

Abstract
The FMR1 gene is polymorphic for the length of CGG trinucleotide repeat expansions in the 5′ untranslated region. Premutation (55–200 CGG repeats) and full mutation (>200 CGG repeats) alleles give rise to their respective disorders by different pathogenic mechanisms: RNA gain of function toxicity leads to fragile X-associated tremor/ataxia syndrome in the premutation range, and transcriptional silencing and absence of fragile X mental retardation protein (FMRP) leads to fragile X syndrome in the full mutation range. However, for the latter, incomplete silencing and/or size-mosaicism might result in some contribution to the disease process from residual mRNA production. To address this possibility, we examined the brains of 3 cases of fragile X syndrome for the presence of intranuclear inclusions in the hippocampal dentate gyrus. We identified low levels (0.1%–1.3%) of intranuclear inclusions in all 3 cases. Quantitative RT-PCR for FMR1 mRNA and immunofluorescence for FMRP revealed low but detectable levels of both RNA and protein in the 3 cases, consistent with the presence of small numbers of inclusions. The intranuclear inclusions were only present in FMRP-immunoreactive cells. The small numbers of inclusions and very low levels of both FMR1 RNA and protein suggest that the clinical course in these 3 subjects would not have been influenced by contributions from RNA toxicity.
Keywords: Autism, FMR1, FXTAS, Fragile X syndrome, Neurodegeneration, Parkinson
INTRODUCTION
The FMR1 gene is polymorphic for CGG trinucleotide repeats on the 5′ untranslated region. In the general population, most individuals harbor FMR1 alleles with fewer than 45 CGG repeats, with a modal value of approximately 30 repeats. For individuals with >200 repeats (full mutation range), the gene generally becomes transcriptionally silent (1–4), leading to fragile X syndrome (FXS), the leading single-gene cause of intellectual impairment and autism (5). CGG-repeat expansions in the premutation range (55–200 repeats) give rise to the leading known single-gene cause of primary ovarian insufficiency (5–7) and the late-onset neurodegenerative disorder, fragile X-associated tremor/ataxia syndrome (FXTAS). FXTAS affects ~40% of males and 8% to 16% of females who carry the premutation identified through known fragile X probands (8–12). Finally, the premutation is also associated with psychiatric problems (13–16), neurodevelopmental problems (5, 7, 17, 18), and immunological symptoms (19–21).
The pathogenesis of premutation-associated disorders, particularly FXTAS, is thought to be mediated by an RNA-toxicity mechanism that involves expression of the expanded CGG-repeat FMR1 mRNA (22–25) (Fig. 1), which is analogous to the pathogenic mechanism causing myotonic dystrophy (DM) in which the expanded CUG-repeat DMPK mRNA sequesters proteins that are involved with splice regulation (26). Thus, the mechanism underlying FXTAS (RNA toxicity) is entirely distinct from the mechanism for FXS, which is due to gene silencing and consequent absence of fragile X mental retardation protein (FMRP) (Fig. 1). FMR1 mRNA is present within the intranuclear inclusions that are the neuropathological hallmark of FXTAS (27–32). Consistent with the sequestration model of DM, functional protein sequestration has now also been observed for FXTAS (27). In this regard, although FXTAS has thus far been identified only in carriers of premutation alleles, it is possible that a minority of individuals with full mutation alleles that retain some transcriptional activity (33–35), or who harbor both full mutation and evident (or occult) premutation alleles (size mosaics) (36), would display neuropathological and/or clinical features of FXTAS, as long as some expanded CGG-repeat mRNA is present to trigger the pathogenic process.
Figure 1.
Sequestration model for fragile X–associated tremor/ataxia syndrome (FXTAS) and possible modes for development of RNA toxicity. The underlying pathogenic basis for FXTAS is believed to be RNA toxicity of the expanded CGG-repeat RNA; the expanded repeat is thought to sequester one or more proteins, thus impairing their normal cellular functions. Although all cases of FXTAS to date have been reported in carriers of premutation alleles (scenario i), there are 3 additional possibilities: (ii) the presence of both full mutation and premutation alleles, but with only premutation alleles active (solid arrow); (iii) a carrier identified as having only full mutation alleles, but with occult premutation alleles that are active; or (iv) a carrier with only full mutation alleles that retain some transcriptional activity. For scenarios i – iii, neuropathology is produced by premutation-expansion mRNA; for scenario iv, mRNA with full mutation CGG-repeat expansions is pathogenic.
MATERIALS AND METHODS
Subjects
Clinical and neuropathological characteristics of the 3 males with FXS have been described in detail elsewhere (37). Their case histories and molecular diagnostic features are summarized here.
