier.32–34 An aliquot of nuclei-free cell lysate was placed in streptavidin-coated wells and incubated with anti-histone-biotin antibody and anti-DNA peroxidase-conjugated antibody for 2 hours at room temperature. After incubation, the sample was removed, and the wells were washed and incubated with 100 μL of the substrate, [2,2′-azino-di[3-ethylbenzthiazolin-sulfonate], and allowed to react at room temperature. Absorbance was read at 405 nm using a plate reader. Results were expressed as absorbance at 405 nm/min/mg protein.
Caspase Assay
Caspases-3 and -9 activities were determined using a kit from Biomol International in accordance with the manufacturer's instructions. At designated time periods, cell monolayers were washed twice with Dulbecco's phosphate-buffered saline (DPBS) and scraped with a rubber policeman. The suspension was briefly centrifuged at 1000g at 4°C, and the supernatant was removed. The cell pellet was lysed in 150 μL ice-cold caspase assay buffer containing 50 mM HEPES, pH 7.4, 0.1% CHAPS, 1 mM dithiothreitol, 0.1 mM EDTA, and 0.1% NP-40. The resultant suspension was centrifuged at 10,000g, 4°C, for 10 minutes, and 10 to 20 μL supernatant was used for caspase assay. The fluorogenic substrate for caspase-9 (Ac-LEHD-AFC) has an excitation at 380 nm and an emission at 505 nm. The fluorogenic substrate for caspase-3 (Ac-DEVD-AFC) has an excitation at 400 nm and an emission at 505 nm. Lysis buffer alone and lysis buffer plus substrate were used as background controls. Activity was expressed as relative fluorescence units (RFU) per milligram of protein per minute.
Transfection
ARPE-19 cells were seeded in six-well plates at a density of 0.35 × 106 cells/well in antibiotic-free normal growth medium to achieve 60% to 70% confluence on day 2. For transfection of each well, 2 to 8 μL siRNA duplex (0.25–1 μg) was diluted in 100 μL DMEM without serum and antibiotics and was labeled as A. Then 2 to 8 μL transfection reagent (FuGENE; Roche) was diluted in serum-free medium and labeled as B. Solutions A and B were mixed together and incubated at room temperature for 30 to 45 minutes. The cell monolayer was rinsed in serum and antibiotic free-DMEM, and the siRNA-transfection mix was added dropwise onto the monolayer and incubated for 12 hours at 37°C. Fresh medium containing serum and antibiotics was added on the monolayer without removing the transfection mixture and was incubated for an additional 12 hours. The cells were serum starved and then treated with 20 μM CPT or 60 μM Nutlin-3. Total cell lysates were prepared for Western blot analysis.
Cell Proliferation
Cell proliferation was measured by analyzing cell doubling time. Doubling-time experiments were carried out over a period of 48 hours, during which serum was present throughout. Equal numbers of cells were first plated in triplicate in six-well plates, and one set was harvested and counted after 24 hours to determine the number of attached cells. The other set of cells was treated with 5 μM CPT, Nutlin-3, or DMSO (vehicle control) in serum-containing medium. Cells were trypsinized and counted after 24 and 48 hours of treatment. Mean doubling time was calculated as described previously.35
Western Blot Analysis
The protocol for Western blot analysis has been described earlier.32–34 Typically, cell monolayers were first washed with ice-cold DPBS and lysed for 10 minutes in ice-cold cell lysis buffer containing protease inhibitors. Lysates were centrifuged at 10,000g for 10 minutes at 4°C, followed by SDS-PAGE. Proteins were transferred to membranes (Immobilon-P; Millipore Bedford, MA) and probed with the indicated antibodies overnight at 4°C in TBS buffer containing 0.1% Tween-20 and 5% nonfat dry milk (blotting grade; Bio-Rad, Hercules, CA). Membranes were subsequently incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 hour, and the immunocomplexes were visualized by the ECL detection system. Densitometric analysis of all Western blots was performed using ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; available at http://rsb.info.nih.gov/ij/index.html). Representative Western blots from three experiments are shown.
Statistical Analysis
All data are expressed as mean ± SE. Experiments were repeated three times, with triplicate samples for each. Analysis of variance and appropriate post hoc testing determined the significance of the differences between means. P < 0.05 was considered significant.
Results
Effects of TNF-α/CHX and Camptothecin on Apoptosis of ARPE-19 and Primary RPE Cells
Earlier studies from our laboratory demonstrated that both TNF-α/CHX and CPT induced apoptosis in intestinal epithelial (IEC-6) cells through death receptor-dependent (extrinsic) and -independent (intrinsic) pathways, respectively.32–38 To determine whether TNF-α/CHX and CPT were capable of inducing apoptosis in human retinal epithelial cells, we compared their effects in ARPE-19 and primary RPE cells with those observed in IEC-6 cells. Consistent with our previous reports,32–38 both TNF-α/CHX and CPT significantly increased DNA fragmentation in IEC-6 cells (Fig. 1A). Conversely, both TNF-α/CHX and CPT failed to increase DNA fragmentation in ARPE-19 and primary RPE cells (Fig. 1A). Because the expression of antiapoptotic Bcl-2 family proteins is known to confer resistance against apoptosis in multiple cell types, we compared Bcl-2 and Bcl-xL expression in these cells. Increased expression of Bcl-2 and Bcl-xL was observed in both the ARPE-19 and the primary RPE human cells compared with IEC-6 cells (Fig. 1B). Bcl-2 (ARPE-19 and primary RPE cells) and Bcl-xL (primary RPE cells; Fig. 1B) had a higher electrophoretic mobility shift, probably suggesting posttranslational processing.
Figure 1.

TNF-α/CHX and CPT-mediated apoptosis in IEC-6, ARPE-19, and primary RPE cells. IEC-6, ARPE-19, and primary RPE cells were grown to confluence for 3 days, serum starved for 24 hours, and treated with a combination of TNF-α/CHX or CPT for 4 hours. DNA fragmentation was measured by ELISA (mean ± SEM; n = 3). *P < 0.05; significantly different compared with untreated cells. (B) IEC-6, ARPE-19, and primary RPE cell lysates were analyzed for the levels of Bcl-2 and Bcl-xL using specific antibodies. Actin was used as an internal loading control.
TNF-α/CHX did not modulate p53 protein levels or p53 protein phosphorylation in IEC-6 cells but increased Mdm2 phosphorylation (Fig. 2A), with simultaneous increases of Akt and ERK1/2 phosphorylation as survival responses (Fig. 2B). Bcl-2 and Bcl-xL protein levels did not change during TNF-α/CHX treatment (Fig. 2A). Increased phosphorylation of proapoptotic JNK in response to TNF-α/CHX (Fig. 2B) caused caspase-3 activation, which eventually resulted in DNA fragmentation (Fig. 1A), consistent with our previous observations.38 In contrast to death receptor-dependent apoptosis, the DNA-damaging agent CPT robustly increased p53 protein levels and phosphorylation in IEC-6 cells (Fig. 2A). In addition, the phosphorylation of Akt, JNK, and ERK1/2 in response to CPT was accompanied by the phosphorylation of Mdm2 (Fig. 2A). Phosphorylated Mdm2 is degraded by ubiquitination.32,39 As a result, p53 levels increased and caused transcriptional upregulation of its downstream targets, including p21Cip1, Mdm2, and Bax (Fig. 2A). Increased levels of Bax led to caspase-3 activation (Fig. 2A) and caused DNA fragmentation (Fig. 1A).
