

Abstract
While there are a plethora of medications that block seizures, these same drugs have little effect on preventing or curing epilepsy. This suggests that the molecular pathways for epileptogenesis are distinct from those that produce acute seizures and therefore will require the identification of novel truly ‘antiepileptic’ therapeutics. Identification and testing of potential antiepileptic drug targets first in animal models and then in humans is thus becoming an important next step in the battle against epilepsy. In focal forms of human epilepsy the battle, however, is complicated by the large and varied types of brain abnormalities capable of producing a state of chronic, recurrent seizures. Unfortunately, once the epileptic state develops, it often persists to produce a life-long seizure disorder that can only be suppressed by anticonvulsant medications, and cured only in some through surgical resection of the seizure focus. While deductive approaches to drug target identification use our current state of knowledge, based mostly on animal models of epileptogenesis, a growing reductionist approach often referred to as systems biology takes advantage of newer high-throughput technologies to profile large numbers and types of molecules simultaneously. Some of these approaches, such as functional genomics, proteomics, and metabolomics have been undertaken in both human and animal epileptic brain tissues and are beginning to hone in on new therapeutic targets. While these methods are highly sensitive, this same sensitivity also produces a high rate of false positives due to variables other than those of interest. The experimental design, therefore, needs to be tightly controlled to reduce these unintended results that can be misleading. Most importantly, epileptogenic targets need to be validated in animal models of epileptogenesis, so that, if successful, these new methods have the potential to identify unbiased, important new therapeutics.
Keywords: epileptogenesis, microarray, genomics, systems biology, epilepsy, genetics
Introduction
Epilepsy is a disabling neurological disorder of recurrent seizures affecting up to 1% of the population [3]. In most patients with partial epilepsy, seizures start in focal brain regions following a wide variety of brain insults, but often no clear histopathological abnormalities are identified [8]. Patients who fail to respond to anticonvulsant medications can greatly benefit from surgery to remove brain regions where seizures originate. These observations suggest that focal epileptic brain regions are both necessary and sufficient to produce clinical seizures. Yet, seizures can occur even without a clear pathologic lesion, suggesting that normal anatomical structures are somehow altered to produce hypersynchronous electrical discharges. Regardless of the original brain insult, epileptic foci show a remarkably similar electrophysiological pattern of localized, abnormal electrical discharges in rhythmic patterns that spread to widespread brain regions to produce clinical seizures. While in young children epileptic foci occur mostly in the neocortex, in adults and older children, these are often in the hippocampus [3].
Little is known about how focal brain regions become and remain epileptic: a process often referred to as epileptogenesis (Fig. 1). While effective in preventing seizures, current seizure medications have shown little promise in blocking epileptogenesis [66]. Consistently, patients who have remained seizure free on anticonvulsant medications have a high risk for seizure recurrence when these medications are discontinued, suggesting that the underlying molecular and cellular networks that develop during the epileptogenic process are maintained in an epileptic focus and are not appreciably altered by current anticonvulsant medications. This highlights the need for novel truly anti-epileptogenic rather than anticonvulsant treatments.
Figure 1. A model for targeted epileptogenesis drug development.

Following a wide array of brain insults, epilepsy develops over a prolonged latent period. During this latent period both cellular, networking, and underlying molecular events occur that create an often permanent state of recurrent seizures. Current medications work mostly as anticonvulsants preventing seizures, but have little proven effects on epileptogenesis. Newer, truly anti-epileptogenic drugs that target either acute or more chronic molecular events during the latent period are needed. One hypothesis, is that feedback between cellular, electrical, and molecular events grows the epileptic focus over time until a threshold is reached that is capable of producing clinical seizures.
A major difficulty in designing new treatments to block epileptogenesis comes from the large number of brain insults capable of producing epilepsy, the long latent period between the insult and the first clinical seizure, as well as our limited knowledge of the epileptogenic process itself. A question that arises is whether there are in fact common molecular and networking events that can explain the epileptogenic process, and that could be used to target therapeutics. It is hard to imagine how common pathways could exist for both severe brain injuries, such as those from traumatic brain injury and stroke, and more subtle developmental abnormalities, such as cortical dysplasias or slow-growing tumors. In the case of brain trauma or stroke, neuronal loss, inflammation, and scar tissue (gliosis) are commonly seen; however, since these pathological entities are not present in all epileptic foci, it is unclear whether they are required for epileptogenesis. On the other hand, if an anti-epileptogenic treatment was developed that targets neuronal loss or inflammation, the treatment may not be effective in patients with slow-growing tumors or cortical dysplasias. Following an injury, the long latent period between the brain insult and the first epileptic seizure in humans raises an equally important question of timing of when to give an anti-epileptogenic treatment. For example, children with febrile seizures commonly have a latency period of at least 2–3 years before their first recurrent epileptic seizure and it often takes months or years following a head injury for recurrent seizures to develop [36]. Treatments at the time of the febrile seizure or acute injury are likely to be mechanistically different than those given during the latent period.
Other than these clinical observations, very little is known about the epileptogenic process in humans. The majority of our knowledge comes from animal models where chronic epileptic conditions can be induced after a wide variety of chemical treatments, injuries, and electrical stimulations [71]. Surprisingly, only recently have long-term recordings been performed in animal models to reveal the progressive network changes that occur during the epileptogenic process [40, 70, 73]. These studies, that are described in more detail in other articles in this issue, demonstrate a progressive increase in interictal spiking and subclinical seizure activity long before spontaneous seizures first appear in both the kainic acid status epilepticus and perinatal infarct models in rats. These findings suggest that long before the first of many recurrent clinical seizures, extensive molecular and functional networking changes have already occurred. One important question that arises, is whether the development of interictal discharges and subclinical seizure activities are either necessary or sufficient to produce clinically significant seizures. A greater understanding of both the electrical networking as well as molecular and cellular processes in animal models of epileptogenesis will be critical for developing as well as testing novel anti-epileptic therapeutics.
Figure 1 suggests a working model of epileptogenesis that can be used as a simplistic guide for developing targeted therapeutics. After a brain insult, cellular and molecular changes ensue, resulting in hypersynchronous neuronal firing. This hypersynchrony grows over time during a very active ‘latent’ period where ongoing feedback occurs between electrical networks and the cellular and molecular changes that progress towards a chronic epileptic state. Eventually, spontaneous seizures occur and the disease remains active throughout life. While current anticonvulsant medications appear to be effective only once full-blown seizures have developed [66], novel truly anti-epileptogenic strategies will need to target earlier, molecular events either during the acute period of brain injury or during the prolonged latent period. A major goal for systems biology, that will be described below, is to identify novel therapeutic targets from human epileptic tissues and animal models that block specific molecular pathways required for these early stages of disease.