Case 1: Patient manifested global developmental delay and was admitted to a residential facility at age 15 years. He was diagnosed with FXS at age 52, displaying on physical exam large, prominent ears, a long narrow face, macroorchidism, and a full scale IQ of 34; at age 65, his full scale IQ had fallen to 22. A chromosomal analysis at 52 years of age yielded 23% fragile-X-positive metaphases. He was a social individual who befriended his caretakers and peers. He repeated words and phrases and required no psychotropic medications. He was diagnosed at age 77 with a gait ataxia and parkinsonism not related to FXS and began treatment with amantadine but without therapeutic benefit. A computed tomography scan of the brain revealed reduced cerebral and cerebellar volumes compared to the general population, and ischemic white matter changes. Within the last 6 months of his life he was diagnosed with dementia and died at 78 years. Genomic analysis of DNA isolated from postmortem brain tissue established his full mutation status, with hypermethylated full mutation alleles (CGG repeats of 339, 486, 619, 755, 938, 1225) (Table).
Table 1.
Molecular Characteristics and Inclusion Neuropathology of the 3 Adults with Fragile X Syndrome
| Case Information | FMR1 mRNA Levels | Inclusions in Dentate Gyrus(c) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CGG | Methylation status | Mean | SEM | Neurons | % | Astrocytes | % | ||
| Case 1 | 339; 486; 619; 755; 938; 1225 | 100%(a) | Cortex | 0.003 | 9.31E-05 | 5 of 3085 | 0.16 | 4 of 900 | 0.44 |
| Cerebellum | 0.008 | 0.00015 | |||||||
| Case 2 | 436 | 100% | Cortex | 0.001 | 7.80E-05 | 10 of 2308 | 0.43 | 7 of 776 | 0.91 |
| Cerebellum | 0.027 | 0.002 | |||||||
| Case 3 | 429; (340–440)(b) | 72% | Cortex | 0.037 | 0.002 | 9 of 1330 | 0.68 | 5 of 365 | 1.34 |
| Cerebellum | 0.016 | 0.001 | |||||||
| Controls | all <40 | -- | Cortex | 1.134 | 0.065 | 0 of 12,895 | 0 of 2925 | ||
| Cerebellum | 0.831 | 0.055 | |||||||
| Negative Control | -- | -- | Cell Line | 0 | 0 | n/a | n/a | ||
Any unmethylated alleles would be present at levels below the detection threshold of our method (~5%).
CGG repeat values in parentheses are unmethylated alleles as measured by Southern Blot.
All Fragile X Syndrome cases had intranuclear inclusions in neurons and astrocytes of the dentate gyrus granule cell layer; none of the control cases (n = 5) had intranuclear inclusions.
Case 2: Patient had experienced developmental delay in early childhood, with severe language deficits and the inability to speak in full sentences. He was hyperactive and anxious throughout childhood and showed excessive rocking, hand-flapping, poor eye contact, tactile defensiveness, and destructive tantrums resulting in a diagnosis of autism. He entered a group home at age 30 years, where he required antipsychotic medications for behavior problems. Death at age 57 years was due to failed resuscitative efforts after choking. Molecular testing of postmortem brain tissue demonstrated the presence of a methylated full mutation allele of 436 CGG repeats (Table).
Case 3: Patient had delayed development and physical features including long face, prominent ears, and macroorchidism, which led to a diagnosis of fragile X syndrome. Abnormal behavioral features included anxiety and hand biting. He was shy but interactive and he was able to speak in full sentences. He had savant skills for trivia and he had a full scale IQ of 57 in adulthood. At age 64 he developed a primary liver neoplasm and died within 3 months. Southern blot analysis of DNA isolated from postmortem brain tissue showed methylation mosaicism, with a hypermethylated full mutation allele of 429 CGG repeats present in 72% of the cells and unmethylated alleles ranging from approximately 340 to 440 CGG repeats present in the remaining cells (Table).
Tissue Samples
Tissue harvest and brain autopsies for all 3 cases were performed in accordance with University of California, Davis, Institutional Review Board-approved protocols. One hemisphere of the fresh brain was cut in coronal blocks that were separately frozen at −80°C. The remaining hemisphere was fixed in 10% phosphate-buffered formalin. The formalin-fixed hemisphere was examined grossly for pathology. For this study, serial sections through the hippocampus proper were analyzed. Data reported are from a single section and standardized in location across cases, but similar results were identified in sections across the hippocampus. Hippocampi were analyzed because FXTAS cases have shown clear evidence of pathologic alterations in this region. Evaluations of FMRP immunoreactivity and colocalization of inclusions within FMRP-reactive cells were carried out in sections from the frontal cortex for comparison with FMR1 mRNA measurements from the frontal cortex because hippocampal tissue was unavailable for RNA or protein measurements.