Figure 2.

TNF-α/CHX- and CPT-mediated signaling in IEC-6 and ARPE-19 cells. IEC-6 and ARPE-19 cells were grown to confluence for 3 days, serum starved for 24 hours, and treated with a combination of TNF-α/CHX or CPT for 4 hours. (A) Cell lysates were separated on SDS-PAGE, and Western blot analysis was carried out using antibodies specific for phospho- and total- p53, Mdm2, p21Cip1, Bax, Bcl-2, Bcl-xL, and active caspase-3. Actin was used as loading control. (B) Cell lysates from this experiment were analyzed for the levels of phospho- and total- JNK1/2, Akt, and ERK1/2 by Western blot analysis.
In comparison to IEC-6 cells, ARPE-19 cells expressed high basal levels of p53 and its downstream target p21Cip1. Higher Mdm2 protein levels were observed in untreated ARPE-19 cells compared with IEC-6 cells (Fig. 2A). The enhanced Bcl-xL and Bcl-2 expression observed in ARPE-19 cells compared with IEC-6 cells (Fig. 2A) was consistent with observations shown in Figure 1B. In addition, ARPE-19 cells had lower basal levels of proapoptotic Bax. Exposure of ARPE-19 cells to TNF-α/CHX decreased the expression of p53 and p21Cip1. Furthermore, TNF-α/CHX increased Akt, Mdm2, and ERK1/2 phosphorylation but had no effect on p53 phosphorylation in ARPE-19 cells. Additionally, TNF-α/CHX increased the phosphorylation of JNK (Fig. 2B) but failed to activate caspase-3 (Fig. 2A) or to induce DNA fragmentation in ARPE-19 cells (Fig. 1A).
CPT increased the expression of p53 and its phosphorylation, similar to our observations of CPT-mediated signaling in IEC-6 cells. However, the phosphorylated p53 band in ARPE-19 cells had a higher electrophoretic mobility shift compared to IEC-6 cells (Fig. 2A), indicating phosphorylation on additional residues or posttranslational modifications of p53. Concomitantly, CPT increased p21Cip1 expression in ARPE-19 cells (panel showing light exposure). In addition, CPT increased Akt, Mdm2, and ERK1/2 phosphorylation but failed to activate proapoptotic JNK (Fig. 2B). Neither Bcl-2 nor Bcl-xL protein levels changed in response to CPT or TNF-α/CHX in ARPE-19 cells (Fig. 2A). Given that the p53-dependent proapoptotic target Bax did not increase in response to CPT and that antiapoptotic Bcl-xL and Bcl-2 protein levels remained elevated, CPT failed to activate caspase-3 (Fig. 2A) and DNA fragmentation (Fig. 1A) in ARPE-19 cells.
Nutlin-3 Inhibits ARPE-19 Proliferation
Because 20 μM CPT increased p53 phosphorylation and protein (Fig. 2A) but failed to induce apoptosis in ARPE-19 cells (Fig. 1A), we tested whether lower concentrations had similar effects on p53 signaling. Treatment of cells with 5 μM CPT or Nutlin-3 for 24 hours significantly increased mean doubling time compared with DMSO-treated control cells (Fig. 3A), indicating decreased rates of proliferation. Both CPT and Nutlin-3 at lower doses had no effect on DNA fragmentation and caspase-3 activity (data not shown). CPT increased p53 phosphorylation and protein levels with a concomitant increase of p21Cip1 (Fig. 3B). Similar to the effect of CPT, Nutlin-3 significantly increased p53 protein with a concomitant upregulation of its downstream target, p21Cip1 (Fig. 3B). Interestingly, p53 phosphorylation in response to CPT was significantly higher than the response of Nutlin-3 (Fig. 3B). Moreover, neither p73 (p53 family protein) nor p27Kip1 protein levels changed in response to CPT or Nutlin-3, indicating a specific involvement of p53/p21Cip1 signaling.
Figure 3.

Effect of CPT and Nutlin-3 on the proliferation of ARPE-19 cells. ARPE-19 cells were trypsinized, and equal numbers of cells were seeded in serum-containing medium. Sixteen to 18 hours later, attached cells were treated with 5 μM DMSO, CPT, or Nutlin-3. After treatment, cells were grown for 24 hours and (A) were trypsinized and counted, and the mean doubling time was plotted. *P < 0.05; significantly different compared with DMSO-treated cells. (B) Cell lysates were analyzed by Western blot for the levels of phospho-53 and total p53, Rb, p73, p21Cip1, and p27. Actin was used as an internal loading control.
Unphosphorylated retinoblastoma tumor suppressor protein (Rb) prevents cell cycle progression by binding with E2F family transcription factors, whereas the phosphorylation of Rb by Cdk/cyclin complexes results in the release of bound E2F to stimulate the transcription of genes involved in DNA synthesis and S-phase progression.40,41 As expected, levels of phosphorylated Rb (p-Rb) were found to be markedly lower in CPT- and Nutlin-3–treated cells with concomitant increases in unphosphorylated Rb (lower band; Fig. 3B). Our results indicate that lower concentrations of both CPT and Nutlin-3 activate p53/p21 signaling and inhibit proliferation.
Higher Doses of Nutlin-3 Induce Apoptosis in ARPE-19 Cells and Primary RPE Cultures
Given that 5 μM Nutlin-3 inhibited proliferation (Fig. 3) but failed to induce apoptosis in ARPE-19 cells, we sought to determine whether higher concentrations of Nutlin-3 were capable of inducing apoptosis. Data presented in Figure 4A show that 20 to 40 μM Nutlin-3 had no effect on apoptosis, as evident by the insignificant amount of DNA fragmentation compared with vehicle-treated controls. Overnight treatment of ARPE-19 cells with 5 to 20 μM Nutlin-3 failed to induce DNA fragmentation (data not shown). However, 60 μM Nutlin-3 significantly increased DNA fragmentation (Fig. 4A) and resulted in the appearance of the active caspases-9 and -3 fragments, indicating cell death (Fig. 4B). Concentrations lower than 60 μM Nutlin-3 failed to activate caspases-9 and -3 (Fig. 4B). Furthermore, treatment of ARPE-19 cells with 60 μM Nutlin-3 induced cell death within 2 hours, as evidenced by the appearance of bright, rounded apoptotic cells and loss of cell-cell contact, both of which significantly increased after 4 hours of exposure to 60 μM Nutlin-3 (Fig. 4C). Enzymatic activities of caspase-9 and caspase-3 increased from a basal level of 12.1 ± 2.3 to 45.3 ± 10.9 and from 87.4 ± 9.2 to 321.1 ± 22.5 RFU/mg protein/min (mean ± SEM; n = 3; P < 0.05), respectively, in response to Nutlin-3 in ARPE-19 cells.