Systems biology of the brain: Both power and limitations
Systems biology can be variously defined, but one appealing definition is the ability to obtain, integrate and analyze complex data from multiple experimental sources using interdisciplinary tools [56]. The goal of this seemingly diffuse approach is to find ways to condense and focus diverse and highly variant types of molecular data into meaningful insights. For epileptogenesis, the most immediate goal is to translate patterns of molecules that characterize the epileptogenic process in a way that turns these insights into effective therapeutics.
Within the past 10 years a number of key technological breakthroughs have led to a virtual explosion of high-throughput molecular data that are being generated from both human epileptic tissues as well as a number of animal models of epilepsy. Methods that simultaneously look at the expression patterns of all forms of a given type of molecule are often referred to ‘-omics.’ Figure 2 summarizes some of the types of molecules that can be profiled using high-throughput approaches including functional genomics, proteomics, and metabolomics. Without going into the technical details of each, functional genomics refers to the high-throughput measurement of gene expression by measuring messenger RNA (mRNA) levels; proteomics refers to the measurement of both protein levels as well as covalent protein modifications such as phosphorylation; and metabolomics is the high-throughput measure of small molecules such as neurotransmitters. However, the molecular complexity of a single mammalian cell goes well beyond these molecules. Even within the genomic field, we are learning that almost half of the human genome produces RNA transcripts that do not code for proteins [17]. These can be broken down into both short RNAs called microRNAs as well as long RNAs referred to as long non-coding RNAs. The functions of these transcripts are still poorly understood; however, there is increasing evidence that these non-coding RNAs have important regulatory roles, including roles in brain function [32, 60]. Other emerging high-throughput molecular methods include the analysis of lipds (lipidomics) [1, 14] and sugars (glycomics) [21]. While all of these molecular components are of critical importance for normal cell function, one the greatest challenges is integrating data from these diverse systems into meaningful ways to hone in on specific pathways and therapeutic targets.
Figure 2. A systems biology model for high-throughput studies of epilepsy.

Within a cell, such as the neuron shown here, genetic differences as well as differences in gene expression (mRNA), protein expression, and small molecule expression (such as neurotransmitters) characterize the epileptic focus. These individual molecular systems must also be placed within the contexts of larger systems including organ, tissue, and cell type. The complexity of this system creates one of the greatest challenges for the study and interpretation of high-throughput molecular profiling methods in epilepsy.
Commercial microarray platforms have become a mainstay for functional genomics and have been the main high-throughput approach used to study epilepsy. They are quite accurate and presently can probe the entire human genome with tens of thousands of specific probes to measure gene expression levels [50]. The standard bioinformatic approach to interpret these results is to create an arbitrary statistical cutoff and lump differentially-expressed genes into functional categories. However, this strength in numbers also generates one of the greatest limitations of this approach: the unintentional identification of hundreds or even thousands of false positive genes and pathways. This creates a high background of unrelated genes and pathways obscuring those of potential interest. For epilepsy, the sheer number of measurements in a single experiment allows the detection of differences not only relevant to the ‘epileptic’ part of the model, but also due to countless other unanticipated variables. This limitation is readily apparent from the growing number of gene expression profiling experiments in both human epileptic tissues and animal models that show surprisingly little agreement (see [69] for a nice review of these studies to date).
Figure 2 also illustrates that for the human brain it is important to place molecular profiling results into the context of higher order systems including organ, tissue, and cell type. For example, to make biological sense of a given gene expression change in a patient with epilepsy, it is important to know where the brain tissue came from (such as frontal or temporal cortex) which cell types (neurons or glia), and which neuronal layers. Furthermore, of particular importance for epilepsy, all of the aforementioned factors need to be linked to the tissue’s electrical abnormalities, such as interictal spikes and seizures. While coupling these higher order ‘systems’ with molecular profiling seems a daunting task, a true systems approach to the disease will require keeping track of and integrating all of these variables within the system. Toward this end, Figure 3 shows a flow chart structure for a database we have developed around our human epilepsy surgery program. This database keeps track of and integrates clinical, electrophysiological, imaging, and molecular data critical for both interpreting the data and generating new research directions [49].
Figure 3. Computational workflow linking human epilepsy surgery to systems biology.

In order to keep track of hundreds of variables that will affect the outcome of human epileptic tissue studies, we have developed a database the links all aspects of the pre-surgical, surgical, and tissue work. These include neuropsychology, electrophysiology, imaging, tissue location, and a number of high-throughput molecular studies derived from these tissues.
Genetics versus functional genomics of epilepsy
Gene expression, in the form of mRNA transcription, is regulated by complex gene structures and transcriptional networks that involve both sequences that code for proteins and regulatory sequences between the coding portions of the genes. A growing list of single gene defects have been identified in families with several rare types of inherited epilepsy. These Mendelian genetic forms of epilepsy can be divided into those that have clear structural brain developmental abnormalities and those that do not [2]. This is an important distinction when thinking about epileptogenesis as the lesion itself may or may not itself be intrinsically hyperexcitable, but may induce epileptogenesis in surrounding more normal brain tissue. Heritable gene defects have highlighted the importance of ion channels and neurotransmitter receptors. These include voltage- and ligand-gated ion channels, such as the alpha-4 subunit of the nicotinic neuronal acetylcholine receptor in autosomal dominant nocturnal frontal lobe epilepsy, the KCNQ2 and KCNQ3 potassium channels in benign familial neonatal convulsions, the CACNG2 calcium channel in a form of spinocerebellar ataxia with epilepsy, and the GABA(A) receptor in another form of generalized epilepsy [2, 24, 55, 65]. In addition to these human mutations, many more epileptic syndromes have been observed in inbred strains of mice. These include a sodium/hydrogen exchanger [23], a calcium channel subunit [15], a ferritin heavy chain [46], and glutamic acid decarboxylase [42].
In addition to Mendelian genetic disorders, nucleotide changes in the genome can lead to sequence changes or single nucleotide polymorphisms (SNPs) that may (synonymous) or may not (non-synonomous) produce amino acid changes, but could alter one’s susceptibility to the disease [24]. A statistically well-powered study of 3445 patient and 6935 controls of European ancestry, however, failed to show any genome-wide significant associations in patients with sporadic forms of focal epilepsy [43]. Specific known gene defects have also not yet been found in a majority of patients with idiopathic epilepsy [18]. The failure of these genetic/genomic approaches to date suggests that, rather than specific gene defects, most sporadic forms of epilepsy result from the interactions of many genes, proteins, and other cellular constituents to produce a hyperexcitable state. Thus, while genetic make-up could change one’s susceptibility, a genetic predisposition does not appear to be required.