FMR1 Genotyping (CGG-Repeat Size Quantification)
Genomic DNA was isolated from 500 mg of frozen cortex and cerebellum from each of the 3 cases using standard methods (Qiagen, Valencia, CA). In Cases 1 and 3, DNA was also extracted from whole blood (3–5 ml). The results from CGG-repeat sizing from whole blood were consistent with those obtained in brain tissue. Repeat size and methylation status were determined by both PCR and Southern Blot analysis using a FluorChem 8800 Image Detection System (Alpha Innotech, San Leandro, CA), as previously described (35). For Southern blot analysis, 10 μg of DNA was digested with EcoRI and NruI, and the Stb12.3 FMR1 genomic sequence, labeled with Dig-11-dUTP, was used as the probe. Genomic DNA was also amplified by PCR using the c and f primers. Details of the DNA testing are as previously described (38, 39).
FMR1 mRNA Level Quantification
Total RNA was isolated from frontal cortex brain tissue using standard procedures (Trizol, Invitrogen, Tigard, OR). Expression levels of the FMR1 gene and of the control gene, glucuronidase, were measured by real-time quantitative fluorescence RT-PCR methods as previously described (33, 34, 37, 38).
Histology and Immunofluorescence Staining
Tissue blocks were processed for paraffin embedding, histochemical staining and immunostaining by routine methodologies, including hematoxylin and eosin (H&E), Congo red, and Luxol fast blue-periodic acid Schiff stains and Bielschowsky impregnation. Immunocytochemical staining using antibodies against ubiquitin (Z0458), glial fibrillary acidic protein (GFAP; Z0334), tau protein (PHF8; A0024), neurofilament (NF; N1591), and β-amyloid precursor protein (β-APP; M0872) (all from Dako, Carpinteria, CA) was performed using automated methods (Dako immunostainer), as previously reported (21, 28, 31, 37). Appropriate positive and negative staining controls were employed for each antibody.
Immunofluorescence staining was performed on sections from the frontal cortex of the 3 FXS cases using antibodies to either the C- or N-terminus of human FMRP (mouse anti-FMRP; Millipore 1C3 clone; chicken anti-FMRP; Aves Labs, Tigard, OR; 40) and anti-ubiquitin. We observed very similar reactivity with the mouse (1C3 clone) and chicken anti-FMRP antibodies in all experiments; this report focuses on results using the latter. Secondary antibodies used were Alexa Fluor 488 (for FMRP; Molecular Targets; Tigard, OR) and Alexa 546 (for ubiquitin; Molecular Targets). Briefly, tissues were deparaffinized through xylene and graded alcohols, treated for antigen retrieval for 30 minutes in Tris-Buffer, pH 8.5 at 90°C, and allowed to return to room temperature (RT) for an additional 30 minutes. The tissue was treated with 0.1% sodium borohydride for 15 minutes, followed by 0.5% H2O2 for 30 minutes with gentle agitation. Tissues were then treated for 60 minutes with a 0.1 M copper (II) sulfate solution in a 100 mM ammonium acetate buffer (pH 5.0) to reduce contributions of autofluorescence from lipofuscin to the immunofluorescence staining. The tissue was then rinsed in 0.1 M phosphate buffered saline for 30 minutes and blocked in 10% normal goat serum in 0.01 M phosphate buffered saline with 0.3% Triton X-100. Blocking solution was used as a diluent for the primary antibodies that were diluted to 1:1000 (v/v; 1C3), 1:20,000 (v/v; chicken anti-FMRP), and 1:1000 (v/v; rabbit anti-ubiquitin). The tissue was incubated overnight at RT in the primary antibody with gentle agitation, then rinsed with 10% normal goat serum and incubated for 1 hour in the secondary antibody cocktail (each secondary antibody 1:2000 v/v in 10% normal goat serum and 0.3% triton X-100) at RT with gentle agitation. Sections were counterstained with DAPI in Vectamount hard set mounting media (Vector Labs, Burlingame, CA) and secured with coverslips. Wild type C57BL/6J mouse and age-matched, non-FXS human control frontal cortex tissue sections were used as positive controls to guide immunostaining. Sections from an Fmr1 knock-out mouse lacking the Fmr1 promoter region and first exon, generated by Willemsen et al (41), were used as negative controls (i.e. Fmr1 gene-deficient with zero FMRP reactivity). All immunofluorescent staining was run in a single batch with human and mouse control sections that omitted the primary antibody as controls. Antibody reactivities were evaluated using a Nikon ECLIPSE E-600 epifluorescence microscope.
