

Abstract
In humans, corticosteroids are often administered prenatally to improve lung development in preterm neonates. Studies in exposed children as well as in children, whose mothers experienced significant stress during pregnancy indicate behavioral problems and possible increased occurrence of epileptic spasms. This study investigated whether prenatal corticosteroid exposure alters early postnatal seizure susceptibility and behaviors. On gestational day 15, pregnant rats were injected i.p. with hydrocortisone (2× 10 mg/kg), betamethasone (2× 0.4 mg/kg) or vehicle. On postnatal day (P)15, seizures were induced by flurothyl or kainic acid (3.5 or 5.0 mg/kg). Horizontal bar holding was determined prior to seizures and again on P17. Performance in the elevated plus maze was assessed on P20-22. Prenatal exposure to betamethasone decreased postnatal susceptibility to flurothyl-induced clonic seizures but not to kainic acid-induced seizures. Prenatal hydrocortisone decreased postnatal weight but did not affect seizure susceptibility. Hydrocortisone alone did not affect performance in behavioral tests except for improving horizontal bar holding on P17. A combination of prenatal hydrocortisone and postnatal seizures resulted in increased anxiety. Prenatal exposure to mineralocorticoid receptor blocker canrenoic acid did not attenuate, but surprisingly amplified the effects of hydrocortisone on body weight and significantly worsened horizontal bar performance. Thus, prenatal exposure to excess corticosteroids alters postnatal seizure susceptibility and behaviors. Specific effects may depend on corticosteroid species.
Keywords: betamethasone, hydrocortisone, kainic acid, flurothyl, anxiety
1. INTRODUCTION
In humans, prenatal (24–34 weeks of gestational age) administration of synthetic corticosteroids (betamethasone or dexamethasone) as well as hydrocortisone is used for prevention of respiratory distress syndrome in those pregnancies threatened with premature delivery (Jobe and Soll 2004, Roberts and Dalziel 2006). The treatment improves lung development and significantly reduces morbidity and mortality of infants if preterm delivery occurs (Guinn, et al. 2001). However both in humans and experimental animals, prenatal surges of corticosteroids or stress during the last third of pregnancy have been repeatedly linked to postnatal behavioral impairments (Aghajafari, et al. 2002, Battin, et al. 2007, Crowther, et al. 2007, Davis, et al. 2006, Diaz, et al. 1997, French, et al. 2004, Lemaire, et al. 2000, Purdy and Wiley 2004, Stott 1973, Vallee, et al. 1999, Vallee, et al. 1997, Welberg and Seckl 2001).
On the other hand, the risks and benefits of the prenatal corticosteroid treatment in terms of seizure susceptibility changes remain unclear. Recently, a clinical study linked higher level of stress experienced by mothers during pregnancy to postnatal occurrence of a seizure disorder, infantile spasms (Shang, et al. 2010). In experimental animals, prenatal treatments with betamethasone or dexamethasone have anticonvulsant effects in the kindling model (Velíšek 2005, Young, et al. 2006). In developing offspring, prenatal betamethasone increases hippocampal expression of anticonvulsant neuropeptide Y (NPY) (Heilig, et al. 1993, Velíšek 2006b), corroborating thus findings of anticonvulsant effects of prenatal betamethasone or dexamethasone exposure in kindling epileptogenesis.
Placental 11beta-hydroxysteroid-dehydrogenase converting in rats corticosterone to much less active 11-dehydro-corticosterone protects the fetus from high levels of maternal glucocortioids. However, if the maternal levels are excessive, natural corticosteroids would eventually cross placental barrier and bind to both glucocorticoid and mineralocorticoid receptors in the fetal brain (Joels 2001, Levitt, et al. 1996, Reul, et al. 1987). Betamethasone and dexamethasone are synthetic hormones with high affinity for glucocorticoid receptors. Synthetic corticosteroids are poor substrate for 11beta-hydroxysteroid-dehydrogenase (Siebe et al. 1993). After systemic administration in pregnant rodents, they easily cross placental barrier and bind to glucocorticoid receptors in the fetal brain (de Kloet 2003, Matthews 2000). For investigation of the effects of prenatal corticosteroid excess, prenatal administration of corticosteroids may represent a convenient and quantifiable model (Barbazanges, et al. 1996, Maccari, et al. 1995).
Here we investigated the effects of prenatal exposure to synthetic corticosteroid betamethasone versus natural hydrocortisone on seizure susceptibility and behaviors in immature rats. Both these corticosteroids are used prenataly in humans (Roberts and Dalziel 2006). Further, we used a mineralocorticoid receptor (MR) antagonist to determine the effects mediated via this receptor system. The hypothesis was that prenatal exposure to synthetic corticosteroids would enhance seizure susceptibility and worsen performance in behavioral tests.
2. EXPERIMENTAL PROCEDURES
2.1. Animals and prenatal treatments
Female, timed pregnant Sprague-Dawley rats were purchased from Taconic Farms. Experiments were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine and carried out according to the Revised Guide for the Care and Use of Laboratory Animals [NIH GUIDE, 25(28), 1996] as well as to the “Principles of laboratory animal care” (NIH publication No. 86-23, revised 1985). All effort has been made to reduce pain and suffering. Rats were kept on regular light:dark cycle (light 07:00–19:00) with free access to food and water. On gestational day 15 (G15), pregnant rats received two doses of betamethasone (0.4 mg/kg injected i.p. each) or two doses of hydrocortisone (10 mg/kg i.p. each) at 08:00 and 18:00 (all drugs were purchased from Sigma, St. Louis, MO, USA, unless stated otherwise). These doses were found effective in previous studies (Velíšek 2006b) and in pilot experiments. Control pregnant rats received two i.p. injections of the equivalent volume of vehicle (saline). Additional pregnant rats were treated at 08:00 and 18:00 with 50 mg/kg of MR receptor antagonist canrenoic acid and 1 hour later with hydrocortisone at 10 mg/kg. Some rats received vehicle (normal saline) instead of canrenoic acid and additional rats received two injections of canrenoic acid followed by a vehicle instead of hydrocortisone. Day of birth was considered zero. On postnatal day 1 (P1), the offspring were counted, sex-identified, and litters were reduced to 10 pups, if possible 5 males and 5 females. The offspring were used in the experiments starting on P15, which in terms of brain development corresponds to human infants (Avishai-Eliner, et al. 2002, Gottlieb, et al. 1977). At P15, body weight of the animals was determined as biological control because prenatal exposure to corticosteroids is associated with the decreases in postnatal body weight (Velíšek 2006b, Welberg and Seckl 2001, Young et al., 2006). Each experimental subgroup contained pups from at least three different litters to minimize the “litter effect”. It should be emphasized that not all the rats were exposed to all the tests. However, rats tested on the horizontal bar on P15 (prior to seizures) were retested on P17 (after the seizures). The experiments with a particular prenatal exposure were always run in parallel with saline-exposed controls. While we attempted to carry the experiments blindly, all animals were weighted prior to testing and the decreased weight gain pointed to those exposed to corticosteroids. Betamethasone and hydrocortisone experiments were carried out during different seasons and therefore, their controls were also run separately. However, initial analysis indicated that there were no differences between the controls for hydrocortisone and betamethasone and thus, control data were combined. In preliminary experiments, cross-fostering was performed in half of the offspring between saline- and hormone-treated mothers since a previous study indicated an attenuating role of cross-fostering on the effects of prenatal exposure (Brabham, et al. 2000). Cross-fostering was then introduced as an additional variable into ANOVA. As we found no main effect of cross-fostering and no interactions with other effects, we did not continue with cross-fostering beyond preliminary experiments.