Figure 4.

Nutlin-3 induces apoptosis in ARPE-19 and primary RPE cells. (A) ARPE-19 cells were grown to confluence for 3 days, serum starved for 24 hours, and treated with 20, 40, and 60 μM Nutlin-3 for 2 hours. DNA fragmentation was measured by ELISA (mean ± SEM). *P < 0.05; significantly different compared with DMSO. (B) Western blot for active caspase-9 and -3 in 0, 20, 40, and 60 μM Nutlin-3–treated lysates. Actin was used as an internal loading control. (C) ARPE-19 cells were treated with DMSO or Nutlin-3 (60 μM) for 2 and 4 hours, and monolayers were photographed under a phase-contrast microscope equipped with a charge-coupled device camera using Image J software. (D) Primary RPE cultures were grown to confluence, serum starved for 24 hours, and treated with 0, 20, 40, and 60 μM Nutlin-3 for 2 hours. Lysates were analyzed by Western blot for the presence of active caspase-9 and -3. Actin was used as an internal loading control.
Given that both ARPE-19 and primary RPE cells showed resistance to various inducers of apoptosis (Fig. 1A), we investigated whether Nutlin-3 was capable of sensitizing primary RPE cells to apoptosis. At a dose of 60 μM, Nutlin-3 activated caspases-9 and -3, as evident by the levels of active caspase fragments (Fig. 4D). Lower doses of Nutlin-3 failed to activate caspases, which was similar to the results obtained with ARPE-19 cells. Treatment of primary RPE cells with 60 μM Nutlin-3 caused a loss of cell-cell contact with the concomitant appearance of bright, round apoptotic cells and increased DNA fragmentation (Figs. 5A, 5B). These observations indicate that inhibition of the Mdm2-p53 interaction induces apoptosis in RPE cells.
Figure 5.

Activation of p53 signaling in response to Nutlin-3. (A) Primary RPE cells were treated with DMSO or Nutlin-3 (60 μM) for 4 hours, and monolayers were photographed under a phase-contrast microscope equipped with a charge-coupled device camera using ImageJ software. (B) DNA fragmentation was measured by ELISA (mean ± SEM). *P < 0.05; significantly different compared with DMSO. (C) Primary RPE cells were treated with DMSO or 60 μM Nutlin-3, and lysates were analyzed for phospho-p53 (Ser15, Ser46), total p53, and Mdm2. (D) Cell extracts were analyzed to determine the levels of Bcl-xL, Bcl-2, PUMA, Noxa, Siva-1, and active caspase-3 by Western blot analysis. Actin was used as a loading control.
Nutlin-3 Upregulates p53-Responsive Targets Leading to Apoptosis
Nutlin-3 increased the phosphorylation of p53 at Ser15 and Ser46 and simultaneously increased p53 expression in primary RPE cells (Fig. 5C). Activation of p53 increased the expression of Mdm2 and several proapoptotic proteins, including PUMA, Noxa, and Siva-1 (Figs. 5C, 5D). Furthermore, Nutlin-3 decreased Bcl-xL and Bcl-2 proteins (Fig. 5D) and thereby sensitized primary RPE cells to apoptosis. Because Nutlin-3 was capable of inducing apoptosis in both primary RPE cells and ARPE-19 cells, we compared Nutlin-3–induced p53 signaling in ARPE-19 cells. Nutlin-3 (60 μM) increased p53 expression within 2 hours; this elevation remained through 4 hours (Fig. 6A). Although ARPE-19 cells expressed p73 protein, 60 μM Nutlin-3 failed to modulate its levels, indicating that Nutlin-3 specifically regulates Mdm2/p53 signaling (Fig. 6A). Increased levels of p53 were accompanied by the upregulation of p53-responsive proapoptotic targets, including Siva-1, Noxa, PUMA, and Bax (Fig. 6A). Bcl-xL protein increased after 2 hours of 60 μM Nutlin-3 treatment, probably as a survival response, but decreased by 4 hours. Similar to Bcl-xL, Bcl-2 protein levels were initially high but decreased through 4 hours of Nutlin-3 treatment (Fig. 6A). Decreased levels of prosurvival Bcl-xL and Bcl-2 and simultaneous increases in proapoptotic Siva-1, Noxa, PUMA, and Bax result in mitochondrial depolarization, cytochrome c release, and caspases-9 and -3 activation and apoptosis.
Figure 6.

Nutlin-3 activates p53 signaling in ARPE-19 cells. (A) ARPE-19 cells were treated with DMSO or Nutlin-3 (60 μM) for 2 and 4 hours. Cell extracts were prepared, and levels of p53, p73, Siva-1, Noxa, PUMA, Bax, Bcl-xL, Bcl-2, active caspase-9, and active caspase-3 proteins were determined using Western blot analysis. Actin was used as an internal loading control. (B) Cell lysates were analyzed for the levels of phospho- and total-Akt and Mdm2 by Western blot analysis. (C) Confluent serum-starved ARPE-19 monolayers were first treated (Treatment a) with DMSO for 3 hours, followed by subsequent treatment (Treatment b) with DMSO (DMSO/DMSO) or (60 μM) Nutlin-3 (DMSO/NUT) for 2 hours. Similarly, cells were treated with (20 μM) CPT for 3 hours, followed by DMSO (CPT/DMSO) or 60 μM Nutlin-3 (CPT/NUT) for 2 hours. DNA fragmentation was measured by ELISA (mean ± SEM; n = 3). *P < 0.05; significantly different compared with DMSO/DMSO. †P < 0.05; significantly different compared with DMSO/Nutlin-3.
Akt participates in apoptotic and survival pathways; therefore, we investigated whether proapoptotic effects of Nutlin-3 correlate with Akt signaling. Nutlin-3 increased Akt phosphorylation, which in turn caused the phosphorylation of Mdm2 (Fig. 6B). p53 activation in response to 60 μM Nutlin-3 led to the upregulation of Mdm2 protein levels within 2 hours that decreased at 4 hours (Fig. 6B). Mdm2 is known to be a substrate for caspase-dependent proteolytic cleavage during apoptosis.42 Data presented in Figure 6B show the appearance of a cleaved Mdm2 fragment within 2 hours that remained throughout the entire 4-hour period, indicating robust activation of caspase-3 and apoptosis in response to 60 μM Nutlin-3.
In subsequent experiments, we sequentially treated cells with DMSO for 3 hours, followed by 60 μM Nutlin-3 for 2 hours (DMSO/Nutlin-3) or administered CPT for 3 hours followed by 60 μM Nutlin-3 for 2 hours (CPT/Nutlin-3). CPT/Nutlin-3 significantly increased DNA fragmentation compared with DMSO/Nutlin-3 (Fig. 6C), indicating that pretreatment with CPT sensitizes ARPE-19 cells to Nutlin-3–induced apoptosis.