Quantification of FMRP and Ubiquitin Reactivity
To quantify the number of FMRP-reactive cells in the FXS cases, 200 cells were counted in layers II–IV of the frontal cortex at 400x magnification starting at the apex of the most prominent gyrus on the slide and proceeding medially into the sulcus, counting every cell in the field of view. FMRP-reactive cells were counted if the immunoreactivity was associated with a DAPI-stained nucleus with a granular appearance and perinuclear pattern. The presence of intranuclear inclusions, as defined by ubiquitin-positive staining, was simultaneously quantified to allow for a direct comparison between the presence or absence of FMRP and presence of inclusions in the same cells. Intranuclear inclusions were defined as a clear, round, prominent ubiquitin signal inside a DAPI-stained nucleus and near a nucleolus. This method also allowed for a semiquantitative calculation of the numbers of FMRP-reactive cells that showed inclusions compared to non-FMRP-reactive cells.
Quantification of Intranuclear Inclusions in the Hippocampus
Because of an extremely low number and irregular distribution of intranuclear inclusions identified in the FXS cases on initial survey, it would not be appropriate to use stereological methods, which depend upon an even distribution of inclusions throughout the region of interest (28). Therefore, to provide a semiquantitative estimate of inclusion numbers, a single coronal hippocampal section at the level of the lateral geniculate nucleus was selected from each case for consistency across cases. Every granule cell and astrocyte in the granule cell layer of the dentate gyrus in the section was counted at 1000x (H&E) to determine a percentage of cells with intranuclear inclusions. The granule cell layer was chosen because it has been shown previously in cases of males with FXTAS that approximately 1% to 7% of granule cells and 20% to 40% astrocytes in the granule cell layer show intranuclear inclusions (28); moreover, the dentate gyrus granule cell layer provided a clear region of interest to guide cell counting that was consistent across cases. Hippocampus sections from 5 age-matched, non-FXS control cases were counted using identical methods and no intranuclear inclusions were observed in any control case. An experimenter blinded to the identity of all slides performed all cell counting.
RESULTS
Gross Examination
Brains of Cases 1 and 3 were grossly normal with mild cortical atrophy deemed appropriate for age. The brain of Case 2 showed moderate ventricular dilatation and prominent atrophy of the hippocampus and amygdala. The cerebella of all 3 cases appeared generally decreased in size. On midline sagittal sectioning of the vermis, both the superior and posterior lobar divisions were smaller compared to those of age-matched control brains. Further analyses of pathological features in the cerebella are reported elsewhere (37).
Light Microscopy
In all 3 cases, the penetrating cortical arterioles of the temporal cortex showed vascular hyalinosis. These vessel walls were negative for amyloid birefringence with Congo red stain, but showed faint staining for β-APP (not shown). Immunostains were also negative in brain parenchyma for β-amyloid, α-synuclein, and tau, i.e. immunostains routinely used for the diagnosis of the common neurodegenerative disorders Alzheimer disease, Lewy body disease, and frontotemporal degenerations (data not shown).
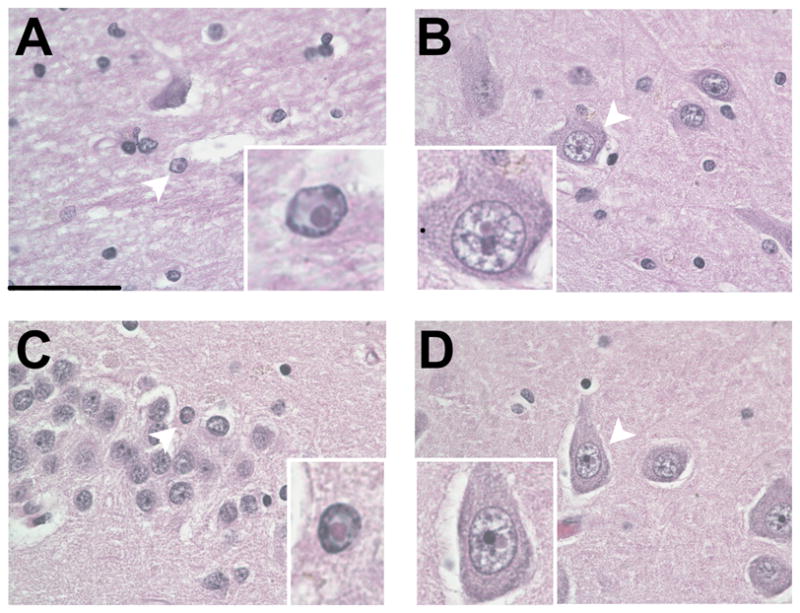
Upon closer inspection of H&E-stained sections of the hippocampus, eosinophilic intranuclear inclusions were identified in neuronal and astrocytic nuclei of the hilus, CA1, and dentate gyrus of all 3 cases (Table). The percentages of granule cells and astrocytes with inclusions identified in these cases of FXS (0.1%–1.3% in astrocytes) are extremely low compared to the reported numbers of inclusions in the dentate gyrus granule cell layer in FXTAS (28, 29). The inclusions were ovoid, hyaline in appearance, between 2 and 5 μm in diameter, most often near the nucleolus (Fig. 2A–D) and stained positively for ubiquitin and were negative for Congo red and periodic acid Schiff and Bielschowsky; thus, they fit all criteria for intranuclear inclusions described previously in FXTAS, as defined by Greco et al (28, 29). Inclusions were also identified in granule cells in the cerebellum but were not quantified (data not shown).