The effects of prenatal betamethasone exposure on postnatal behaviors without seizure experience have been reported previously (Velíšek 2006b).
2.2. Seizure induction and parameters
2.2.1. Flurothyl seizures
were induced in an air-tight chamber (volume 9.4 l). The rat was gently placed in the chamber and flurothyl delivery has been initiated at a constant rate of 40 μl per minute onto a pad of filter paper (Sperber and Moshé 1988). Flurothyl (SynQuest Laboratories, Inc., Alachua, FL, USA) is a liquid convulsant ether with low boiling point temperature. Therefore, it easily evaporates from the filter paper and is inhaled by the tested rat. Flurothyl induces two different primarily generalized seizures (Velíšek 2006a): clonic seizures of face and forelimbs with preserved righting ability, and tonic-clonic seizures of all four limbs after the righting ability has been lost. In immature rats, flurothyl-induced clonic and tonic-clonic seizures develop in very fast succession (Velíšková, et al. 1996). Latency to onset of clonic and tonic-clonic seizures was determined. From this data we calculated thresholds for seizure types in terms of amount of infused flurothyl required for the induction of that particular seizure (Lánský, et al. 1997).
2.2.2. Kainic acid (KA) seizures
were induced by a solution of KA in PBS (final pH adjusted to 5.5) injected intraperitoneally. The following doses were used: 3.5 and 5.0 mg/kg (always in 10 ml/kg). In addition a 16 mg/kg dose was used in some hydrocortisone-exposed rats. Kainic acid elicits automatic behaviors, in infant rats mostly consisting of scratching (Stafstrom, et al. 1992, Velíšková, et al. 1988). Further, continuous clonic seizures (status epilepticus) develop usually after loss of righting (Albala, et al. 1984). Latency to onset of the first automatism and status epilepticus was recorded.
2.3. Behavioral testing
2.3.1. Bar holding
on the horizontal bar tests motor capabilities and gross motor behavior defects in highly motivating environment (Murphy, et al. 1995). During the test, the rat is positioned perpendicularly on the horizontal bar (1.5 cm diameter, positioned 50 cm above the padded floor) with the front paws reaching the bar. Once the rat grasps the bar with the hindpaws, the timer is started. Bar holding was investigated with a criterion of 2 minutes on P15 (prior any seizure challenge) and on P17 (i.e., 2 days after seizure testing). Three trials have been performed on each day and the best (longest) result entered the statistics (Velíšek 2006b).
2.3.2. Elevated-plus maze test
was administered on P20-22. The rats were tested in one session per day, for total of three sessions. Elevated-plus maze consists of two open arms and two closed arms connected with a 10×10 cm platform. The maze utilizes natural fear of the rats from open spaces (File 1993, Treit, et al. 1993), testing thus innate anxiety (unconditional fear related to human panic disorder (Zangrossi, et al. 2001)). The rat is positioned at the end of an open arm facing outwards and the latency to enter the distant third of the closed arm is determined. This latency (transfer latency; TL) (Hliňák and Krejčí 2002, Pellow and File 1986, Velíšek 2006b) has inverse relationship to innate anxiety: The longer the latency, the less anxiety is present. Repeated exposure to the elevated-plus maze test also involves a significant memory retention component because the rat can remember position of the safe (enclosed) area. Therefore, it is possible to calculate a memory retention index from the transfer latency during repeated exposures, which is indicative of learning and memory. Retention index (RI) was calculated as RI=log(TLP20)−log(TLP22). RI values less than zero indicate impaired memory retention, value of zero marks no memory retention, and values above zero indicate that learning and memory retention was present over the trials.
2.4. Statistics
All data were first analyzed using two-way ANOVA (factors: prenatal exposure; sex). In case there was no main effect of sex and no interaction between the main effects, data for both sexes were combined. In relevant analyses, body weight was introduced as a covariate. Since we were interested in the effects of prenatal exposure, then in the absence of other main effects or interactions, the presentation of the results is focused on the prenatal exposure effect. Post-hoc analysis after ANOVA was performed by the Fisher Protected Least Square Degree (PLSD) test. In case of two-group comparisons, Student’s t-test was used. Level of significance was preset at p<0.05 adjusted for multiple group comparisons if necessary.
3. RESULTS
3.1. Body weight
Prenatal hydrocortisone exposure on G15 (n=86) significantly decreased body weight on P15 compared to saline-exposed controls (n=75; ANOVA F(1,157)=4.069; *p<0.05; Figure 1A). Females (n=72) had significantly lower body weight than males (n=89; ANOVA F(1,157)=7.801; p<0.05). There was no interaction between prenatal treatment and sex of the subjects.
Figure 1. Prenatal corticosteroid exposure decreases postnatal weight.

(A) Exposure to both hydrocortisone (left) and betamethasone (right) on gestational day 15 significantly decreased postnatal weight on P15; *p<0.05.
(B) In different groups of prenatally exposed rats, hydrocortisone (HC) again decreased postnatal body weight. When canrenoic acid (CAN; an mineralocorticoid receptor antagonists) was co-administered with hydrocortisone, the weight loss was further amplified. Canrenoic acid applied in control arrangement had no such effect on the body weight; *p<0.05 versus 2x saline + 2x saline group or 2x CAN + 2x saline group; #p<0.05 versus 2x saline + 2x HC group.
Prenatal exposure to betamethasone on G15 (n=31) also significantly lowered body weight on P15 compared to saline-exposed animals (n=30; Figure 1A; ANOVA F(1,57)=6.318; *p<0.05). While there was a consistent trend in females (n=32) to lower body weight compared to males (n=29), in this particular setting there was no significant difference and no interaction.