Inhibition of Src and PI3-Kinase Potentiates Nutlin-3–Induced Apoptosis
Because Nutlin-3 activated Akt/Mdm2 signaling, we investigated whether the inhibition of Akt by a specific PI3-kinase inhibitor (LY294002) and Src-kinase inhibitor (PP2) would increase the apoptotic response. Both LY294002 and PP2 inhibited Nutlin-3–mediated phosphorylation of Akt (Fig. 7B, lanes 2, 4, 6). Although LY294002 and PP2 had no cytotoxic effect, combining LY294002 or PP2 with Nutlin-3 significantly increased DNA fragmentation (Fig. 7A). We next determined whether LY294002 and PP2 increased apoptosis in response to Nutlin-3 by modulating the levels of p53 protein and its phosphorylation. LY294002 did not significantly alter the Nutlin-3–induced increase in p53 phosphorylation and protein (Fig. 7B, lanes 2, 4), suggesting the involvement of additional pathways. Conversely, cotreatment of cells with PP2 and Nutlin-3 increased p53 phosphorylation compared with the effect of Nutlin-3 alone (Fig. 7B, lanes 2, 6). However, the increase in p53 protein in response to PP2 + Nutlin-3 was less than to Nutlin-3 alone (Fig. 7B, lanes 2, 6). Both LY294002 and PP2 inhibited Nutlin-3–induced Mdm2 phosphorylation; however, the extent of inhibition was greater in cells treated with PP2. LY294002 decreased Mdm2 protein levels, which further decreased in response to PP2 (Fig. 7B, lanes 1, 5). Nutlin-3 decreased Mdm2 protein levels, which further decreased after LY294002 or PP2 with Nutlin-3, indicating enhanced apoptosis.
Figure 7.

LY294002 and PP2 sensitizes ARPE-19 cells to Nutlin-3–induced apoptosis. (A) Serum-starved ARPE-19 cell monolayers were pretreated with or without Src kinase inhibitor (PP2; 10 μM) and PI3-kinase inhibitor (LY294002; 10 μM) for 1 hour, followed by treatment with 60 μM Nutlin-3 for 3 hours. An equal volume of DMSO was used as a control. Samples were analyzed for DNA fragmentation using ELISA (mean ± SEM; n = 3). *P < 0.05; significantly different compared with Nutlin-3. (B) Western blot analysis for the levels of phospho- and total- p53, Mdm2, Akt, and Bcl-2 are shown. Actin was used as a loading control.
Since LY294002 and PP2 blocked Nutlin-3–induced Akt phosphorylation and potentiated Nutlin-3–induced apoptosis, we investigated whether Bcl-2 expression and its phosphorylation are involved in regulating apoptotic pathways in ARPE-19 cells. Consistent with our previous observations in Figure 6, Nutlin-3 decreased Bcl-2 expression. In addition, Nutlin-3 increased apoptosis (Fig. 7A) and Bcl-2 phosphorylation at Ser70 (Fig. 7B, lanes 1, 2). LY294002 increased Bcl-2 phosphorylation without changing its expression. On the other hand, PP2 increased Bcl-2 phosphorylation and decreased its expression. However, combining LY294002 or PP2 with Nutlin-3 further increased the phosphorylation of Bcl-2 Ser70 and decreased Bcl-2 levels (Fig. 7B), suggesting that the inhibition of Akt causes the accumulation of phosphorylated Bcl-2 and, thereby, alters its antiapoptotic function.
Cycloheximide Inhibits Nutlin-3–Induced Apoptosis
Cycloheximide (CHX), a protein synthesis inhibitor, decreases the synthesis of p53 and several of its transcriptional targets, including Bax and p21Cip1, and modulates p53-dependent apoptosis in various cell types, including intestinal epithelial cells.32 CHX inhibited basal and CPT-induced p53 expression and phosphorylation but had no effect on caspase-3 activation, as judged by the lack of active fragments (Fig. 8A). To determine whether CHX modulates Nutlin-3–induced apoptosis, we pretreated ARPE-19 cells with CHX followed by treatment with 60 μM Nutlin-3. CHX completely blocked Nutlin-3–induced p53 upregulation and decreased the expression far below the basal level (Fig. 8B). Although p73 was detected in ARPE-19 cells, both CPT and Nutlin-3 failed to alter its levels. Unlike p53, pretreatment with CHX had no effect on p73.
Figure 8.
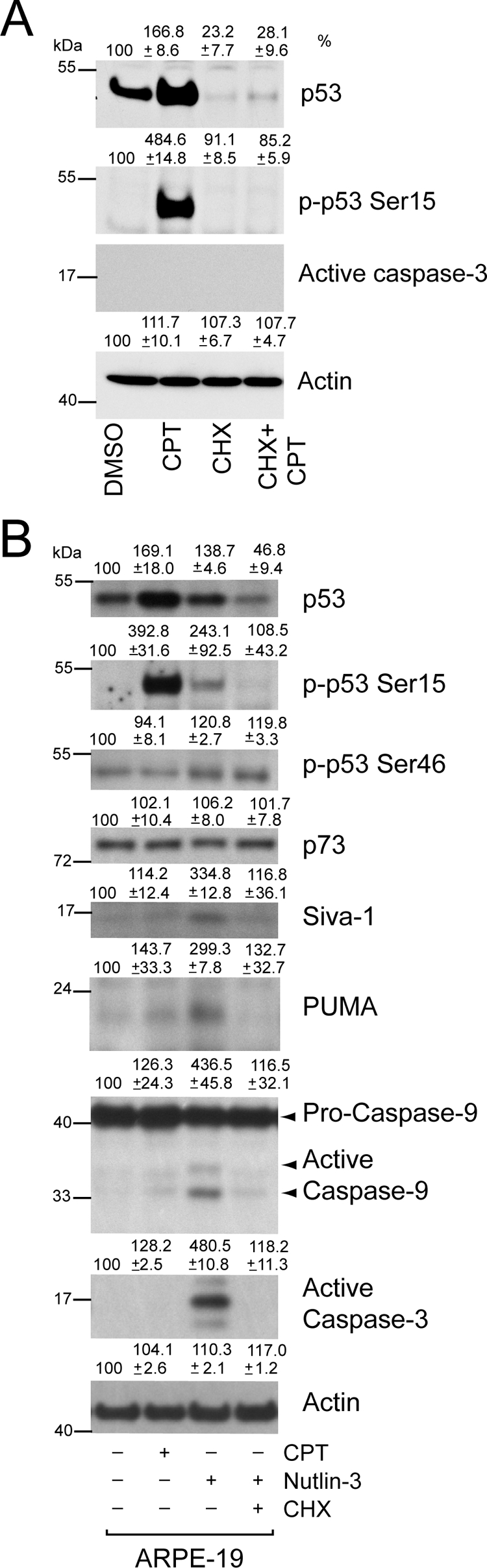
Cycloheximide inhibits Nutlin-3–induced apoptosis. (A) Confluent serum-starved ARPE-19 cells were pretreated with CHX for 1 hour, followed by treatment with or without 20 μM CPT for 3 hours. Western blot for changes in protein levels for phospho-53 and total p53, active caspase-3, and actin are shown. (B) Serum-starved cells were pretreated with CHX for 1 hour, followed by treatment with 60 μM Nutlin-3 for 2 hours. Cells were simultaneously treated with 20 μM CPT for 3 hours, and DMSO-treated cells were used as control. Western blot analysis for changes in protein levels for phospho-53 and total p53, p73, PUMA, Siva-1, and active caspase-9 and caspase-3 are shown.