Figure 2.

Intranuclear inclusions in cases of fragile X syndrome (FXS). (A) Intranuclear inclusion in an astrocyte in the hippocampal hilus in Case 1. (B) Intranuclear inclusions in a neuron in the hilus of Case 2. (C) Granule cell with an intranuclear inclusion in the dentate gyrus of Case 2. (D) Pyramidal neuron in CA3c with an intranuclear inclusion in Case 3. All plates 1000x original magnification; the scale bar for large images is 50 μm. Insets are optical magnifications of the cells containing inclusions, indicated by white arrowheads.
FMRP and Ubiquitin Immunofluorescent Staining
To verify whether there was detectable tissue expression of FMRP, we performed immunofluorescence staining on 5-μm-thick paraffin sections from frontal cortex. Positive staining was present in all 3 cases based on FMRP-positive staining surrounding DAPI-stained nuclei; approximately 15% of cells were positive in Cases 1 and 2 and 22% of cells in Case 3 (Fig. 3A–C, F). In contrast, the Fmr1 knock-out mouse negative control showed no detectable FMRP immunoreactivity (Fig. 3D) and positive control sections from wild type mice and age-matched control cases without FXS showed FMRP immunoreactivity in a clear majority of cells (>80%, non-FXS controls; Fig. 3E). The non-FXS human control sections from which primary antibody was omitted and the control tissues that were not incubated with primary antibody did not show high levels of lipofuscin-related fluorescence, indicating that the copper (II) sulfate treatment was sufficient to reduce autofluorescence that might have confounded interpretation of the results in these tissues. The results, including all controls, were confirmed using 2 separate antibodies targeting either the C- or N-terminal regions of FMRP and did not differ depending on which anti-FMRP antibody was used. It was also noted that the pattern of FMRP reactivity within cells appeared distinct in the cases with FXS compared to control tissue. In the 3 FXS cases, FMRP staining appeared to display a more prominent perinuclear pattern rather than the typical, broader distribution throughout the soma and proximal portion of the apical dendrites observed in age-matched control cases stained in parallel (Fig. 3E). Case 1 showed FMRP reactivity in ~11% of cells counted, and all showed a perinuclear staining pattern. Additionally, the red ubiquitin immunofluorescence in Case 1 was relatively high in the cytoplasm, often adjacent to green FMRP immunofluorescence, resulting in a yellow-green cytoplasmic staining pattern (Fig. 3Aa). Intranuclear inclusions were clearly visible as bright, round, red puncta within a DAPI-stained nucleus in only FMRP-reactive cells. Case 2 showed FMRP reactivity similar to Case 1, with a clear perinuclear pattern in 14% of the cells counted as well as ubiquitin reactivity in the cytoplasm; although in Case 2, the ubiquitin reactivity did not appear consistently adjacent to the FMRP reactivity. Intranuclear inclusions were clearly visible as bright, round, red puncta within a DAPI-stained nucleus in only FMRP-reactive cells. Case 3 showed a greater number of FMRP-reactive cells (22%), which was expected because this case was shown to have methylation mosaicism. The ubiquitin signal in Case 3 was lower than that in the other 2 cases, but was also constrained to a perinuclear pattern. Interestingly, there was a subset of cells in Case 3 that demonstrated a relatively normal FMRP reactivity pattern, such that the cytoplasm was filled with FMRP reactivity and there was a hint of straining into the apical dendrites. An example with an intranuclear inclusion is shown in Fig. 3Cc. In all 3 FXS cases, ubiquitin-positive intranuclear inclusions were present only in cells showing FMRP reactivity (Fig. 3A–C, F). Quantification of the number of FMRP-reactive cells and intranuclear inclusions is tabulated in Fig, 3F. It is important to note that intranuclear inclusions were only present in FMRP-reactive cells in the FXS cases and never present in non-FXS control cases.
Figure 3.

Intranuclear inclusions colocalize with fragile X mental retardation protein (FMRP)–reactive cells in fragile X syndrome (FXS). (A) Ubiquitin (red) and FMRP (green) immunoreactivity in Case 1. (a) Optical enlargement of inclusions in FMRP-reactive cells. (B) Ubiquitin and FMRP immunoreactivity in Case 2. (b) Optical enlargement of inclusions in FMRP-reactive cells. C: FMRP immunoreactivity in Case 3. (c) Optical enlargement of inclusions in FMRP-reactive cells. (D) Fmr1 knockout mouse negative control. (E) Non-FXS control case stained for ubiquitin and FMRP. Ubiquitin immunoreactivity is not sufficiently high to be visualized. (F) Quantification of FMRP-reactive cells and ubiquitin-positive intranuclear inclusions in 200 cells counted. In no cases were inclusions identified in FMRP-non-reactive cells, nor were inclusions identified in non-FXS control cases. All tissues were treated to reduce lipofuscin-related fluorescence. Scale bar: 100 μm in A applies to A-E.