Both prenatal exposures with saline+hydrocortisone (n=10) and canrenoic acid+hydrocortisone (n=10) significantly decreased body weight (Figure 1B; ANOVA F(3,42)=76.824; *#p<0.05) compared to both saline+saline-exposed controls (n=10; post-hoc Fisher PLSD test; *p<0.05) or canrenoic acid+saline-exposed rats (n=20; post-hoc Fisher PLSD test; #p<0.05). The females (n=22) in this experiments had again significantly lower body weight than males (n=28; ANOVA F(1,42)=7.816; p<0.05; not illustrated), however there was no interaction between prenatal treatment and sex.
3.2. Seizure susceptibility
3.2.1. Flurothyl seizures
In rats prenatally exposed to hydrocortisone (n=15), there was no effect of prenatal exposure on susceptibility to flurothyl-induced clonic (F(1,25)=0.723; p>0.05) and tonic-clonic (F(1,25)=1.771; p>0.05; Figure 2A) seizures compared to saline-exposed controls (n=14). There was no effect of sex and no interaction of main effects, therefore male and female data were combined for presentation. Prenatal exposure to betamethasone (n=7) significantly increased threshold for flurothyl-induced clonic seizures, i.e., there was an anticonvulsant effect compared to saline-exposed controls (n=6; Figure 2B; Student’s t-test; *p<0.05). Despite a trend, there was no significant effect of prenatal betamethasone exposure on tonic-clonic flurothyl seizure threshold.
Figure 2. Prenatal exposure to betamethasone, but not hydrocortisone, decreases susceptibility to flurothyl-induced clonic seizures.

(A) Prenatal exposure to hydrocortisone had no effects on susceptibility to either clonic or tonic-clonic flurothyl-induced seizures compared to controls. (B) Prenatal exposure to betamethasone significantly increased amount of flurothyl required to induce clonic seizures compared to controls (increased flurothyl seizure threshold = anticonvulsant effect). There was no effect on the tonic-clonic seizure threshold.
*p<0.05 compared to controls.
3.2.2. Kainic acid-induced seizures
In prenatally hydrocortisone-exposed rats after the 3.5 mg/kg KA dose (n=24) and saline-exposed controls (n=15), there was no effect of prenatal exposure on the latency to onset of first automatisms (F(1,35)=0.022; p>0.05; Figure 3A), no effect of sex, and no interaction. Similarly latency to onset of status epilepticus was not affected by either prenatal exposure (F(1,35)=1.488; p>0.05; Figure 3A) or sex, and there was no interaction of the effects. After the dose of 5.0 mg/kg of KA (n=24) versus controls (n=19), there was again no effect of prenatal exposure on the latency to onset of automatisms (F(1,39)=2.202; p>0.05; Figure 3B), no sex difference, and no interaction of the effects. In the latency to onset of status epilepticus there was no effect of prenatal exposure (F(1,38)=1.751; p>0.05; Figure 3B), no sex difference, and no interaction. Dose-response for both automatisms and status epilepticus in hydrocortisone-exposed rats and in controls was investigated by adding a group of rats injected with 16 mg/kg of KA (hydrocortisone-exposed n=7; saline-exposed n=11). Two-way ANOVA was used (factors: prenatal exposure; dose of KA). There was a significant effect of KA dose on the latency to onset of the first automatism (F(2,93)=17.685; *p<0.05), but no effect of prenatal exposure was present (F(1,93)=0.003; p>0.05), and there was no interaction. There were similar findings for status epilepticus: Only the effect of KA dose was significant (F(2,83)=58.716; *p<0.05; Figure 3C), but no effect of prenatal exposure (F(1,83)=1.330; p>0.05) or interaction of the effects were observed.
Figure 3. Prenatal exposure to either betamethasone or hydrocortisone does not alter susceptibility to kainic acid-induced seizures.

(A, B) There were no effects of prenatal hydrocortisone exposure on automatisms and status epilepticus induced by 3.5 mg/kg (C) or 5.0 mg/kg (D) of kainic acid.
(C) Overview of the KA dose-response for automatisms and status epilepticus in prenatally hydrocortisone- and saline-exposed rats. In addition to the 3.5 and 5.0 mg/kg doses, the dose of 16.0 mg/kg of KA was used. Two-way ANOVA revealed the effects of the KA dose for both symptoms (<-*-> p<0.05; significant difference only in the horizontal direction indicated by the arrows across the KA doses), but not of the prenatal exposure (vertical direction).
(D, E) Prenatal exposure to betamethasone did not change latency to onset of automatisms or status epilepticus induced by 3.5 mg/kg (A) or 5.0 mg/kg (B) of kainic acid compared to controls.
In prenatally betamethasone-exposed rats after 3.5 mg/kg dose of KA (n=23), there was no effect of prenatal exposure on latency to onset of automatisms compared to controls (n=21; F(1,38)=0.026; p>0.05; Figure 3D), no sex difference, and no interaction. Similarly, latency to onset of status epilepticus was not affected by prenatal exposure (F(1,38)=1.108; p>0.05; Figure 3D) or sex. For the 5.0 mg/kg dose of KA, group size was smaller (saline-exposure, n=7; betamethasone-exposure, n=8). Therefore combined male and female data were evaluated using Student’s t-test. Despite small anticonvulsant trends, there was no significant effect of prenatal betamethasone exposure on latency to onset of first automatisms (p>0.05) or status epilepticus (p>0.05; Figure 3E).
The rats surviving seizures (see Table 1) were followed in additional behavioral tests. Prenatal hydrocortisone exposure significantly enhanced lethal effects of 3.5 mg/kg dose of KA.
Table 1.
Survival through P17 after seizures on P15 and prenatal exposure on G15
| prenatal exposure G15 | seizure induction on P15 | ||
|---|---|---|---|
| flurothyl | kainic acid 3.5 mg/kg | kainic acid 5.0 mg/kg | |
| saline | 25/25 | 29/30 | 17/33 |
| betamethasone 2× 0.4 mg/kg | 7/7 | 21/24 | 5/8 |
| hydrocortisone 2× 10 mg/kg | 23/23 | 7/14 | 13/25 |
|
| |||
| statistics: χ2, p | n/a | 15.7; 0.0004* | 0.329; 0.8483 |
First number in the field marks the number of rats surviving on P17 while the second number indicates the number of rats tested in the particular seizure test on P15. All rats experiencing fluorthyl seizures survived. On the other hand, prenatal hydrocortisone treatment significantly decreased survival rate after seizures induced by 3.5 mg/kg of kainic acid i.p.