As discussed above, the field of systems biology focuses not on heritable differences in nucleotide sequence, but instead on the study of gene, protein, metabolite, and other molecular expression unique to epileptic tissues. Even prior to the development of high-throughput, pan-genomic methods, there have been a number of gene expression changes noted from human and animal epileptic tissues [54]. In human tissue, altered expression of genes has been noted for GABA and glutamate receptors [12, 13, 25, 26, 41, 72, 76], sodium channels [51] and excitatory amino acids and their synthetic enzymes and transporters [57, 62, 67]. Gene expression changes in BDNF and other neurotrophins that may “regulate” the epileptic state have also been identified [27, 29, 45, 68].
With the development of cDNA and oligonucleotide microarrays that consist of thousands of DNA probes immobilized onto a solid surface, large numbers of gene expression changes can be probed simultaneously through functional genomics. Functional genomic studies to date have examined both human tissues and animal models. The remainder of this article will focus predominantly on these studies. One thing that will emerge from this discussion is the surprisingly poor overlap between different studies in different systems. Putting together all of the published functional genomic studies to date, Wang et al. found only 53 genes consistently differentially expressed in more than 2 studies and only 2 genes (BDNF and S100) consistently differentially expressed across both human and animal studies[69]. Therefore, another major focus of this article will be to explore the reasons for this poor concordance in order to both make sense of existing data as well as improve study design for the future.
Human tissue functional genomics to identify epileptogenic drug targets
One of the major problems in developing treatments for human brain disorders has been the poor translation of therapeutics in animal models back to patients. This is not unexpected when one considers both the length of time human brain disorders take to develop as well as the profound anatomical differences between the highly sulcated human brain and the simple rodent brain that is used for most preclinical studies. For epilepsy, both of these differences are of considerable importance with often long intervals between a brain insult and the first epileptic seizure in humans. One of the unique features of human epilepsy that makes it particularly attractive for high-throughput molecular profiling is that it is one of a few brain disorders where fresh, rather than postmortem, tissues are readily available. This is because medically refractory patients can greatly improve their seizure control following surgical procedures that remove regions where seizures first start. This surgical approach yields abundant, fresh tissue samples from both neocortical and hippocampal regions that are exceptionally well-characterized because of the extreme care that goes into the identification of what tissue to remove for a good surgical outcome. Freshness of the tissue significantly reduces RNA degradation that is common in post-mortem tissue samples [64].
Fresh human tissue samples from patients with medically refractory epilepsy is thus an incredible resource for both understanding and developing targeted anti-epileptogenic therapeutics. That being said, the degree of tissue complexity, relationship of epileptic tissues to structural lesions, availability and appropriateness of ‘control tissues,’ and relationship of the molecular profiling to the underlying electrical activities are all important variables that can lead to vast differences in gene expression. Since the goal of these studies is to identify genes and pathways linked to epilepsy, knowing the electrical properties of the tissues is critical. Seizures in animals produce rapid and marked changes in gene expression [11, 30]. Therefore, it is important to know both the time interval from the most recent seizure as well as more recent interictal activities of the tissues prior to their surgical removal. One of the greatest conceptual limitations of human tissue functional genomic studies for epileptogenesis is whether patterns of gene expression in fully mature epileptic regions will shed light on the epileptogenic process that likely occurred years prior to the surgery. While it is possible that epileptogenic genes and pathways are no longer activated in the chronic disease state, it is also possible that they remain elevated in the chronic interictal state and could be potential drug targets. After all, one has to explain why focal regions of the brain are maintained in a chronically hyperexcitable, and often life-long, state that is prone to produce spontaneous seizures.
For human functional genomic studies, tissue histopathology can also have far-reaching effects. Studies from both human and animal models reveal that epileptic foci do not always have pathologic abnormalities [8]. Even in patients with focal lesions, it is often the surrounding, normal-appearing region, rather than the structural abnormalities themselves, that are the main epileptic generators [6, 20]. Consistently, we have identified a small, but highly reproducible group of gene expression changes in neocortical seizure onset zones that was entirely independent of whether or not there was a nearby, associated lesion [59]. In many ways, it makes sense that the more the normal brain architecture is disrupted from a lesion, the less likely it will be able to produce epileptic activities that may require a functional organizational network. This is particularly true for structural lesions that are not intrinsically electrically active, such as metastatic tumors, infarcts, and glia scars. However, when these abnormal brain regions are included in functional genomic analyses, the resulting expression changes could be biased towards genes that reflect unique features of the lesion rather than the epileptogenic process. In these instances, functional genomic studies of brain lesions can also generate important clues for understanding human brain pathologies that range from cortical dysplasias [25], to hemimegalencephaly [10], to tuberous sclerosis [26].
The above discussion hopefully makes it clear that one of the most critical aspects of what comes out of a high-throughput experiment is what goes in to the experimental design [50]. Even a conceptually well-controlled experimental design may still generate gene expression changes that reflect unintentional variables. The goal is to reduce the unintentional variables by knowing as much as possible about the tissue. A parallel approach is to reduce unintentional variables by having sufficient statistical power though both biological and technical replicates. While most studies contain a number of biological replicates (multiple patients), because of the high cost of microarrays as well as newer high-throughput sequencing methods, most studies use a single measurement per patient without experimental (technical) replicates. Without technical replicates, only global expression changes for the group can be determined without any statistical confidence for each patient.
Perhaps the most important aspect of functional genomic study design is the selection of control tissue to act as a reference point for hundreds or thousands of gene expression levels in the epileptic tissue. An ideal control tissue should be identical to the epileptic tissue in every way except for the variable under study: epileptic activity and/or tissue pathology. For studies of temporal lobe epilepsy, such an ideal control tissue is not available from within the same patient as only one temporal lobe is ever removed. Therefore, most functional genomic studies of the human temporal lobe have used age-matched postmortem temporal lobe tissues from non-epileptic patients (Reviewed by [5]). Thus far, there has been very little agreement in the pattern of differentially expressed genes between these studies. However, some of the pathways implicated include gliosis, cellular stress, immune response, and synaptic plasticity pathways. While these pathways could be directly relevant for epilepsy and epileptogenesis, many of the identified genes could have arisen from other variables of less interest inherent to this experimental design that compares epileptic temporal lobe samples to post-mortem controls. These include differences between fresh and postmortem tissues, genetic background differences between patients and controls, medication effects produced by anticonvulsants taken by epileptic patients, as well as differences in the tissue pathology.