Levels of phosphorylated p53 in response to Nutlin-3 were significantly lower than CPT-induced p53 phosphorylation. CHX inhibited Nutlin-3–induced p53 phosphorylation at Ser15. Phosphorylation of p53 at Ser46 is known to regulate the apoptotic activity of p53.43 CPT failed to stimulate p53 Ser46 phosphorylation in ARPE cells. Nutlin-3 increased the phosphorylation of p53 Ser46, which strongly correlated with its ability to induce apoptosis in these cells. Of particular importance is the role of CHX in modulating p53 Ser46 phosphorylation. CHX failed to inhibit p53 Ser46 phosphorylation (Fig. 8B), indicating CHX specifically inhibits the synthesis of proteins involved in the regulation of p53 Ser15 phosphorylation. In addition, CHX decreased the expression of Siva-1 and PUMA protein levels and concomitantly prevented the activation of caspases-9 and -3. CPT failed to induce the expression of PUMA and Siva-1 protein levels and consequently had no effect on the activation of caspase-3 (Figs. 8A, 8B).
Silencing of p53 Inhibits Nutlin-3–Mediated Apoptosis
Data presented thus far have suggested a role of p53 in Nutlin-3–mediated cytotoxicity. We examined the effect of inhibiting p53 expression by siRNA. We optimized experimental conditions to maximize inhibition of p53 gene expression by siRNA transfection. Specificity of p53 knockdown was confirmed by a significant decrease in total p53 protein in cells transfected with p53 siRNA but not in cells transfected with control siRNA (Fig. 9A). The levels of p73 did not change under these experimental conditions, indicating that siRNA transfection was specific for p53 and had no off-target effects on other members of the p53 family. Knockdown of p53 inhibited the expression of basal p21Cip1, Bcl-2 and partially inhibited Bcl-xL proteins (Fig. 9A), supporting the conclusion that p53 predominantly regulates p21Cip1 and Bcl-2 expression as downstream targets. Consistent with our previous observations in Figure 5B, Nutlin-3 upregulated the expression of PUMA and Siva-1, which, in turn, caused caspase-3 activation (Fig. 9B). CPT failed to modulate levels of PUMA and Siva-1 and, in turn, had no effect on caspase-3 activation and apoptosis (Fig. 9B, lanes 1, 2). Conversely, the inhibition of p53 accumulation in response to Nutlin-3 in cells transfected with p53 siRNA significantly blocked the upregulation of Siva-1 and PUMA (Fig. 9B, lanes 3, 6) and, consequently, inhibited caspase-3 activation and apoptosis. These results indicate that Nutlin-3–induced apoptosis is p53 dependent.
Figure 9.

Knockdown of p53 by gene-specific siRNA inhibits Nutlin-3–induced apoptosis. (A) ARPE-19 cells were transfected with control or p53-specific siRNA. Cell lysates were analyzed by Western blot for the levels of p53, p73, p21Cip1, Bcl-2, and Bcl-xL using specific antibodies. (B) Cells transfected with control or p53 siRNA were treated with 20 μM CPT for 3 hours or with 60 μM Nutlin-3 for 2 hours. Cell extracts were analyzed to determine the levels of PUMA, Siva-1, active caspase-3, and actin by Western blot analysis.
Discussion
Although primary RPE cells do not undergo apoptosis under normal conditions, apoptosis of intestinal epithelial cells is required for mucosal homeostasis. These differences can support the physiological need for long-term survival of primary RPE cells as opposed to the rapid turnover of intestinal epithelial cells. ARPE-19 cells, a spontaneously arising primary RPE cell line, and primary RPE cells were insensitive to cytokine and DNA damage-induced apoptosis compared with IEC-6 cells (Fig. 1A), likely because of the high expression of endogenous Bcl-xL and Bcl-2 proteins (Fig. 1B) and the decreased expression of proapoptotic Bax (Fig. 2A). We used primary cultures of RPE cells and compared them with ARPE-19 cultures to demonstrate that both cell types were resistant to apoptosis and expressed comparable levels of antiapoptotic Bcl-2 and Bcl-xL, thus indicating the presence of similar prosurvival defense mechanisms. Several posttranslational mechanisms, including phosphorylation and ubiquitination, are known to regulate Bcl-244 and Bcl-xL antiapoptotic function.45 Another plausible explanation is that the suppression of death-induced signaling complex formation by Bcl-xL prevents caspase-8 activation.46 Increased phosphorylation of Bcl-2 promotes survival in several cell lines, including intestinal epithelial cells.37 On the other hand, Neuroprotectin D1 dephosphorylates Bcl-xL at Ser62 during oxidative stress and promotes the survival of ARPE-19 cells,26 indicating that the phosphorylation status of various Bcl-2 family proteins could potentially regulate primary RPE survival. Phosphorylation within the flexible loop of Bcl-2 induces a conformational change that dictates its function.47 Some studies have shown that the phosphorylation of Bcl-2 at Ser70 inhibits its antiapoptotic effect.48,49 It has been suggested that Bcl-xL and Bcl-2 heterodimerize with proapoptotic proteins Bax and Bad at the outer mitochondrial membrane, which is known to inhibit the release of cytochrome c (which activates Apaf-1 and leads to capase-9 activation)50 and, therefore, preserves mitochondrial integrity. Phosphorylation and other posttranslational modifications of Bcl-2 in response to diverse stimuli are known to disrupt the association of Bcl-2 with other BH3 domain-only proteins, thereby freeing the sequestered proapoptotic proteins.47,48 As a result, Bcl-2 failed to protect cells from apoptosis. Nutlin-3 increased the phosphorylation of Bcl-2 at Ser70 and sensitized both the ARPE-19 and the primary RPE cells to apoptosis. Furthermore, the inhibition of Akt phosphorylation in response to Nutlin-3 further increased the phosphorylation of Bcl-2 at Ser70 and potentiated the death of ARPE-19 cells, suggesting a compelling correlation between the phosphorylation of Bcl-2 and the induction of apoptosis. Increased phosphorylation of Bcl-2 may result because of the activation of kinase or the inhibition of phosphatase. Because Bcl-2 phosphorylation increased in the presence of LY294002 and PP2, it is unlikely that Src or PI3-kinase phosphorylates Bcl-2. However, several kinases, including JNK, are known to regulate Bcl-2 phosphorylation at Ser70.37,44 Therefore, we predict that increased Bcl-2 phosphorylation and accumulation is probably mediated through the activation of kinases other than Src/PI3-kinase or the decreased activity of phosphatases.