3.3. Behavioral assessment after seizures
3.3.1. Horizontal bar holding
Body weight was not involved in the horizontal bar holding (see below), therefore it was not used as a covariate. First, the rats were tested in horizontal bar holding on P15 prior to seizure test. Prenatal exposure (levels: saline; n=82, betamethasone; n=30, and hydrocortisone; n=62) did not alter horizontal bar holding performance on P15 (ANOVA F(2,173)=0.691; p>0.05; Figure 4A). There was no effect of sex and there was no interaction of factors. Combination of postnatal seizures with prenatal exposure to corticosteroids did not affect horizontal bar holding (ANOVA F(2,144)=0.593; p>0.05; Figure 4A). Although there was an additional factor of seizure (levels: KA 3.5, KA 5.0 and flurothyl), statistical analysis did not show any main effect of seizure, or sex, and there was no interaction of the effects.
Figure 4. Combination of prenatal exposure to hydrocortisone and postnatal seizures decreases transfer latency in the elevated-plus maze.
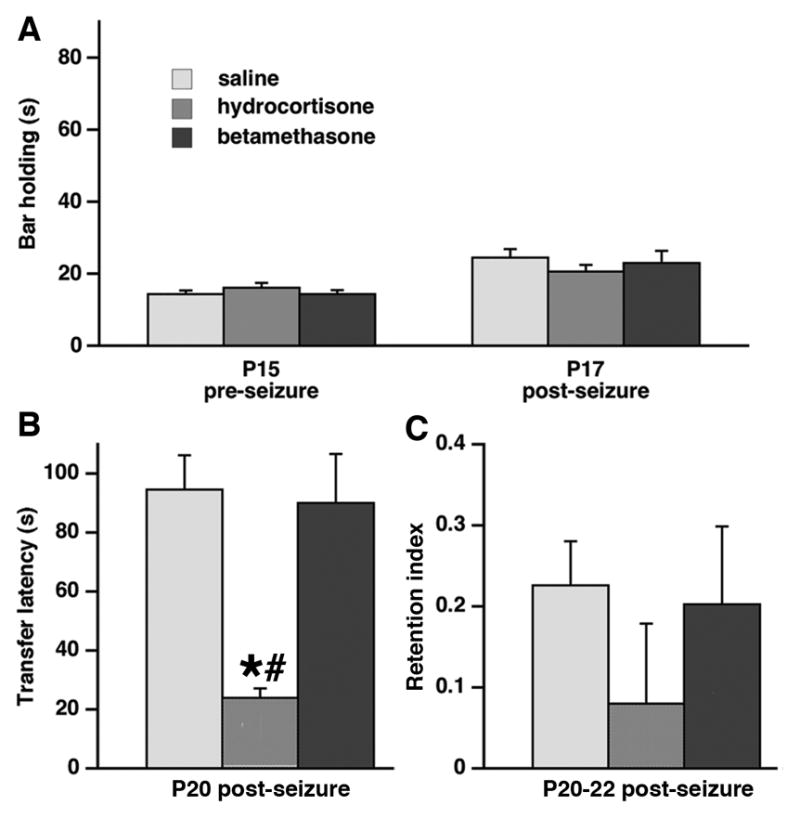
(A) There was no effect of either prenatal hydrocortisone or prenatal betamethasone exposure on horizontal bar performance on P15 (prior to seizure challenge) or on P17 (tested after the P15 seizure experience).
(B) Only prenatal exposure to hydrocortisone in combination with P15 seizures significantly decreased transfer latency in the elevated plus maze assessed on P20 (*p<0.05 versus controls; #p<0.05 versus prenatal betamethasone-exposure) indicating increased anxiety.
(C) Both prenatal hydrocortisone and betamethasone exposure in combination with P15 seizures did not alter retention index determined on P20-22 in the elevated plus maze compared to saline-exposed controls.
3.3.2. Elevated plus maze: Transfer latency
Transfer latency is indicative of innate anxiety. On P20, there was a significant effect of prenatal exposure on the transfer latency (ANOVA F(2,114)=3.286; p<0.05; Figure 4B). As there was no effect of sex, seizure type, and no interaction, these factors were removed from analysis. Fisher PLSD post-hoc test determined that transfer latency significantly decreased in rats prenatally exposed to hydrocortisone (n=13) compared to prenatal saline exposure (n=71; *p<0.05) as well as compared to prenatal betamethasone exposure (n=33; #p<0.05). These findings are consistent with increased anxiety after prenatal hydrocortisone exposure combined with postnatal seizure experience.
3.3.3. Elevated plus maze: Retention index
Retention index calculated from repeated testing (P22 and P20) in the elevated plus maze serves as a measure of memory retention. In rats experiencing seizures on P15, there was no significant effect of prenatal exposure on the memory retention index (ANOVA F(2,114)=0.517; p>0.05; Figure 4C). The group sizes were identical as in 3.3.2. There was no effect of sex, seizure, and no interaction. This finding indicates that memory retention was unaffected by a combination of prenatal corticosteroid exposure and postnatal seizure experience.
3.4. Behavioral assessment in rats prenatally exposed to hydrocortisone but not subjected to seizures
These experiments were performed only in prenatally hydrocortisone-exposed rats or in the rats exposed to a combination of canrenoic acid and hydrocortisone. Holding on the horizontal bar may be affected by body weight. Body weight was introduced as a covariate in the ANOVA because body weight was altered by prenatal exposure to corticosteroids (see Figure 1). However, there was no effect of this covariate. On P15 in all seizure-naïve rats, prenatal exposure to hydrocortisone (n=91) did not affect performance on the horizontal bar (ANOVA F(1,176)=0.026; p>0.05; Figure 5A) compared to saline controls (n=81). There was no effect of sex and no interactions between any of the factors. The test was repeated on P17 in those rats not subjected to seizure challenge. Interestingly, on P17 those rats prenatally exposed to hydrocortisone (and not subjected to seizures; n=17) had significantly better horizontal bar performance than prenatally saline-exposed rats (n=10; ANOVA F(1,25)=9.623; p<0.05; Figure 5B). There was again no effect of sex and no interaction between factors. The rats were also tested in the elevated plus maze on P20-22. On P20, there was no effect of prenatal exposure to hydrocortisone (n=26) on the transfer latency (ANOVA F(1,45)=0.325; p>0.05; Figure 5C) compared to controls (n=21), no effect of sex, and no interaction between the factors. Finally, using repeated testing in the elevated plus maze between P20-22, the retention index was determined. Similar to transfer latency, prenatal exposure to hydrocortisone (n=25) had no effect on the retention index compared to prenatal saline exposure (n=21; ANOVA F(1,44)=0.407; p>0.05; Figure 5D). Again, there was no effect of sex and no interaction.
Figure 5. Prenatal hydrocortisone exposure without seizure experience improves horizontal bar holding in the repeated test.

(A) Bar holding determined on P15 was not affected by prenatal hydrocortisone exposure compared to controls.