An alternative experimental design that we and others have used in neocortical studies to reduce the contributions of these unwanted variables is to compare gene expression between two or more different brain regions within the same patient [4, 38, 58, 59]. This removes the unwanted variables of tissue freshness, genetic differences, and medication effects and allows for a more focused profiling experiment that can ask for expression differences linked primarily to electrical and pathological differences. Patients undergoing a 2-stage surgical procedure that includes long-term subdural electrical recordings are ideal for this type of study. Subdural or depth recordings are often regarded as the ‘gold standard’ for precise electrical localization of epileptic brain regions. These recordings highlight the extensive electrical abnormalities of the mature human epileptic brain that go well beyond seizures. While seizures are relatively rare, interictal discharges are quite frequent. While the exact relationship of ongoing interictal activities to seizures is unclear [63], quantitation of interictal discharges have been used with varying success to identify regions of seizure onset and predict surgical outcomes [7]. Neuronal activity is a critical force that shapes nervous system development and plasticity [19, 44, 52]. It thus seems likely that ongoing ictal and interictal epileptic activities will have profound effects on the functional and structural changes that could lead to and maintain hyperexcitability. Consistently, individual genes encoding neurotransmitter receptors, ion channels, amino acid transporter, transcription factors and neurotrophic factors have been found to be differentially expressed in various animal models of epilepsy and in human epileptic brain tissues using both for individual genes and using high-throughput methods [10, 11, 22, 28, 30, 33, 53, 54].
Using an internally controlled experimental design for high-throughput gene expression profiling of paired human neocortical regions from 5 patients, we found a highly consistent group of 11 genes differentially expressed between seizure onset zones and nearby ‘control’ regions [59]. Control regions were defined as not having seizure onset or spread and minimal interictal spiking. While anatomical differences are also likely to contribute to expression changes, those genes that change regardless of the exact anatomical location are likely to be fundamental to the epileptic process itself. All tissue samples, including controls, were evaluated by long term subdural electrode recordings and had no abnormal pathology except for some with mild gliosis. Genes were identified that showed significant changes both within each patient as well as across multiple patients. Several of these genes, including immediate early gene transcription factors EGR-1, c-fos and EGR-2, are well known to play important roles in signaling pathways involved in synaptic plasticity. The pattern of gene activation also suggested the involvement of the transcription factor CREB, that was consistently activated in seizure onset zones within specific neuronal lamina (II-IV). Further validation of these genes as biomarkers of human epileptic foci was obtained using quantitative reverse-transcriptase PCR that showed these genes are highly accurate markers of seizure onset zones from 17 patients with highly different neocortical pathologies.
We also asked whether the activation of these genes correlated with the degree of epileptic activity. While gene induction did not correlate very well with a patient’s seizure frequency, gene expression correlated precisely with interictal activity [58, 59]. Taken together, these findings describe a robust relationship between human interictal spiking and gene expression changes of transcription factors and other genes that could contribute to the epileptic state of the tissue. Figure 4 presents a hypothesis on how interictal activity could strengthen and maintain abnormal synaptic structures that produce the hypersynchrony required for interictal spiking and seizures. Ongoing work in our lab is extending this experimental model to pan-genomic arrays, proteomic, and metabolomic studies. Subsequent proof that pathways identified in this manner are required for epileptogenesis will require further testing in animal models as a precursor to drug development.
Figure 4. Model of epileptogenesis from genes to pathways identified from functional genomics studies.
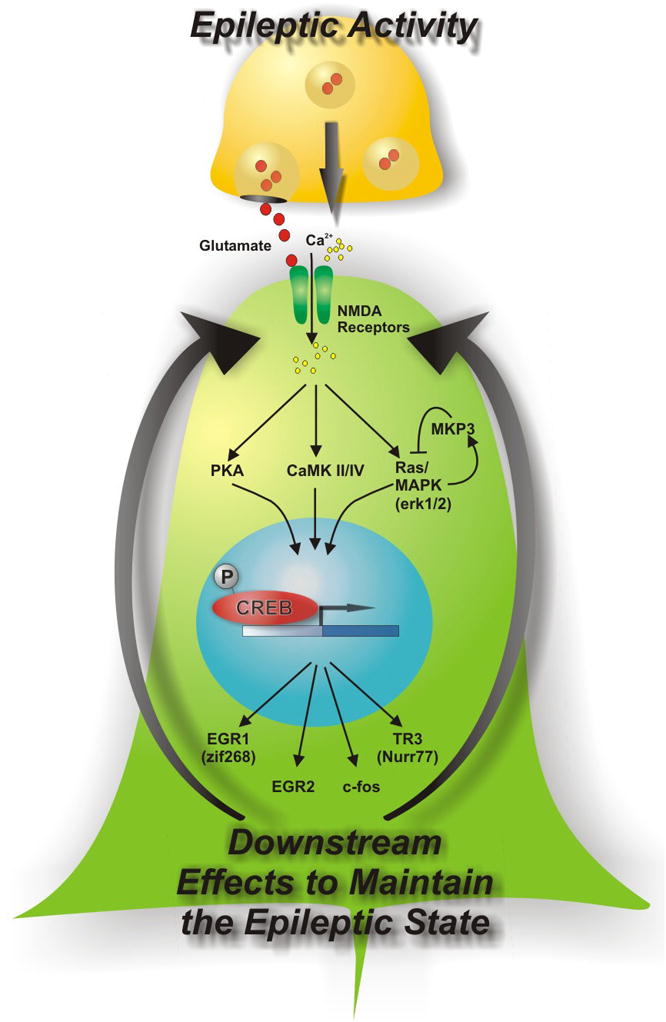
Based on the pattern of gene activations in human neocortical regions of seizure onset, this model describes how ongoing epileptic activities could create a state of heightened signaling through a number of pathways including the CREB transcription factor [59]. Some of these pathways that are known to activate CREB signaling such as protein kinase A (PKA), calcium calmodulin kinase II (CAMKII), or mitogen activated protein kinase (MAPK) could be targeted for anti-epileptic drug development.
Two other studies used a similar, but not identical, internally-controlled experimental design. One study compared cortical regions showing the presence or absence of interictal activity and found a set of differentially-expressed genes including transferrin, multiple GABA system-related genes, and many oligodendrocyte genes [4]. The differential expression of oligodendrocyte genes raises the question whether other variables such as the proportion of white matter in the two samples may underlie some of these changes and will require follow up studies that look closely at the histopathology of the brain tissues used for RNA sampling. Another study compared entorhinal cortex to lateral temporal cortex in patients with temporal lobe epilepsy, but without correlating electrical recordings to precise tissue locations [38]. This study implicated 16 genes that represented neurotransmitter and complement systems. Thus, even within these three fairly similar human cortical studies, a lack of overlap can easily be accounted for by differences in experimental design and further studies will be needed to find common pathways and therapeutic targets.