Genotyping and mRNA Levels
The diagnosis of FXS in the 3 cases was conducted using PCR techniques. Southern blot analysis clearly showed the presence of methylation mosaicism in Case 3 while no apparent mosaicism was detected in the other cases. However, the detection limit of the Southern blot analysis may have masked a very small percentage of cells carrying an unmethylated premutation or full mutation allele (<5%, Table). Measurements of FMR1 mRNA expression in both frontal cortex and cerebellum revealed very low expression levels (Table), consistent with the presence of a small percent of unmethylated alleles and therefore mosaicism. In addition, we observed correspondingly low FMR1 mRNA expression (<2%, Table), in agreement with the very low percentages of cells bearing intranuclear inclusions (<1%) and showing FMRP-positive immunofluorescence (<1%). Importantly, in the present study it is possible to make comparisons between the FMR1 mRNA levels, FMRP levels and the presence of inclusions because the quantification of FMR1 mRNA levels, FMRP immunoreactivity and the presence of intranuclear inclusions were all made in the frontal cortex.
DISCUSSION
We have detected small numbers of intranuclear inclusions in granule cell and astrocytic cell populations in the dentate gyrus of the hippocampus of 3 older adult cases with FXS. We were also able to identify the presence of intranuclear inclusions in the frontal cortex of each case with FXS. Although the presence of such inclusions is generally considered to be restricted to the premutation range (wherein the FMR1 gene is active and producing elevated levels of expanded CGG-repeat containing FMR1 mRNA) it had remained a formal possibility that either residual transcriptional activity of unmethylated full mutation alleles (methylation mosaicism) (34) or the existence of small numbers of transcriptionally-active premutation alleles (size mosaicism) could give rise to occasional inclusion-bearing cells in FXS (Fig. 1). Indeed, mice with unmethylated, full mutation alleles do possess small numbers of intranuclear inclusions (42). Interestingly, the Fmr1 gene remains fully unmethylated above 200 CGG repeats in CGG knock-in mice (42, 43). The present study provides the first report of intranuclear inclusions in full mutation carriers with FXS, supporting the idea that even low expression levels of premutation- or full-mutation length FMR1 mRNA are sufficient to promote intranuclear inclusion formation. In the CGG knock-in mouse model, the existence of inclusions for alleles with more than 200 CGG repeats is consistent with an RNA-based mechanism for cellular toxicity and intranuclear inclusion formation (25, 26), since the Fmr1 gene retains transcriptional activity (42). Importantly, the mouse model suggests that the premutation- and full-mutation length alleles themselves, and not necessarily elevated Fmr1 expression levels, are sufficient to result in inclusion formation, albeit at relatively low levels (42).
The RNA-based model of inclusion formation raises the expectation that there is at least residual transcriptional activity in the 3 FXS cases. Indeed, measurement of bulk FMR1 mRNA yielded low but detectable levels of FMR1 mRNA in the frontal cortex of each of the FXS cases studied. Using immunofluorescent staining we also detected very low levels of FMRP immunoreactivity as a measure of gene activity in the same brain regions where the FMR1 mRNA levels were quantified (i.e. frontal cortex). The residual FMR1 mRNA and FMRP protein expression do not permit definition of the type of mosaicism giving rise to residual expression and inclusion formation; however, Southern blot analysis of these 3 cases suggests the presence of both size-and methylation-mosaicism in at least one of them, i.e. Case 3.
The absence of ubiquitin-positive intranuclear inclusions in cells negative for FMRP reactivity is important because it supports the models proposing that the expanded (premutation or full mutation) FMR1 mRNA is critical for inclusion formation. Based on a semiquantitative analysis it appears that within the population of FMRP-reactive cells, the numbers of intranuclear inclusions are similar to those previously reported in FXTAS cases (i.e. 10%–20%). This finding is important because the relatively low number of inclusions in the hippocampi of these FXS cases most likely reflects the relative sparseness of FMRP reactivity seen in the cortex.