(B) When the horizontal bar holding was repeated on P17, those rats exposed prenatally to hydrocortisone significantly improved performance compared to controls (*p<0.05).
(C) There was no effect of prenatal hydrocortisone exposure on transfer latency determined in the elevated plus maze on P20.
(D) Prenatal hydrocortisone exposure did not affect memory retention index calculated from the transfer latency on P20 and P22 in the elevated plus maze.
(E) Prenatal co-administration of canrenoic acid (CAN) and hydrocortisone (HC) significantly decreased performance on the horizontal bar compared to both prenatally saline-exposed controls (*p<0.05) and to controls injected with canrenoic acid and saline (#p<0.05).
Pretreatment with canrenoic acid (ANOVA F(3,34)=7.865; p<0.05; Figure 5E) significantly affected bar holding. Post-hoc tests determined that the group prenatally treated with canrenoic acid+hydrocortisone (n=10) suffered impairments in bar holding compared to all other groups (*p<0.05). Body weight as a covariate in ANOVA did not show any effect of on the horizonatal bar performance. Similarly, there was no effect of sex (ANOVA F(1,30)=0.120; p>0.05) and no interaction between the factors. Please note that the horizontal bar performance in the P15 rats prenatally-exposed to double saline+saline (n=10), double saline+hydrocortisone (n=10) or double canrenoic acid+saline (n=20; Figure 5E) was not different from those rats exposed to a single injection of saline or hydrocortisone on P15 (Figure 5A).
4. DISCUSSION
Main results of this study are: (1) Prenatal betamethasone exposure alters threshold for flurothyl-induced clonic seizures in P15 rats, yet it is without effects on kainic acid-induced seizures. Prenatal hydrocortisone exposure did not affect seizure susceptibility. (2) Combination of prenatal hydrocortisone exposure and postnatal seizures results in increases in anxiety behaviors. (3) Mineralocorticoid receptor antagonist canrenoic acid co-administered with prenatal hydrocortisone amplifies the decrease in body weight gain induced by hydrocortisone and impairs the horizontal bar performance on P15. (4) Prenatal exposure to hydrocortisone improves horizontal bar holding in repeated test on P17 in seizure naïve rats.
If, in this study, any effect or prenatal corticosteroid exposure on seizure susceptibility was found, it was the anticonvulsant effect after prenatal betamethasone exposure, i.e., the susceptibility to seizures was decreased. At least two previous studies are consistent with this finding, revealing decreases in seizure susceptibility in immature rats in both hippocampal kindling and maximal electroshock seizure models after prenatal betamethasone (Velíšek 2005, Young et al., 2006). Similarly, another synthetic corticosteroid dexamethasone decreased seizure susceptibility (had anticonvulsant effects) in the kindling model (Young et al., 2006). While the mechanisms of these effects of prenatally administered corticosteroids are unclear, they may involve enhanced peptidergic inhibition (Velíšek 2006b) and changes in other systems such as glucocorticoid receptors (this effect is restricted to the exposure during the last third of rat pregnancy) or even hypothalamus-pituitary-adrenal axis (Shoener, et al. 2006, Welberg and Seckl 2001, Young et al., 2006).
Betamethasone and dexamethasone are synthetic hormones with high affinity for glucocorticoid receptors. They easily cross placental barrier and bind to glucocorticoid receptors in the fetal brain (de Kloet 2003, Matthews 2000). Natural corticosteroids bind to both glucocorticoid and mineralocorticoid receptors in the brain (Joels 2001, Levitt et al., 1996, Reul et al., 1987). By the feedback pathways, corticosteroids decrease the release of hypothalamic corticotropin-releasing hormone (CRH) (Chen, et al. 2004), a strong convulsant agent in the immature brain (Baram and Schultz 1991) suggesting moderate decreases in seizure susceptibility. However, natural corticosteroids can be metabolized in the brain and some of their metabolites may mediate proconvulsant effects (McInnes, et al. 2004). The net effect on seizure susceptibility may therefore be dependent on corticosteroid species. While after betamethasone (dexamethasone) the suppression of hypothalamic CRH may prevail with resulting decreases in seizure susceptibility, use of hydrocortisone may boost production of proconvulsant metabolites counteracting suppression of CRH. We can speculate that higher doses of hydrocortisone may even reverse the net effect into proconvulsant. This speculation is supported by enhancement of lethal effects of kainic acid (3.5 mg/kg) by prenatal hydrocortisone. It should be emphasized that although hydrocortisone (cortisol) is a natural human corticosteroid, it is not present in the rat and this may account for no overall effects on susceptibility to KA-induced seizures compared to stress (Frye and Bayon 1999).
An interesting question is whether the effect of prenatal betamethasone exposure on seizure susceptibility is model-specific? Until recently and including this study, prenatal betamethasone exposure was associated with anticonvulsant effects, i.e., decreased seizure susceptibility, in three models of seizures: kindling (Velíšek 2005, Young et al., 2006), maximal electroshock (Young et al., 2006), and flurothyl (this study). This study also demonstrated that there was no effect of prenatal betamethasone on kainic acid-induced seizures. Betamethasone did not affect seizures induced by either 3.5 or 5.0 mg/kg of kainic acid suggesting true lack of betamethasone efficacy. Finally, in addition to those findings, our recent study indicated that prenatal betamethasone exposure had significant and unique proconvulsant effects in spasms induced by NMDA in infant rats (Velíšek, et al. 2007). These results taken together suggest that effects of prenatal betamethasone on seizure susceptibility are indeed model-specific. The mechanisms beyond these differences are still elusive and one may only speculate that specific molecular changes induced by betamethasone (such as in the NMDA receptor subunit expression) may be the culprit.
There was a significant decrease in body weight after prenatal corticosteroid exposure irrespective of the corticosteroid used. This finding is consistent with many previous studies (Carlos, et al. 1992, Scheepens, et al. 2003, Velíšek 2006b, Welberg and Seckl 2001, Young et al., 2006). Thus, a decrease in postnatal weight gain represents a simple and reliable indicator of biological activity of prenatally administered corticosteroids. Interestingly, a decrease in postnatal body weight gain was amplified after prenatal pretreatment with mineralocorticoid receptor antagonist canrenoic acid suggesting that in this case, canrenoic acid may have a positive feed-back.