Animal model functional genomics to identify epileptogenic drug targets
While the greatest strength of human studies lies in their ‘closeness’ to the human condition, their greatest limitation comes from the unproven assumption that genes, proteins, molecules, and pathways identified to be differentially expressed in patients with fully matured epileptic disorders will generate therapeutic targets critical for epileptogenesis. Towards this end there have been many studies using functional genomic methods in animal (mostly rodent) models of epilepsy [69]. While these models may be better than human tissue studies to follow the molecular changes during epileptogenesis, very little concordance in differentially expressed genes has been found. Each animal model has its unique electrical and histological features that may explain much of this variability. For example, functional genomic studies have been performed both on acute seizure models as well as long-term models of epileptogenesis. Many models start with the generation of prolonged, acute seizures using methods that produce status epilepticus, such as the systemic administration of kainic acid or pilocarpine, or maximal electroshock treatment. An advantage of these models is that gene expression profiling experiments can be done both acutely after the status, as well as weeks later during the epileptogenic process. Not surprisingly, immediately after the induction of status epilepticus, dramatic changes in gene expression coincide with histopathological changes related to tissue injury, death, and inflammation [33, 37, 39]. Subsequent, long-term time points have demonstrated gene expression profiles that continue to show inflammatory changes, but also reflect synaptic plasticity changes that can differ in animals that later develop spontaneous seizures [11, 34, 35, 37, 47, 61].
For each model, one has to ask whether the resulting expression changes shed light on the epileptogenic process or whether they are due to concurrent cellular processes that occur after the initial brain insult, such as cell death, gliosis, and inflammation. A major challenge is paring down these long lists of genes to pathways critical for epileptogenesis that could be used as novel targets for drug development. One limitation of current long-term animal models is that each individual animal can have a different time course of epileptogenesis thereby making results from arbitrary time points for functional genomic measurements at 2 weeks or 2 months after the insult, difficult to interpret. To overcome this type of variance, one could either look at individual animals or pool large groups of animals together to average out the differences. The problem with this latter approach is that it could also mask important changes. Recently, investigators have begun long term video EEG monitoring of animal models of epileptogenesis that are shedding important new light on the ictal and interictal patterns during epileptogenesis [40, 70, 73]. These studies used either kainic acid or focal ischemia to describe the relationships between interictal and ictal activities that will be critical for future high-throughput studies of epileptogenesis. In fact, there is a strong suggestion from this work that interictal activity builds up prior to spontaneous seizures and may even have a causative role. Because of this work together with our findings of consistent gene expression changes in human epileptic neocortex that relate best to interictal activity, we have optimized a rat model of epileptogenesis using tetanus toxin that produces a progressive build up of interictal spiking without significant brain injury and relatively few spontaneous seizures [9]. High-throughput functional genomic and other systems biology studies tailored to individual animals undergoing long-term EEG monitoring may be the best way to link expression changes to each animal’s unique time course during epileptogenesis. Furthermore, once drug candidates for epileptogenesis are identified from human and animal studies, such focused animal models will be needed for testing therapeutics.
Other types of molecular profiling of epileptic tissues
While functional genomics has been the most widely used systems biology method to study epilepsy, there are other classes of biological molecules that can now be profiled in high-throughput ways. Not surprisingly, these have the same limitations as functional genomics beholden to experimental design and have added limitations in that these methods are still in their infancy. For example, a handful of proteomic studies have begun to look at human cerebral cortex [31], hippocampus [75], and human cerebrospinal fluid from patients with temporal lobe epilepsy [74]. Proteomics not only measures the amount of a given protein, but also whether there are any modifications of a protein such as phosphorylation. Similar to the functional genomic studies, there was essentially no overlap of the differentially expressed proteins in these studies. For example, one study used 2-dimensional gels followed by mass spectrometry to identify 57 differentially expressed proteins in rat hippocampus 12 and 72 hours after pilocarpine-induced status epilepticus [48]. The measure of small molecules such as amino acids and neurotransmitters can be measured from tissues in a high-throughput manner using either NMR spectroscopy or mass spectroscopy methods referred to as metabolomics. A single metabolomics study after pentylenetetrazole-induced seizures using proton NMR spectroscopy showed brain region dependent metabolite levels [16]. Just as the number of functional genomic studies have grown, proteomic, metabolomic, and other ‘omic’ studies will also increase in numbers, but will be subject to the same limitations in experimental design. One of the most exciting and challenging prospects for systems biology is to integrate all of these molecular profiling methods together as a means to look for common pathways across multiple molecular species as well as to place these back into the complex structure of the brain.
Conclusions and future perspectives
High-throughput systems approaches to study epilepsy are now a stable part of our experimental armamentarium to identify novel drug targets for epileptogenesis. However, given their power, sensitivity, and the huge financial investments made into these technologies, surprisingly few novel therapeutics have emerged thus far. One of the major difficulties for both human epilepsy as well as for animal models comes from the many diverse ways that epilepsy can develop. An important take home message is, because of the sensitivity, there is a high likelihood of false positive results due to any and all variables that are not controlled for in the experimental design. In many cases, results from functional genomic studies essentially validate what is already known and seen histopathologically, with major changes observed in immune system, cell death, neurotransmitters, and synaptic plasticity genes. What is needed are ways to see the forest through the trees with improved experimental design that allows a reductionist approach which focuses on epileptogenic variables and eliminates others. Towards this end, a few commonly differentially expressed genes and pathways are emerging in both human and animal functional genomic studies. One hypothesis we have raised is that ongoing feedback between interictal activity and synaptic plasticity pathways, that include CREB activation, may be a central and targetable pathway for epileptogenesis. The growth of other high-throughput molecular profiling methods such as proteomics and metabolomics, will provide additional ways to reduce complexity through the identification of common epileptogenic pathways shared by multiple molecular components within the system.