Activation of p53 occurs by stabilization and posttranslational modifications in response to stress signals. IEC-6 cells expressed low basal levels of p53, which increased in response to CPT, leading to the increased expression of Bax and apoptosis (Fig. 2A). Conversely, confluent serum-starved ARPE-19 cells had high basal expression of p53 and p21Cip1 proteins (Fig. 2A). CPT robustly increased p53 protein levels and phosphorylation in both cell types (Fig. 2A). However, it failed to induce p53-dependent expression of PUMA, Noxa, and Bax (Figs. 8B, 9B), which caused mitochondrial depolarization. Therefore, no effect was observed on caspase-9 and -3 activation in ARPE-19 cells. Different results were reported by Nair et al.,13 who found that 0.1 μM CPT for 48 hours resulted in caspase-3 activation and cell death in ARPE-19 cells. The difference in the response of cells to CPT between our findings and those of Nair et al.13 appeared to be due to differences in cell cycle status. Serum-starved, quiescent, confluent ARPE-19 cells treated with 20 μM CPT for varying times failed to undergo apoptosis. However, cells treated with a low concentration of CPT (5 μM) at the time of plating (during proliferation) significantly increased mean doubling time (Fig. 3A). In addition, low doses of CPT significantly induced p53 and its downstream target, p21Cip1, indicating the predominant effects were on cell cycle regulators rather than on apoptotic inducers (Fig. 3B). Others have found that 100 μM CPT treatment for 4 hours failed to induce significant apoptosis, and prolonged exposure for 72 hours was required to induce significant cytotoxicity.51 The confluent, serum-starved, quiescent ARPE-19 monolayers used in our model are more representative of those found in an intact retina. Under normal physiologic conditions, primary RPE cells are constantly exposed to oxidant-mediated injuries, causing a mild, stresslike increase of p53-dependent signaling that may prime these cells to be protected from the toxicity of oxidative stress-induced cell death. Preconditioning ARPE-19 cells with sublethal doses of hydrogen peroxide increased the cellular resistance to subsequent toxic exposures.52 Thus, it is reasonable to posit that increased expression of p53 and p21Cip1 in ARPE-19 cells is crucial to modulate proliferation and to safeguard these cells from external stress. Both cell cycle arrest and senescence require complete engagement of p53 and Rb-mediated signaling pathways.53 p53-Mediated induction of p21Cip1 inhibits the cyclin-dependent kinase activity required for the phosphorylation of Rb protein, which releases bound E2F and leads to the resumption of cell cycle progression.40,41 Our results demonstrate that a low dose of Nutlin-3 (5 μM) for 24 hours upregulates p53 and causes a block in cell proliferation, primarily because of the accumulation of the Cdk inhibitor p21Cip1, which decreases levels of phosphorylated Rb (Fig. 3B). Nutlin-3 affects senescence in mouse fibroblasts54 and proliferation and differentiation in preosteoclasts55 by a similar p53-dependent mechanism.
We have demonstrated that Nutlin-3 (60 μM) increases the phosphorylation and expression of p53 in primary RPE (Fig. 5C) and ARPE-19 (Figs. 6A, 7B, 8B) cells, which is similar to the responses of CPT (Figs. 2A, 8A). Increased expression of p53 upregulates the expression of proapoptotic proteins PUMA, Bax, Noxa, and Siva-1 (Figs. 5D, 6A) and leads to apoptosis (Figs. 4A, 5B), suggesting that similar apoptotic pathways exists in both the ARPE-19 and primary RPE cells and that the inhibition of Mdm2 binding with p53 causes p53 activation and thereby sensitizes these cells to apoptosis. Furthermore, our results suggest a transition of p53-dependent signaling from cell cycle arrest to apoptosis that is affected by the duration and concentration of Nutlin-3. The concentrations of Nutlin-3 used in our studies were significantly higher than the 5- to 10-μM doses used by other investigators to induce apoptosis in retinoblastoma cells.18,56 Given that retinoblastoma cells rarely contain p53 mutations but have an inactivated Rb pathway, lower doses of Nutlin-3 may be able to efficiently antagonize the Mdm2-p53 interaction and cause cell death.18 We have recently reported that 5 μM Nutlin-3 is capable of inducing apoptosis in IEC-6 cells expressing wild-type p53.32 Cooperative interactions of Rb and p53 regulate senescence and apoptosis in various cell types.57 ARPE-19 cells have wild-type p53 and Rb, suggesting a synergistic role of Rb/p53 signaling, which probably does not permit p53 activation with low doses of Nutlin-3. In addition, the ability of Nutlin-3 to induce the apoptosis of retinoblastoma cells was shown to be independent of p53 phosphorylation.56 However, Nutlin-3 increased p53 phosphorylation in primary RPE and ARPE-19 cells during apoptosis (Figs. 5C, 7B, 8B). Nutlin-3–mediated p53 stabilization and phosphorylation were sufficient to induce cell death, suggesting that the consequences of p53 activation by the Mdm2 inhibitor were fundamentally different from those of other pathways driven by genotoxic stress and DNA damage.
Nutlin-3 decreased the levels of prosurvival proteins Bcl-xL and Bcl-2 in primary RPE (Fig. 5D) and ARPE-19 (Fig. 6A) cells. A shift in the ratio of antiapoptotic versus proapoptotic proteins alters mitochondrial outer membrane potential and induces caspases-9 and -3 activation, culminating in apoptosis. Interestingly, pretreatment of cells with CPT followed by Nutlin-3 potentiates cell death (Fig. 6C), suggesting that previous modifications of p53 in response to CPT amplify the ability of Nutlin-3 to cause cell death. One possible explanation is that preferential binding of p53 to the p21Cip1 promoter under basal conditions predominantly regulates proliferation and participates in enhanced survival responses. Nutlin-3 treatment shifts the binding of p53 to the promoters of PUMA, Bax, Noxa, and Siva-1, resulting in apoptosis. The ability of Nutlin-3 to increase PUMA and Siva-1 expression was inhibited by CHX, which, in turn, led to decreased caspases-9 and -3 activation and protected cells from apoptosis (Fig. 8B). A similar role for CHX protection against okadaic acid-induced cytotoxicity reported in ARPE-19 cells58 suggests that rapid de novo synthesis of proteins is necessary to induce cell death. Our p53 siRNA studies further confirmed the participation of p53 signaling in the apoptosis of ARPE-19 cells. Abrogation of p53 signaling prevented Nutlin-3 from inducing PUMA and Siva-1 and, in turn, attenuated caspase-3 activation (Fig. 9B), strongly reinforcing our hypothesis that Nutlin-3–induced apoptosis in ARPE-19 cells requires the activation of p53. Furthermore, p53 siRNA significantly decreased p21Cip1 and Bcl-2 expression but had a marginal effect on Bcl-xL, indicating that under basal conditions, Bcl-2 is predominantly regulated by p53. However, it can be argued that the decrease of Bcl-2/Bcl-xL expression in cells transfected with p53 siRNA should increase sensitivity to Nutlin-3 because Bcl-2/Bcl-xL binds proapoptotic BH3 proteins and sequesters them in the cytoplasm.59 Instead, the inhibition of these proteins significantly protected ARPE-19 cells from Nutlin-3–induced apoptosis. Because p53 siRNA inhibits the synthesis of PUMA and Siva-1 proteins, Nutlin-3 fails to induce apoptosis. Our data indicate that in the absence of functional p53, these changes are not sufficient to sensitize cells to apoptosis. The siRNA studies also suggest that p53 regulates the expression of proteins involved in proliferation as well as apoptosis. However, the outcome of p53 modulation depends on stimulus-specific, posttranslational modification of p53.