Prenatal hydrocortisone exposure did not affect behavioral performance on the horizontal bar or in the elevated plus maze. Yet, horizontal bar performance was impaired after a combination of prenatal hydrocortisone and canrenoic acid. Co-administration of mineralocorticoid receptor antagonist canrenoic acid along with hydrocortisone was planned to identify the effects mediated through the hydrocortisone action on mineralocorticoid receptors. The explanation of this effect of canrenoic acid on hydrocortisone actions may come from the hypothalamus-pituitary-adrenal feedback mechanisms, which is mediated by mineralocorticoid receptors (Atkinson, et al. 2008). However, in canrenoic acid-pretreated rats this feedback was eliminated, and we can speculate that this may possibly induce further release of natural corticosteroids responsible for the amplification of the hydrocortisone effects. Alternative explanation is that the effects of corticosteroids and their antagonists may include crossover activity at progesterone receptors (Rupprecht, et al. 1993). Additionally, the presumed specific mineralocorticoid antagonists may act on mineralocorticoid/glucocorticoid heterodimers (Trapp, et al. 1994) further complicating the outcome and interpretation of the data.
While we did not find an effect of prenatal exposure on the retention index, a marker of memory retention, it should be mentioned that this marker represents an indirect measure, and is further affected by additional variables such as anxiety status, locomotor activity, and seizure history. These variables likely contributed to the large variability of data and may affect also seizure susceptibility outcome. More specific memory tests are required for reliable information of prenatal corticosteroid exposure influence on memory status. Yet our finding is supported by a recent study, which did not show any effect of prenatal betamethasone or dexamethasone exposure on Morris Water Maze performance in adult rats (Emgard, et al. 2007).
In the previous study in seizure-naïve rats, we found an increased transfer latency in the elevated plus maze after prenatal betamethasone exposure consistent with decreased anxiety (Velíšek 2006b). In this study the acute seizure experience eliminated the effect of betamethasone exposure on anxiety. On the other hand, combination of prenatal hydrocortisone with seizures in this study significantly increased anxiety in those rats. These finding indicate that there may be complex interactions between prenatal corticosteroid exposure (steroid-specific) and postnatal seizure experience (acute seizure vs. chronic epilepsy) on the expression of anxiolytic molecules such as NPY (Reibel, et al. 2001). We can further speculate about participation of other molecules (such as glucocortioids themselves), since glucocorticoids have anxiogenic features (Tronche, et al. 1999). Hippocampus also contains corticotropin-releasing hormone (CRH) (Chen et al., 2004, Yan, et al. 1998). Expression of CRH may be also increased by prenatal corticosteroid exposure (Bosch, et al. 2007, Shoener et al., 2006) resulting in changes in anxiety because CRH is a potent anxiogenic molecule (Stenzel-Poore, et al. 1994).
In conclusion, we found model-specific effects of prenatal betamethasone exposure on seizure susceptibility and increased anxiety after hydrocortisone combined with seizure experience. The results indicate that effects of prenatal exposure to corticosteroids on seizure susceptibility may be seizure syndrome specific, therefore studies in humans seeking the effects of prenatal corticosteroids (or stress) on epilepsy without defining a specific syndrome may not return any positive results (Li, et al. 2008).
Acknowledgments
Supported by the NIH grants NS-41366, NS-059504, and NS-072966 from NINDS, by the grant #6-FY08-214 from the March of Dimes Birth Defect Foundation, and by the Albert Einstein College of Medicine. There were no other financial interests. The author is grateful to Ms. Zunju Hu for outstanding technical assistance and to Dr. Jana Velíšková for very critical comments on the manuscript.
ABBREVIATIONS
- CAN
canrenoic acid
- HC
hydrocortisone
- HPA
hypothalamus-pituitary-adrenal
- KA
kainic acid
- MR
mineralocorticoid
- NPY
neuropeptide Y
- P
postnatal day
- PBS
phosphate-buffered saline
- RI
retention index
Footnotes
Current affiliation: Department of Cell Biology & Anatomy, Department of Pediatrics, New York Medical College, Valhalla, NY, USA.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aghajafari F, Murphy K, Matthews S, Ohlsson A, Amankwah K, Hannah M. Repeated doses of antenatal corticosteroids in animals: a systematic review. Am J Obstet Gynecol. 2002;186:843–849. doi: 10.1067/mob.2002.121624. [DOI] [PubMed] [Google Scholar]
- Albala BJ, Moshé SL, Okada R. Kainic-acid-induced seizures: a developmental study. Dev Brain Res. 1984;13:139–148. doi: 10.1016/0165-3806(84)90085-3. [DOI] [PubMed] [Google Scholar]
- Atkinson HC, Wood SA, Castrique ES, Kershaw YM, Wiles CC, Lightman SL. Corticosteroids mediate fast feedback of the rat hypothalamic-pituitary-adrenal axis via the mineralocorticoid receptor. Am J Physiol Endocrinol Metab. 2008;294:E1011–1022. doi: 10.1152/ajpendo.00721.2007. [DOI] [PubMed] [Google Scholar]
- Avishai-Eliner S, Brunson KL, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci. 2002;25:518–524. doi: 10.1016/s0166-2236(02)02241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baram TZ, Schultz L. Corticotropin-releasing hormone is a rapid and potent convulsant in the infant rat. Dev Brain Res. 1991;61:97–101. doi: 10.1016/0165-3806(91)90118-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbazanges A, Piazza PV, Le Moal M, Maccari S. Maternal glucocorticoid secretion mediates long-term effects of prenatal stress. J Neurosci. 1996;16:3943–3949. doi: 10.1523/JNEUROSCI.16-12-03943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battin MR, Bevan C, Harding JE. Repeat doses of antenatal steroids and hypothalamic-pituitary-adrenal axis (HPA) function. Am J Obstet Gynecol. 2007;197:40 e41–46. doi: 10.1016/j.ajog.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Bosch OJ, Musch W, Bredewold R, Slattery DA, Neumann ID. Prenatal stress increases HPA axis activity and impairs maternal care in lactating female offspring: implications for postpartum mood disorder. Psychoneuroendocrinology. 2007;32:267–278. doi: 10.1016/j.psyneuen.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Brabham T, Phelka A, Zimmer C, Nash A, Lopez JF, Vazquez DM. Effects of prenatal dexamethasone on spatial learning and response to stress is influenced by maternal factors. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1899–1909. doi: 10.1152/ajpregu.2000.279.5.R1899. [DOI] [PubMed] [Google Scholar]