Acknowledgments
This work was supported by grants from NIH/NINDS R01NS045207 and R01NS058802 (JAL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adibhatla RM, Hatcher JF. Role of Lipids in Brain Injury and Diseases. Future Lipidol. 2007;2:403–422. doi: 10.2217/17460875.2.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrade DM. Genetic basis in epilepsies caused by malformations of cortical development and in those with structurally normal brain. Hum Genet. 2009;126:173–93. doi: 10.1007/s00439-009-0702-1. [DOI] [PubMed] [Google Scholar]
- 3.Annegers JF. The Epidemiology of Epilepsy. In: Wyllie E, editor. The Treatment of Epilepsy: Principles and Practices. Lea & Febiger; Philadelphia: 1993. pp. 157–164. [Google Scholar]
- 4.Arion D, Sabatini M, Unger T, Pastor J, Alonso-Nanclares L, Ballesteros-Yanez I, Garcia Sola R, Munoz A, Mirnics K, DeFelipe J. Correlation of transcriptome profile with electrical activity in temporal lobe epilepsy. Neurobiol Dis. 2006;22:374–87. doi: 10.1016/j.nbd.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 5.Aronica E, Gorter JA. Gene expression profile in temporal lobe epilepsy. Neuroscientist. 2007;13:100–8. doi: 10.1177/1073858406295832. [DOI] [PubMed] [Google Scholar]
- 6.Asano E, Chugani DC, Muzik O, Shen C, Juhasz C, Janisse J, Ager J, Canady A, Shah JR, Shah AK, Watson C, Chugani HT. Multimodality imaging for improved detection of epileptogenic foci in tuberous sclerosis complex. Neurology. 2000;54:1976–84. doi: 10.1212/wnl.54.10.1976. [DOI] [PubMed] [Google Scholar]
- 7.Asano E, Muzik O, Shah A, Juhasz C, Chugani DC, Sood S, Janisse J, Ergun EL, Ahn-Ewing J, Shen C, Gotman J, Chugani HT. Quantitative interictal subdural EEG analyses in children with neocortical epilepsy. Epilepsia. 2003;44:425–34. doi: 10.1046/j.1528-1157.2003.38902.x. [DOI] [PubMed] [Google Scholar]
- 8.Babb TL, Pretorius JK. Pathologic Substrates of Epilepsy. In: Wyllie E, editor. The Treatment of Epilepsy: Principles and Practices. Lea & Febiger; Philadelphia: 1993. pp. 55–70. [Google Scholar]
- 9.Barkmeier DT, Loeb JA. An animal model to study the clinical significance of interictal spiking. Clin EEG Neurosci. 2009;40:234–8. doi: 10.1177/155005940904000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baybis M, Lynch D, Lee A, Patel A, McKhann G, 2nd, Chugani D, WJK, Aronica E, Crino PB. Altered expression of neurotransmitter-receptor subunit and uptake site mRNAs in hemimegalencephaly. Epilepsia. 2004;45:1517–24. doi: 10.1111/j.0013-9580.2004.16204.x. [DOI] [PubMed] [Google Scholar]
- 11.Becker AJ, Chen J, Zien A, Sochivko D, Normann S, Schramm J, Elger CE, Wiestler OD, Blumcke I. Correlated stage- and subfield-associated hippocampal gene expression patterns in experimental and human temporal lobe epilepsy. Eur J Neurosci. 2003;18:2792–802. doi: 10.1111/j.1460-9568.2003.02993.x. [DOI] [PubMed] [Google Scholar]
- 12.Blumcke I, Becker AJ, Klein C, Scheiwe C, Lie AA, Beck H, Waha A, Friedl MG, Kuhn R, Emson P, Elger C, Wiestler OD. Temporal lobe epilepsy associated up-regulation of metabotropic glutamate receptors: correlated changes in mGluR1 mRNA and protein expression in experimental animals and human patients. J Neuropathol Exp Neurol. 2000;59:1–10. doi: 10.1093/jnen/59.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Brooks-Kayal AR, Shumate MD, Jin H, Lin DD, Rikhter TY, Holloway KL, Coulter DA. Human neuronal gamma-aminobutyric acid(A) receptors: coordinated subunit mRNA expression and functional correlates in individual dentate granule cells. J Neurosci. 1999;19:8312–8. doi: 10.1523/JNEUROSCI.19-19-08312.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown HA, Murphy RC. Working towards an exegesis for lipids in biology. Nat Chem Biol. 2009;5:602–6. doi: 10.1038/nchembio0909-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burgess DL, Jones JM, Meisler MH, Noebels JL. Mutation of the Ca2+ channel beta subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 1997;88:385–92. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- 16.Carmody S, Brennan L. Effects of pentylenetetrazole-induced seizures on metabolomic profiles of rat brain. Neurochem Int. 56:340–4. doi: 10.1016/j.neuint.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Carninci P, Hayashizaki Y. Noncoding RNA transcription beyond annotated genes. Curr Opin Genet Dev. 2007;17:139–44. doi: 10.1016/j.gde.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Cavalleri GL, Lynch JM, Depondt C, Burley MW, Wood NW, Sisodiya SM, Goldstein DB. Failure to replicate previously reported genetic associations with sporadic temporal lobe epilepsy: where to from here? Brain. 2005;128:1832–40. doi: 10.1093/brain/awh524. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Sochivko D, Beck H, Marechal D, Wiestler OD, Becker AJ. Activity-induced expression of common reference genes in individual cns neurons. Lab Invest. 2001;81:913–6. doi: 10.1038/labinvest.3780300. [DOI] [PubMed] [Google Scholar]
- 20.Chugani DC, Chugani HT, Muzik O, Shah JR, Shah AK, Canady A, Mangner TJ, Chakraborty PK. Imaging epileptogenic tubers in children with tuberous sclerosis complex using alpha-[11C]methyl-L-tryptophan positron emission tomography. Ann Neurol. 1998;44:858–66. doi: 10.1002/ana.410440603. [DOI] [PubMed] [Google Scholar]
- 21.Comelli EM, Head SR, Gilmartin T, Whisenant T, Haslam SM, North SJ, Wong NK, Kudo T, Narimatsu H, Esko JD, Drickamer K, Dell A, Paulson JC. A focused microarray approach to functional glycomics: transcriptional regulation of the glycome. Glycobiology. 2006;16:117–31. doi: 10.1093/glycob/cwj048. [DOI] [PubMed] [Google Scholar]
- 22.Coulter DA. Epilepsy-associated plasticity in gamma-aminobutyric acid receptor expression, function, and inhibitory synaptic properties. Int Rev Neurobiol. 2001;45:237–52. doi: 10.1016/s0074-7742(01)45013-6. [DOI] [PubMed] [Google Scholar]
- 23.Cox GA, Lutz CM, Yang CL, Biemesderfer D, Bronson RT, Fu A, Aronson PS, Noebels JL, Frankel WN. Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice [published erratum appears in Cell 1997 Dec 12;91(6):861] Cell. 1997;91:139–48. doi: 10.1016/s0092-8674(01)80016-7. [DOI] [PubMed] [Google Scholar]