In summary, our results show that p53 regulates cell-cycle checkpoints by way of p21Cip1 and promotes cell survival by way of Bcl-2 expression. CPT activates p53 without causing apoptosis, suggesting that DNA damage–induced p53 activation preferentially participates in the regulation of cell cycle checkpoints. However, inhibition of the Mdm2-p53 interaction leads to the activation of p53, which stimulates PUMA, Noxa, Bax, and Siva-1, decreases Bcl-2 and Bcl-xL expression, and triggers primary RPE cell death. Based on our studies, Nutlin-3–mediated p53 activation can circumvent the resistance of primary RPE cells to apoptosis in response to chemotherapeutic drugs and could have potential advantages as a therapeutic agent in the treatment of proliferative disorders of the retinal pigment epithelium.
Acknowledgments
The authors thank Gregg Short and Danny Morse (Department of Physiology, University of Tennessee Health Science Center, Memphis, TN) for help in preparing the figures and Mary Jane Viar and Rebecca West for valuable suggestions and technical assistance.
Footnotes
Supported by National Institute of Diabetes and Digestive and Kidney Disease Grant DK-16505, National Eye Institute Vision Core Grant P30EY013080, and an unrestricted grant from Research to Prevent Blindness. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or of Research to Prevent Blindness.
Disclosure: S. Bhattacharya, None; R.M. Ray, None; E. Chaum, None; D.A. Johnson, None; L.R. Johnson, None
References
- 1. Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881 [DOI] [PubMed] [Google Scholar]
- 2. Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology. 2010;25:8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pastor JC. Proliferative viteroretinopathy: an overwiew. Surv Ophthalomol. 1998;43:3–18 [DOI] [PubMed] [Google Scholar]
- 4. Lee SC, Known OW, Seong GJ, Kim SH, Ahn JE, Kay ED. Epitheliomesenchymal transdifferentiation of cultured RPE cells. Opthalmic Res. 2001;33:80–86 [DOI] [PubMed] [Google Scholar]
- 5. Ando A, Ueda M, Uyama M, Masu Y, Ito S. Enhancement of dedifferentiation and myoid differentiation of retinal pigment epithelial cells by platelet derived growth factor. Br J Ophthalmol. 2000;84:1306–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hinton DR, He S, Lopez PF. Apoptosis in surgically excised choroidal neovascular membranes in age-related macular degeneration. Arch Ophthalmol. 1998;116:203–209 [DOI] [PubMed] [Google Scholar]
- 7. Dunaief JL, Dentchev T, Ying GS, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442 [DOI] [PubMed] [Google Scholar]
- 8. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233 [DOI] [PubMed] [Google Scholar]
- 9. Xu Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 2003;10:400–403 [DOI] [PubMed] [Google Scholar]
- 10. Green DR, Kroemer G. Cytoplasmic functions of the tumor suppressor p53. Nature. 2009;458:1127–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poyurovsky MV, Katz C, Laptenko O, et al. The C terminus of p53 binds the N-terminal domain of Mdm2. Nature Struct Mol Biol. 2010;17:982–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Velikay M, Stolba U, Wedrich A, Datlinger P, Akramian J, Binder S. The antiproliferative effect of fractionized radiation therapy: optimization of dosage. Doc Ophthalmol. 1994;87:265–269 [DOI] [PubMed] [Google Scholar]
- 13. Nair AR, Schliekelman M, Thomas MB, Wakefield J, Jurgensen S, Ramabhadran R. Inhibition of p53 by lentiviral-mediated shRNA abrogates G1 arrest and apoptosis in retinal pigmented epithelial cell line. Cell Cycle. 2005;4:697–703 [DOI] [PubMed] [Google Scholar]
- 14. Yang Y, Ludwig RL, Jensen JP, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7:547–559 [DOI] [PubMed] [Google Scholar]
- 15. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53-pathway by small-molecule antagonists of Mdm2. Science. 2004;303:844–848 [DOI] [PubMed] [Google Scholar]
- 16. Van-Maerken T, Speleman F, Vermeulen J, et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006;66:9646–9655 [DOI] [PubMed] [Google Scholar]
- 17. Coll-Mulet L, Iglesias-Serret D, Santidrian AF, et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood. 2006;107:4109–4114 [DOI] [PubMed] [Google Scholar]
- 18. Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66 [DOI] [PubMed] [Google Scholar]
- 19. Jiang YL, Escano MF, Sasaki R, et al. Ionizing radiation induces a p53-dependent apoptotic mechanism in ARPE-19 cells. Jpn J Ophthalmol. 2004;48:106–114 [DOI] [PubMed] [Google Scholar]
- 20. Westlund BS, Cai B, Zhou J, Sparrow JR. Involvement of c-Abl, p53 and the MAPK kinase JNK in the cell death program initiated in A2E-laden ARPE-19 cells by exposure to blue light. Apoptosis. 2009;14:31–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharma A, Sharma R, Chaudhary P, et al. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch Biochem Biophys. 2008;480:85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Park SE, Song JD, Kim KM, et al. Diphenyleneiodonium induces ROS-independent p53 expression and apoptosis in human RPE cells. FEBS Lett. 2007;581:180–186 [DOI] [PubMed] [Google Scholar]
- 23. Yang P, McKay BS, Allen JB, Jaffe GJ. Effect of NF-kappa B inhibition on TNF-alpha-induced apoptosis in human RPE cells. Invest Ophthalmol Vis Sci. 2004;45:2438–2446 [DOI] [PubMed] [Google Scholar]
- 24. Honda S, Hjelmeland LM, Handa JT. The use of hyperoxia to induce mild oxidative stress in RPE cells in vitro. Mol Vis. 2001;7:63–70 [PubMed] [Google Scholar]
- 25. Bernstein PS, Delori FC, Richer S, van Kuijk FJ, Wenzel AJ. The value of measurement of macular carotenoid pigment optical densities and distributions in age-related macular degeneration and other retinal disorders. Vision Res. 2010;50:716–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Antony R, Lukiw WJ, Bazan NG. Neuroprotectin D1 induces dephosphorylation of Bcl-xL in a PP2A-dependent manner during oxidative stress and promotes retinal pigment epithelial cell survival. J Biol Chem. 2010;285:18301–18308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang N, Peairs JJ, Yang P, et al. The importance of Bcl-xL in the survival of human RPE cells. Invest Ophthalmol Vis Sci. 2007;48:3846–3853 [DOI] [PubMed] [Google Scholar]