- Carlos RQ, Seidler FJ, Slotkin TA. Fetal dexamethasone exposure alters macromolecular characteristics of rat brain development: a critical period for regionally selective alterations? Teratology. 1992;46:45–59. doi: 10.1002/tera.1420460108. [DOI] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ. Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience. 2004;126:533–540. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther CA, Doyle LW, Haslam RR, Hiller JE, Harding JE, Robinson JS. Outcomes at 2 years of age after repeat doses of antenatal corticosteroids. N Engl J Med. 2007;357:1179–1189. doi: 10.1056/NEJMoa071152. [DOI] [PubMed] [Google Scholar]
- Davis EP, Townsend EL, Gunnar MR, Guiang SF, Lussky RC, Cifuentes RF, Georgieff MK. Antenatal betamethasone treatment has a persisting influence on infant HPA axis regulation. J Perinatol. 2006;26:147–153. doi: 10.1038/sj.jp.7211447. [DOI] [PubMed] [Google Scholar]
- de Kloet ER. Hormones, brain and stress. Endocr Regul. 2003;37:51–68. [PubMed] [Google Scholar]
- Diaz R, Fuxe K, Ogren SO. Prenatal corticosterone treatment induces long-term changes in spontaneous and apomorphine-mediated motor activity in male and female rats. Neuroscience. 1997;81:129–140. doi: 10.1016/s0306-4522(97)00141-3. [DOI] [PubMed] [Google Scholar]
- Emgard M, Paradisi M, Pirondi S, Fernandez M, Giardino L, Calza L. Prenatal glucocorticoid exposure affects learning and vulnerability of cholinergic neurons. Neurobiol Aging. 2007;28:112–121. doi: 10.1016/j.neurobiolaging.2005.11.015. [DOI] [PubMed] [Google Scholar]
- File SE. The interplay of learning and anxiety in the elevated plus-maze. Behav Brain Res. 1993;58:199–202. doi: 10.1016/0166-4328(93)90103-w. [DOI] [PubMed] [Google Scholar]
- French NP, Hagan R, Evans SF, Mullan A, Newnham JP. Repeated antenatal corticosteroids: effects on cerebral palsy and childhood behavior. Am J Obstet Gynecol. 2004;190:588–595. doi: 10.1016/j.ajog.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Frye CA, Bayon LE. Prenatal stress reduces the effectiveness of the neurosteroid 3 alpha,5 alpha-THP to block kainic-acid-induced seizures. Dev Psychobiol. 1999;34:227–234. [PubMed] [Google Scholar]
- Gottlieb A, Keydor I, Epstein HT. Rodent brain growth stages. An analytical review. Biol Neonate. 1977;32:166–176. doi: 10.1159/000241012. [DOI] [PubMed] [Google Scholar]
- Guinn DA, Atkinson MW, Sullivan L, Lee M, MacGregor S, Parilla BV, Davies J, Hanlon-Lundberg K, Simpson L, Stone J, Wing D, Ogasawara K, Muraskas J. Single vs Weekly Courses of Antenatal Corticosteroids for Women at Risk of Preterm Delivery: A Randomized Controlled Trial. JAMA. 2001;286:1581–1587. doi: 10.1001/jama.286.13.1581. [DOI] [PubMed] [Google Scholar]
- Heilig M, McLeod S, Brot M, Heinrichs SC, Menzaghi F, Koob GF, Britton KT. Anxiolytic-like action of neuropeptide Y: mediation by Y1 receptors in amygdala, and dissociation from food intake effects. Neuropsychopharmacology. 1993;8:357–363. doi: 10.1038/npp.1993.35. [DOI] [PubMed] [Google Scholar]
- Hliňák Z, Krejčí I. MK-801 induced amnesia for the elevated plus-maze in mice. Behav Brain Res. 2002;131:221–225. doi: 10.1016/s0166-4328(01)00347-3. [DOI] [PubMed] [Google Scholar]
- Jobe AH, Soll RF. Choice and dose of corticosteroid for antenatal treatments. Am J Obstet Gynecol. 2004;190:878–881. doi: 10.1016/j.ajog.2004.01.044. [DOI] [PubMed] [Google Scholar]
- Joels M. Corticosteroid actions in the hippocampus. J Neuroendocrinol. 2001;13:657–669. doi: 10.1046/j.1365-2826.2001.00688.x. [DOI] [PubMed] [Google Scholar]
- Lánský P, Velíšková J, Velíšek L. An indirect method for absorption rate estimation: Flurothyl-induced seizures. Bull Math Biol. 1997;59:569–579. doi: 10.1007/BF02459466. [DOI] [PubMed] [Google Scholar]
- Lemaire V, Koehl M, Le Moal M, Abrous DN. Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc Natl Acad Sci U S A. 2000;97:11032–11037. doi: 10.1073/pnas.97.20.11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt NS, Lindsay RS, Holmes MC, Seckl JR. Dexamethasone in the last week of pregnancy attenuates hippocampal glucocorticoid receptor gene expression and elevates blood pressure in the adult offspring in the rat. Neuroendocrinology. 1996;64:412–418. doi: 10.1159/000127146. [DOI] [PubMed] [Google Scholar]
- Li J, Vestergaard M, Obel C, Precht DH, Christensen J, Lu M, Olsen J. Prenatal stress and epilepsy in later life: a nationwide follow-up study in Denmark. Epilepsy Res. 2008;81:52–57. doi: 10.1016/j.eplepsyres.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Maccari S, Piazza PV, Kabbaj M, Barbazanges A, Simon H, Le Moal M. Adoption reverses the long-term impairment in glucocorticoid feedback induced by prenatal stress. J Neurosci. 1995;15:110–116. doi: 10.1523/JNEUROSCI.15-01-00110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews SG. Antenatal glucocorticoids and programming of the developing CNS. Pediatr Res. 2000;47:291–300. doi: 10.1203/00006450-200003000-00003. [DOI] [PubMed] [Google Scholar]
- McInnes KJ, Kenyon CJ, Chapman KE, Livingstone DE, Macdonald LJ, Walker BR, Andrew R. 5alpha-reduced glucocorticoids, novel endogenous activators of the glucocorticoid receptor. J Biol Chem. 2004;279:22908–22912. doi: 10.1074/jbc.M402822200. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Rick JT, Milgram NW, Ivy GO. A simple and rapid test of sensorimotor function in the aged rat. Neurobiol Learn Mem. 1995;64:181–186. doi: 10.1006/nlme.1995.1057. [DOI] [PubMed] [Google Scholar]
- Pellow S, File SE. Anxiolytic and anxiogenic drug effects on exploratory activity in an elevated plus-maze: a novel test of anxiety in the rat. Pharmacol Biochem Behav. 1986;24:525–529. doi: 10.1016/0091-3057(86)90552-6. [DOI] [PubMed] [Google Scholar]
- Purdy IB, Wiley DJ. Perinatal corticosteroids: A review of research. Part I: Antenatal administration. Neonatal Netw. 2004;23:15–30. doi: 10.1891/0730-0832.23.2.15. [DOI] [PubMed] [Google Scholar]