It should be noted that the presence of rare intranuclear inclusions in a predominately full mutation background in the context of FXS does not imply that such intranuclear inclusions are indicators of any clinically important neurodegenerative process similar to that seen in FXTAS. Rather, the presence of intranuclear inclusions in the context of FMR1 gene expression (albeit at a reduced level) most likely reflects cellular dysfunction within the small number of cells that harbor the unmethylated full mutation- and/or premutation-sized alleles. An important consideration for future studies of individuals with the full mutation and FXS is whether continued transcription of unmethylated full mutation alleles (34) could result in a distinct, but related, neurodegenerative process to FXTAS, or whether such unmethylated alleles contribute to susceptibility or generation of a completely separate neurodegenerative disorder unrelated to FXTAS. Although movement disorders have been reported in 38.6% of aging individuals (males; n = 44) with FXS (44), at present there is no evidence that such additional neurological diseases are related to mosaicism of the FMR1 gene. Moreover, no pathological anatomical features have been reported in FXS cases that would suggest features indicative of a movement disorder (44). Importantly, Parkinsonism was reported in 9% of the males (n = 44) in the study of Utari et al (44). For the current study it is likely that Case 1 had parkinsonian features (37), but these were unrelated to the methylation mosaicism on the FMR1 gene.
Furthermore, it should be emphasized that none of the 3 FXS cases demonstrated FXTAS symptomatology, including peripheral neuropathy or intention tremor; nor did they have Bergmann gliosis or white matter hyperintensity in the middle cerebellar peduncles. The lack of FXTAS features, in conjunction with the clear presence of FXS symptoms, strongly suggests these FXS cases did not manifest neurodegenerative features similar to those of FXTAS. Further clinical-molecular studies of aging patients with the full mutation, both with and without mosaicism, are needed in order to assess the importance and the functional role of rare intranuclear inclusion formation in patients with FXS. The fact that inclusions, although few, can be found in some cases of FXS despite extremely low levels of full mutation FMR1 mRNA would suggest that elevated FMR1 mRNA levels are not directly responsible for inclusion formation; rather, even low levels of FMR1 mRNA may be sufficient to support the formation of inclusions.
Acknowledgments
This work was supported by National Institute of Health (NIH) grants HD036071, HD056031, NS044299, AG024488, HD002274, MH077554, MH078041, RL1 AG032115, RL1 NS062411, and RL1 AG032119; the National Fragile X Foundation, and the M.I.N.D. Institute. This work was also made possible by a Roadmap Initiative grant (UL1 DE019583) from the National Institute of Dental and Craniofacial Research (NIDCR) in support of the NeuroTherapeutics Research Institute (NTRI) consortium; and by a grant (UL1 RR024146) from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
References
- 1.Fu YH, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell. 1991;67:1047–58. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 2.Pieretti M, Zhang FP, Fu YH, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–22. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 3.Verkerk AJMH, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 4.Snow K, Doud LK, Hagerman R, et al. Analysis of a CGG sequence at the FMR-1 locus in fragile X families and in the general population. Am J Hum Genet. 1993;53:1217–28. [PMC free article] [PubMed] [Google Scholar]
- 5.Chonchaiya W, Schneider A, Hagerman RJ. Fragile X: a family of disorders. Adv Pediatr. 2009;56:165–86. doi: 10.1016/j.yapd.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wittenberger MD, Hagerman RJ, Sherman SL, et al. The FMR1 premutation and reproduction. Fertil Steril. 2007;87:456–65. doi: 10.1016/j.fertnstert.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Chonchaiya W, Utari A, Pereira GM, et al. Broad clinical involvement in a family affected by the fragile X premutation. J Dev Behav Pediatr. 2009;30:544–51. doi: 10.1097/DBP.0b013e3181c35f25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coffey SM, Cook K, Tartaglia N, et al. Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146A:1009–16. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez-Revenga L, Gomez-Anson B, Munoz E, et al. FXTAS in Spanish patients with ataxia: support for female FMR1 premutation screening. Mol Neurobiol. 2007;35:324–8. doi: 10.1007/s12035-007-0020-3. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, et al. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009;17:1359–62. doi: 10.1038/ejhg.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez-Revenga L, Santos MM, Sanchez A, et al. Screening for FXTAS in 95 Spanish patients negative for Huntington disease. Genet Test. 2008;12:135–9. doi: 10.1089/gte.2007.0074. [DOI] [PubMed] [Google Scholar]
- 12.Jacquemont S, Hagerman RJ, Leehey MA, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291:460–9. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 13.Bourgeois JA, Coffey SM, Rivera SM, et al. A review of fragile X premutation disorders: expanding the psychiatric perspective. J Clin Psychiatry. 2009;70:852–62. doi: 10.4088/JCP.08m04476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bourgeois JA, Cogswell JB, Hessl D, et al. Cognitive, anxiety and mood disorders in the fragile X-associated tremor/ataxia syndrome. Gen Hosp Psychiatry. 2007;29:349–56. doi: 10.1016/j.genhosppsych.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourgeois JA, Farzin F, Brunberg JA, et al. Dementia with mood symptoms in a fragile X premutation carrier with the fragile X-associated tremor/ataxia syndrome: clinical intervention with donepezil and venlafaxine. J Neuropsychiatry Clin Neurosci. 2006;18:171–7. doi: 10.1176/jnp.2006.18.2.171. [DOI] [PubMed] [Google Scholar]