- 24.Crino PB. Gene expression, genetics, and genomics in epilepsy: some answers, more questions. Epilepsia. 2007;48(Suppl 2):42–50. doi: 10.1111/j.1528-1167.2007.01066.x. [DOI] [PubMed] [Google Scholar]
- 25.Crino PB, Duhaime AC, Baltuch G, White R. Differential expression of glutamate and GABA-A receptor subunit mRNA in cortical dysplasia. Neurology. 2001;56:906–13. doi: 10.1212/wnl.56.7.906. [DOI] [PubMed] [Google Scholar]
- 26.Crino PB, Trojanowski JQ, Dichter MA, Eberwine J. Embryonic neuronal markers in tuberous sclerosis: single-cell molecular pathology. Proc Natl Acad Sci U S A. 1996;93:14152–7. doi: 10.1073/pnas.93.24.14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeFelipe J, Marco P, Sola RG, Sanchez-Blazquez P, Garzon J. Local changes in GTP-binding protein immunoreactivities in human epileptogenic neocortex. Exp Brain Res. 1998;119:153–8. doi: 10.1007/s002210050328. [DOI] [PubMed] [Google Scholar]
- 28.Doherty J, Dingledine R. The roles of metabotropic glutamate receptors in seizures and epilepsy. Curr Drug Target CNS Neurol Disord. 2002;1:251–60. doi: 10.2174/1568007023339355. [DOI] [PubMed] [Google Scholar]
- 29.Elisevich K, Rempel SA, Smith BJ, Edvardsen K. Hippocampal connexin 43 expression in human complex partial seizure disorder. Exp Neurol. 1997;145:154–64. doi: 10.1006/exnr.1997.6467. [DOI] [PubMed] [Google Scholar]
- 30.Elliott RC, Miles MF, Lowenstein DH. Overlapping microarray profiles of dentate gyrus gene expression during development- and epilepsy-associated neurogenesis and axon outgrowth. J Neurosci. 2003;23:2218–27. doi: 10.1523/JNEUROSCI.23-06-02218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eun JP, Choi HY, Kwak YG. Proteomic analysis of human cerebral cortex in epileptic patients. Exp Mol Med. 2004;36:185–91. doi: 10.1038/emm.2004.26. [DOI] [PubMed] [Google Scholar]
- 32.Fineberg SK, Kosik KS, Davidson BL. MicroRNAs potentiate neural development. Neuron. 2009;64:303–9. doi: 10.1016/j.neuron.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 33.French PJ, O’Connor V, Voss K, Stean T, Hunt SP, Bliss TV. Seizure-induced gene expression in area CA1 of the mouse hippocampus. Eur J Neurosci. 2001;14:2037–41. doi: 10.1046/j.0953-816x.2001.01818.x. [DOI] [PubMed] [Google Scholar]
- 34.Gorter JA, van Vliet EA, Aronica E, Breit T, Rauwerda H, Lopes da Silva FH, Wadman WJ. Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci. 2006;26:11083–110. doi: 10.1523/JNEUROSCI.2766-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorter JA, Van Vliet EA, Rauwerda H, Breit T, Stad R, van Schaik L, Vreugdenhil E, Redeker S, Hendriksen E, Aronica E, Lopes da Silva FH, Wadman WJ. Dynamic changes of proteases and protease inhibitors revealed by microarray analysis in CA3 and entorhinal cortex during epileptogenesis in the rat. Epilepsia. 2007;48(Suppl 5):53–64. doi: 10.1111/j.1528-1167.2007.01290.x. [DOI] [PubMed] [Google Scholar]
- 36.Herman ST. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59:S21–6. doi: 10.1212/wnl.59.9_suppl_5.s21. [DOI] [PubMed] [Google Scholar]
- 37.Hunsberger JG, Bennett AH, Selvanayagam E, Duman RS, Newton SS. Gene profiling the response to kainic acid induced seizures. Brain Res Mol Brain Res. 2005;141:95–112. doi: 10.1016/j.molbrainres.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Jamali S, Bartolomei F, Robaglia-Schlupp A, Massacrier A, Peragut JC, Regis J, Dufour H, Ravid R, Roll P, Pereira S, Royer B, Roeckel-Trevisiol N, Fontaine M, Guye M, Boucraut J, Chauvel P, Cau P, Szepetowski P. Large-scale expression study of human mesial temporal lobe epilepsy: evidence for dysregulation of the neurotransmission and complement systems in the entorhinal cortex. Brain. 2006;129:625–41. doi: 10.1093/brain/awl001. [DOI] [PubMed] [Google Scholar]
- 39.Jimenez-Mateos EM, Hatazaki S, Johnson MB, Bellver-Estelles C, Mouri G, Bonner C, Prehn JH, Meller R, Simon RP, Henshall DC. Hippocampal transcriptome after status epilepticus in mice rendered seizure damage-tolerant by epileptic preconditioning features suppressed calcium and neuronal excitability pathways. Neurobiol Dis. 2008;32:442–53. doi: 10.1016/j.nbd.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 40.Kadam SD, White AM, Staley KJ, Dudek FE. Continuous electroencephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. J Neurosci. 30:404–15. doi: 10.1523/JNEUROSCI.4093-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kapur J, Lothman EW, DeLorenzo RJ. Loss of GABAA receptors during partial status epilepticus. Neurology. 1994;44:2407–8. doi: 10.1212/wnl.44.12.2407. [DOI] [PubMed] [Google Scholar]
- 42.Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1997;94:14060–5. doi: 10.1073/pnas.94.25.14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kasperaviciute D, Catarino CB, Heinzen EL, Depondt C, Cavalleri GL, Caboclo LO, Tate SK, Jamnadas-Khoda J, Chinthapalli K, Clayton LM, Shianna KV, Radtke RA, Mikati MA, Gallentine WB, Husain AM, Alhusaini S, Leppert D, Middleton LT, Gibson RA, Johnson MR, Matthews PM, Hosford D, Heuser K, Amos L, Ortega M, Zumsteg D, Wieser HG, Steinhoff BJ, Kramer G, Hansen J, Dorn T, Kantanen AM, Gjerstad L, Peuralinna T, Hernandez DG, Eriksson KJ, Kalviainen RK, Doherty CP, Wood NW, Pandolfo M, Duncan JS, Sander JW, Delanty N, Goldstein DB, Sisodiya SM. Common genetic variation and susceptibility to partial epilepsies: a genome-wide association study. Brain. doi: 10.1093/brain/awq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–8. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- 45.Kyin R, Hua Y, Baybis M, Scheithauer B, Kolson D, Uhlmann E, Gutmann D, Crino PB. Differential cellular expression of neurotrophins in cortical tubers of the tuberous sclerosis complex. Am J Pathol. 2001;159:1541–54. doi: 10.1016/S0002-9440(10)62539-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lakaye B, de Borman B, Minet A, Arckens L, Vergnes M, Marescaux C, Grisar T. Increased expression of mRNA encoding ferritin heavy chain in brain structures of a rat model of absence epilepsy. Exp Neurol. 2000;162:112–20. doi: 10.1006/exnr.2000.7303. [DOI] [PubMed] [Google Scholar]