- 28. Yang P, Peairs JJ, Tano R, Zhang N, Tyrell J, Jaffe GJ. Caspase-8-mediated apoptosis in human RPE cells. Invest Ophthalmol Vis Sci. 2007;48:3341–3349 [DOI] [PubMed] [Google Scholar]
- 29. Chaum E. Comparative analysis of the uptake and expression of plasmid vectors in human ciliary and retinal pigment epithelial cells in vitro. J Cell Biochem. 2001;83:671–677 [DOI] [PubMed] [Google Scholar]
- 30. Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169 [DOI] [PubMed] [Google Scholar]
- 31. Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine: characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhattacharya S, Ray RM, Johnson LR. Role of polyamines in p53-dependent apoptosis of intestinal epithelial cells. Cell Signal. 2009;21:509–522 [DOI] [PubMed] [Google Scholar]
- 33. Bhattacharya S, Ray RM, Johnson LR. Basic helix-loop-helix protein E47-mediated p21Waf1/Cip1 gene expression regulates apoptosis of intestinal epithelial cells. Biochem J. 2007;407:243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ray RM, Bhattacharya S, Johnson LR. EGFR plays a pivotal role in the regulation of polyamine-dependent apoptosis in intestinal epithelial cells. Cell Signal. 2007;19:2519–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan Q, Viar MJ, Ray RM, Johnson LR. Putrescine does not support the migration and growth of IEC-6 cells. Am J Physiol Gastrointest Liver Physiol. 2000;278:G49–G56 [DOI] [PubMed] [Google Scholar]
- 36. Bhattacharya S, Ray RM, Johnson LR. Decreased apoptosis in polyamine depleted IEC-6 cells depends on Akt-mediated NF-kappaB activation but not GSK3beta activity. Apoptosis. 2005;10:759–776 [DOI] [PubMed] [Google Scholar]
- 37. Ray RM, Bhattacharya S, Johnson LR. Protein phosphatase 2A regulates apoptosis in intestinal epithelial cells. J Biol Chem. 2005;280:31091–31100 [DOI] [PubMed] [Google Scholar]
- 38. Bhattacharya S, Ray RM, Johnson LR. Polyamines are required for activation of c-Jun NH2—terminal kinase and apoptosis in response to TNF-α in IEC-6 cells. Am J Physiol Gastrointest Liver Physiol. 2003;285:G980–G991 [DOI] [PubMed] [Google Scholar]
- 39. Oren M. Decision making by p53: life death and cancer. Cell Death Differ. 2003;10:431–442 [DOI] [PubMed] [Google Scholar]
- 40. Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677 [DOI] [PubMed] [Google Scholar]
- 41. Ahlander J, Bosco G. The Rb/E2F pathway and regulation of RNA processing. Biochem Biophys Res Commun. 2009;384:280–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen L, Marechal V, Moreau J, Levine AJ, Chen J. Proteolytic cleavage of the Mdm2 oncoprotein during apoptosis. J Biol Chem. 1997;272:22966–22973 [DOI] [PubMed] [Google Scholar]
- 43. Oda K, Arakawa H, Tanaka T, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46 phosphorylated p53. Cell. 2000;102:849–862 [DOI] [PubMed] [Google Scholar]
- 44. Ruvolo PP, Deng X, May WS. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia. 2001;15:515–522 [DOI] [PubMed] [Google Scholar]
- 45. Basu A, Haldar S. Identification of a novel Bcl-xL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett. 2003;538:41–47 [DOI] [PubMed] [Google Scholar]
- 46. Wang X, Zhang J, Kim HP, Wang Y, Choi AM, Ryter SW. Bcl-xL disrupts death-inducing signal complex formation in plasma membrane induced by hypoxia/reoxygenation. FASEB J. 2004;18:1826–1833 [DOI] [PubMed] [Google Scholar]
- 47. Gross A, McDonnell JM, Korsmeyer SJ. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911 [DOI] [PubMed] [Google Scholar]
- 48. Haldar S, Jena N, Croce CM. Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci U S A. 1995;92:4507–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Basu A, DuBois G, Haldar S. Posttranslational modifications of Bcl-2 family members-a potential therapeutic target for human malignancy. Front Biosci. 2006;11:1508–1521 [DOI] [PubMed] [Google Scholar]
- 50. Huang DC, Strasser A. BH3-only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842 [DOI] [PubMed] [Google Scholar]
- 51. Hueber A, Esser P, Heimann K, Kociok N, Winter S, Weller M. The topoisomerase I inhibitors, camptothecin and beta-lapachone, induce apoptosis of human retinal pigment epithelial cells. Exp Eye Res. 1998;67:525–530 [DOI] [PubMed] [Google Scholar]
- 52. Jarrett SG, Boulton ME. Antioxidant up-regulation and increased nuclear DNA protection play key roles in adaptation to oxidative stress in epithelial cells. Free Radic Biol Med. 2005;38:1382–1391 [DOI] [PubMed] [Google Scholar]
- 53. Dimri GP. What has senescence got to do with cancer? Cancer Cell. 2005;7:505–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Efeyan A, Ortega-Molina A, Velasco-Miguel S, Herranz D, Vassilev LT, Serrano M. Induction of p53-dependent senescence by the Mdm2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 2007;67:7350–7357 [DOI] [PubMed] [Google Scholar]
- 55. Zauli G, Rimondi E, Corallini F, Fadda R, Capitani S, Secchiero P. Mdm2 antagonist Nutlin-3 suppresses the proliferation and differentiation of human pre-osteoclasts through a p53-dependent pathway. J Bone Miner Res. 2007;22:1621–1630 [DOI] [PubMed] [Google Scholar]
- 56. Elison JR, Cobrinik D, Claros N, Abramson DH, Lee TC. Small molecule inhibition of HDM2 leads to p53-mediated cell death in retinoblastoma cells. Arch Ophthalmol. 2006;124:1269–1275 [DOI] [PubMed] [Google Scholar]
- 57. Bennett MR, Macdonald K, Chan SW, Boyle JJ, Weissberg PL. Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res. 1998;82:704–712 [DOI] [PubMed] [Google Scholar]
- 58. Kaarniranta K, Ryhanen T, Sironen RK, et al. Geldenamycin activates Hsp70 response and attenuates okadaic acid-induced cytotoxicity in human retinal pigment epithelial cells. Brain Res Mol Brain Res. 2005;137:126–131 [DOI] [PubMed] [Google Scholar]
- 59. Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403 [DOI] [PubMed] [Google Scholar]