- 16.Bourgeois JA, Seritan AL, Casillas EM, et al. Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J Clin Psychiatry. 2011;72:175–82. doi: 10.4088/JCP.09m05407blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chonchaiya W, Nguyen DV, Au J, et al. Clinical involvement in daughters of men with fragile X-associated tremor/ataxia syndrome. Clin Genet. 2010;78:39–46. doi: 10.1111/j.1399-0004.2010.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farzin F, Perry H, Hessl D, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27:S138–S44. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 19.Chonchaiya W, Tassone F, Ashwood P, et al. Autoimmune disease in mothers with the FMR1 premutation is associated with seizures in their children with fragile X syndrome. Hum Genet. 2010;128:540–8. doi: 10.1007/s00439-010-0882-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Coffey S, Lua LL, et al. FMR1 premutation in females diagnosed with multiple sclerosis. J Neurol Neurosurg Psychiatry. 2009;80:812–4. doi: 10.1136/jnnp.2008.160960. [DOI] [PubMed] [Google Scholar]
- 21.Greco CM, Tassone F, Garcia-Arocena D, et al. Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch Neurol. 2008;65:1114–6. doi: 10.1001/archneur.65.8.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tassone F, Beilina A, Carosi C, et al. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. RNA. 2007;13:555–62. doi: 10.1261/rna.280807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tassone F, Hagerman PJ. Expression of the FMR1 gene. Cytogenet Genome Res. 2003;100:124–8. doi: 10.1159/000072846. [DOI] [PubMed] [Google Scholar]
- 24.Tassone F, Hagerman RJ, Taylor AK, et al. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Gen. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Arocena D, Hagerman PJ. Advances in understanding the molecular basis of FXTAS. Hum Mol Genet. 2010;19:R83–9. doi: 10.1093/hmg/ddq166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol. 2010;67:291–300. doi: 10.1002/ana.21948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sellier C, Rau F, Liu Y, et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248–61. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greco CM, Berman RF, Martin RM, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–55. doi: 10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- 29.Greco CM, Hagerman RJ, Tassone F, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–71. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- 30.Iwahashi CK, Yasui DH, An HJ, et al. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–71. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 31.Tassone F, Hagerman RJ, Garcia-Arocena D, et al. Intranuclear inclusions in neural cells with premutation alleles in fragile X-associated tremor/ataxia syndrome. J Med Genet. 2004;42:e43. doi: 10.1136/jmg.2003.012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–5. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- 33.Tassone F, Hagerman RJ, Loesch DZ, et al. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am J Med Genet. 2000;94:232–7. doi: 10.1002/1096-8628(20000918)94:3<232::aid-ajmg9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 34.Tassone F, Hagerman RJ, Taylor AK, et al. A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J Med Genet. 2001;39:453–6. doi: 10.1136/jmg.38.7.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tassone F, Hagerman RJ, Chamberlain WD, et al. Transcription of the FMR1 gene in individuals with fragile X syndrome. Am J Med Genet. 2000;97:195–203. doi: 10.1002/1096-8628(200023)97:3<195::AID-AJMG1037>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 36.Nolin SL, Glicksman A, Houck GE, Jr, et al. Mosaicism in fragile X affected males. Am J Med Genet. 1994;51:509–12. doi: 10.1002/ajmg.1320510444. [DOI] [PubMed] [Google Scholar]
- 37.Greco CM, Navarro CS, Hunsaker MR, et al. Neuropathological features in the hippocampus and cerebellum of three older men with fragile X syndrome. Mol Autism. 2011;2:2. doi: 10.1186/2040-2392-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filipovic-Sadic S, Sah S, Chen L, et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem. 2010;56:409–18. doi: 10.1373/clinchem.2009.136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tassone F, Pan R, Amiri K, et al. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–9. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwahashi C, Tassone F, Hagerman RJ, et al. A quantitative ELISA assay for the fragile X mental retardation 1 protein. J Mol Diagn. 2009;11:281–9. doi: 10.2353/jmoldx.2009.080118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, et al. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis. 2006;21:549–55. doi: 10.1016/j.nbd.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 42.Brouwer JR, Huizer K, Severijnen LA, et al. CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome. J Neurochem. 2008;107:1671–82. doi: 10.1111/j.1471-4159.2008.05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Entezam A, Biacsi R, Orrison B, et al. Regional FMRP deficits and large repeat expansions into the full mutation range in a new Fragile X premutation mouse model. Gene. 2007;405:125–34. doi: 10.1016/j.gene.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Utari A, Adams E, Berry-Kravis E, et al. Aging in fragile X syndrome. J Neurodev Disord. 2010;2:70–6. doi: 10.1007/s11689-010-9047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]