- 47.Lauren HB, Lopez-Picon FR, Brandt AM, Rios-Rojas CJ, Holopainen IE. Transcriptome analysis of the hippocampal CA1 pyramidal cell region after kainic acid-induced status epilepticus in juvenile rats. PLoS One. 5:e10733. doi: 10.1371/journal.pone.0010733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu XY, Yang JL, Chen LJ, Zhang Y, Yang ML, Wu YY, Li FQ, Tang MH, Liang SF, Wei YQ. Comparative proteomics and correlated signaling network of rat hippocampus in the pilocarpine model of temporal lobe epilepsy. Proteomics. 2008;8:582–603. doi: 10.1002/pmic.200700514. [DOI] [PubMed] [Google Scholar]
- 49.Loeb JA. A human systems biology approach to discover new drug targets in epilepsy. Epilepsia. 2010;51:171–177. doi: 10.1111/j.1528-1167.2010.02635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loeb JA, Beaumont TL. What goes in is what comes out: How to design and implement a successful microarray experiment. In: Krawetz S, editor. Bioinformatics for systems biology. Springer; New York: 2009. pp. 209–226. [Google Scholar]
- 51.Lombardo AJ, Kuzniecky R, Powers RE, Brown GB. Altered brain sodium channel transcript levels in human epilepsy. Brain Res Mol Brain Res. 1996;35:84–90. doi: 10.1016/0169-328x(95)00194-w. [DOI] [PubMed] [Google Scholar]
- 52.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 53.Miyata H, Chiang AC, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–9. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- 54.Mody I. Ion channels in epilepsy. Int Rev Neurobiol. 1998;42:199–226. doi: 10.1016/s0074-7742(08)60611-x. [DOI] [PubMed] [Google Scholar]
- 55.Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- 56.Parker A, McCaffery I, Patterson S. Examining molecular biology in humans. Biotechniques. 2009;46:358–60. doi: 10.2144/000113141. [DOI] [PubMed] [Google Scholar]
- 57.Rakhade SN, Loeb JA. Focal reduction of neuronal glutamate transporters in human neocortical epilepsy. Epilepsia. 2008;49:226–36. doi: 10.1111/j.1528-1167.2007.01310.x. [DOI] [PubMed] [Google Scholar]
- 58.Rakhade SN, Shah AK, Agarwal R, Yao B, Asano E, Loeb JA. Activity-dependent gene expression correlates with interictal spiking in human neocortical epilepsy. Epilepsia. 2007;48(Suppl 5):86–95. doi: 10.1111/j.1528-1167.2007.01294.x. [DOI] [PubMed] [Google Scholar]
- 59.Rakhade SN, Yao B, Ahmed S, Asano E, Beaumont TL, Shah AK, Draghici S, Krauss R, Chugani HT, Sood S, Loeb JA. A common pattern of persistent gene activation in human neocortical epileptic foci. Ann Neurol. 2005;58:736–47. doi: 10.1002/ana.20633. [DOI] [PubMed] [Google Scholar]
- 60.Sen CK, Roy S. miRNA: licensed to kill the messenger. DNA Cell Biol. 2007;26:193–4. doi: 10.1089/dna.2006.0567. [DOI] [PubMed] [Google Scholar]
- 61.Sharma AK, Searfoss GH, Reams RY, Jordan WH, Snyder PW, Chiang AY, Jolly RA, Ryan TP. Kainic acid-induced F-344 rat model of mesial temporal lobe epilepsy: gene expression and canonical pathways. Toxicol Pathol. 2009;37:776–89. doi: 10.1177/0192623309344202. [DOI] [PubMed] [Google Scholar]
- 62.Sherwin AL, Vernet O, Dubeau F, Olivier A. Biochemical markers of excitability in human neocortex. Can J Neurol Sci. 1991;18:640–4. doi: 10.1017/s0317167100032868. [DOI] [PubMed] [Google Scholar]
- 63.Staley K, Hellier JL, Dudek FE. Do interictal spikes drive epileptogenesis? Neuroscientist. 2005;11:272–6. doi: 10.1177/1073858405278239. [DOI] [PubMed] [Google Scholar]
- 64.Stan AD, Ghose S, Gao XM, Roberts RC, Lewis-Amezcua K, Hatanpaa KJ, Tamminga CA. Human postmortem tissue: what quality markers matter? Brain Res. 2006;1123:1–11. doi: 10.1016/j.brainres.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Steinlein OK. Genetic mechanisms that underlie epilepsy. Nat Rev Neurosci. 2004;5:400–8. doi: 10.1038/nrn1388. [DOI] [PubMed] [Google Scholar]
- 66.Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10–3. doi: 10.1111/j.1528-1167.2008.02005.x. [DOI] [PubMed] [Google Scholar]
- 67.Tessler S, Danbolt NC, Faull RL, Storm-Mathisen J, Emson PC. Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience. 1999;88:1083–91. doi: 10.1016/s0306-4522(98)00301-7. [DOI] [PubMed] [Google Scholar]
- 68.Vernet O, Yong VW, Rostworowski K, Fadich M, Olivier A, Sherwin AL. Protein kinase C activity and intracellular distribution in surgically excised human epileptic neocortex. Brain Res. 1991;547:319–22. doi: 10.1016/0006-8993(91)90978-5. [DOI] [PubMed] [Google Scholar]
- 69.Wang YY, Smith P, Murphy M, Cook M. Global expression profiling in epileptogenesis: does it add to the confusion? Brain Pathol. 20:1–16. doi: 10.1111/j.1750-3639.2008.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.White A, Williams PA, Hellier JL, Clark S, Edward Dudek F, Staley KJ. EEG spike activity precedes epilepsy after kainate-induced status epilepticus. Epilepsia. 51:371–83. doi: 10.1111/j.1528-1167.2009.02339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.White HS. Animal models of epileptogenesis. Neurology. 2002;59:S7–S14. doi: 10.1212/wnl.59.9_suppl_5.s7. [DOI] [PubMed] [Google Scholar]
- 72.White R, Hua Y, Scheithauer B, Lynch DR, Henske EP, Crino PB. Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann Neurol. 2001;49:67–78. doi: 10.1002/1531-8249(200101)49:1<67::aid-ana10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 73.Williams PA, White AM, Clark S, Ferraro DJ, Swiercz W, Staley KJ, Dudek FE. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–12. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xiao F, Chen D, Lu Y, Xiao Z, Guan LF, Yuan J, Wang L, Xi ZQ, Wang XF. Proteomic analysis of cerebrospinal fluid from patients with idiopathic temporal lobe epilepsy. Brain Res. 2009;1255:180–9. doi: 10.1016/j.brainres.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 75.Yang JW, Czech T, Gelpi E, Lubec G. Extravasation of plasma proteins can confound interpretation of proteomic studies of brain: a lesson from apo A-I in mesial temporal lobe epilepsy. Brain Res Mol Brain Res. 2005;139:348–56. doi: 10.1016/j.molbrainres.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 76.Ying Z, Babb TL, Comair YG, Bingaman W, Bushey M, Touhalisky K. Induced expression of NMDAR2 proteins and differential expression of NMDAR1 splice variants in dysplastic neurons of human epileptic neocortex. J Neuropathol Exp Neurol. 1998;57:47–62. doi: 10.1097/00005072-199801000-00007. [DOI] [PubMed] [Google Scholar]