- Reibel S, Nadi S, Benmaamar R, Larmet Y, Carnahan J, Marescaux C, Depaulis A. Neuropeptide Y and epilepsy: varying effects according to seizure type and receptor activation. Peptides. 2001;22:529–539. doi: 10.1016/s0196-9781(01)00347-3. [DOI] [PubMed] [Google Scholar]
- Reul JM, van den Bosch FR, de Kloet ER. Differential response of type I and type II corticosteroid receptors to changes in plasma steroid level and circadian rhythmicity. Neuroendocrinology. 1987;45:407–412. doi: 10.1159/000124766. [DOI] [PubMed] [Google Scholar]
- Roberts D, Dalziel S. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Database Syst Rev. 2006;3:CD004454. doi: 10.1002/14651858.CD004454.pub2. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Reul JM, van Steensel B, Spengler D, Soder M, Berning B, Holsboer F, Damm K. Pharmacological and functional characterization of human mineralocorticoid and glucocorticoid receptor ligands. Eur J Pharmacol. 1993;247:145–154. doi: 10.1016/0922-4106(93)90072-h. [DOI] [PubMed] [Google Scholar]
- Scheepens A, van de Waarenburg M, van den Hove D, Blanco CE. A single course of prenatal betamethasone in the rat alters postnatal brain cell proliferation but not apoptosis. J Physiol. 2003;552:163–175. doi: 10.1113/jphysiol.2003.043414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang NX, Zou LP, Zhao JB, Zhang F, Li H. Association between prenatal stress and infantile spasms: a case-control study in China. Pediatr Neurol. 2010;42:181–186. doi: 10.1016/j.pediatrneurol.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Shoener JA, Baig R, Page KC. Prenatal exposure to dexamethasone alters hippocampal drive on hypothalamic-pituitary-adrenal axis activity in adult male rats. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1366–1373. doi: 10.1152/ajpregu.00757.2004. [DOI] [PubMed] [Google Scholar]
- Sperber EF, Moshé SL. Age-related differences in seizure susceptibility to flurothyl. Dev Brain Res. 1988;39:295–297. doi: 10.1016/0165-3806(88)90033-8. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Thompson JL, Holmes GL. Kainic acid seizures in the developing brain: status epilepticus and spontaneous recurrent seizures. Brain Research. Developmental Brain Research. 1992;65:227–236. doi: 10.1016/0165-3806(92)90184-x. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW. Overproduction of corticotropin-releasing factor in transgenic mice: a genetic model of anxiogenic behavior. J Neurosci. 1994;14:2579–2584. doi: 10.1523/JNEUROSCI.14-05-02579.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott DH. Follow-up study from birth of the effects of prenatal stresses. Dev Med Child Neurol. 1973;15:770–787. doi: 10.1111/j.1469-8749.1973.tb04912.x. [DOI] [PubMed] [Google Scholar]
- Trapp T, Rupprecht R, Castren M, Reul JM, Holsboer F. Heterodimerization between mineralocorticoid and glucocorticoid receptor: a new principle of glucocorticoid action in the CNS. Neuron. 1994;13:1457–1462. doi: 10.1016/0896-6273(94)90431-6. [DOI] [PubMed] [Google Scholar]
- Treit D, Menard J, Royan C. Anxiogenic stimuli in the elevated plus-maze. Pharmacol Biochem Behav. 1993;44:463–469. doi: 10.1016/0091-3057(93)90492-c. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Vallee M, MacCari S, Dellu F, Simon H, Le Moal M, Mayo W. Long-term effects of prenatal stress and postnatal handling on age- related glucocorticoid secretion and cognitive performance: a longitudinal study in the rat. Eur J Neurosci. 1999;11:2906–2916. doi: 10.1046/j.1460-9568.1999.00705.x. [DOI] [PubMed] [Google Scholar]
- Vallee M, Mayo W, Dellu F, Le Moal M, Simon H, Maccari S. Prenatal stress induces high anxiety and postnatal handling induces low anxiety in adult offspring: correlation with stress-induced corticosterone secretion. J Neurosci. 1997;17:2626–2636. doi: 10.1523/JNEUROSCI.17-07-02626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velíšek L. Prenatal betamethasone exposure suppresses kindling epileptogenesis in immature rats. In: Corcoran ME, Moshé SL, editors. Kindling. Vol. 6. Springer; New York: 2005. pp. 11–17. [Google Scholar]
- Velíšek L. Models of Chemically-Induced Acute Seizures. In: Pitkanen A, Schwartzkroin PA, Moshé SL, editors. Models of Seizures and Epilepsy. Elsevier; Amsterdam: 2006a. pp. 127–152. [Google Scholar]
- Velíšek L. Prenatal exposure to betamethasone decreases anxiety in developing rats: Hippocampal neuropeptide Y as a target molecule. Neuropsychopharmacology. 2006b;31:2140–2149. doi: 10.1038/sj.npp.1301016. [DOI] [PubMed] [Google Scholar]
- Velíšek L, Jehle K, Asche S, Velíšková J. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann Neurol. 2007;61:109–119. doi: 10.1002/ana.21082. [DOI] [PubMed] [Google Scholar]
- Velíšková J, Velíšek L, Mareš P. Epileptic phenomena produced by kainic acid in laboratory rats during ontogenesis. Physiol Bohemoslov. 1988;37:395–405. [PubMed] [Google Scholar]
- Velíšková J, Velíšek L, Nunes M, Moshé S. Developmental regulation of regional functionality of substantia nigra GABAA receptors involved in seizures. Eur J Pharmacol. 1996;309:167–173. doi: 10.1016/0014-2999(96)00341-x. [DOI] [PubMed] [Google Scholar]
- Welberg LAM, Seckl JR. Prenatal stress, glucocorticoids and the programming of the brain. J Neuroendocr. 2001;13:113–128. doi: 10.1046/j.1365-2826.2001.00601.x. [DOI] [PubMed] [Google Scholar]
- Yan XX, Toth Z, Schultz L, Ribak CE, Baram TZ. Corticotropin-releasing hormone (CRH)-containing neurons in the immature rat hippocampal formation: light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus. 1998;8:231–243. doi: 10.1002/(SICI)1098-1063(1998)8:3<231::AID-HIPO6>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young NA, Teskey GC, Henry LC, Edwards HE. Exogenous antenatal glucocorticoid treatment reduces susceptibility for hippocampal kindled and maximal electroconvulsive seizures in infant rats. Exp Neurol. 2006;198:303–312. doi: 10.1016/j.expneurol.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Zangrossi H, Jr, Viana MB, Zanoveli J, Bueno C, Nogueira RL, Graeff FG. Serotonergic regulation of inhibitory avoidance and one-way escape in the rat elevated T-maze. Neurosci Biobehav Rev. 2001;25:637–645. doi: 10.1016/s0149-7634(01)00047-1. [DOI] [PubMed] [Google Scholar]