

Abstract
Recent technological advances in the field of chromosome conformation capture are facilitating tremendous progress in the ability to map the three-dimensional (3D) organization of chromosomes at a resolution of several Kb and at the scale of complete genomes. Here we review progress in analyzing chromosome organization in human cells by building 3D models of chromatin based on comprehensive chromatin interaction datasets. We describe recent experiments that suggest that long-range interactions between active functional elements are sufficient to drive folding of local chromatin domains into compact globular states. We propose that chromatin globules are commonly formed along chromosomes, in a cell type specific pattern, as a result of frequent long-range interactions among active genes and nearby regulatory elements. Further, we speculate that increasingly longer range interactions can drive aggregation of groups of globular domains. This process would yield a compartmentalized chromosome conformation, consistent with recent observations obtained with genome-wide chromatin interaction mapping.
Introduction
Genomes are confined inside the densely crowded environment of the cell nucleus. Despite the presence of significant cell-to-cell variation, chromosomes are not randomly organized and levels of organization can be discerned at different scales. Two types of constraints shape the conformation of chromosomes [1]. First, extrinsic constraints include steric features such as the dimensions of the nucleus, the limited ability of chromosomes to intermingle, and associations between chromosomal loci with the nuclear envelope. Second, intrinsic constraints are imposed by the local structure of the chromatin fiber, which can vary from open to condensed and by the formation of loops through long-range interactions between genomic elements.
In recent years there has been a dramatic increase in our ability to study the structure of chromosomes particularly through the development of the chromosome conformation capture (3C) technology [2] and its high-throughput modifications that include several related 4C methods [3-6], 5C [7], Hi-C [8] and ChIA-PET [9]. These 3C-based methods utilize formaldehyde cross-linking to capture interacting loci, followed by DNA fragmentation and proximity-based ligation to convert interacting loci into unique ligation products [Fig. 1, and reviewed in [1]]. Combining these methods with next generation sequencing techniques has enabled the mapping of interactions at high resolution and at a genome-wide scale. 3C-based assays are particularly powerful in revealing the presence of chromatin loops that impose intrinsic constraints on chromosome conformation [10-13].
Figure 1. 3C-based methods.
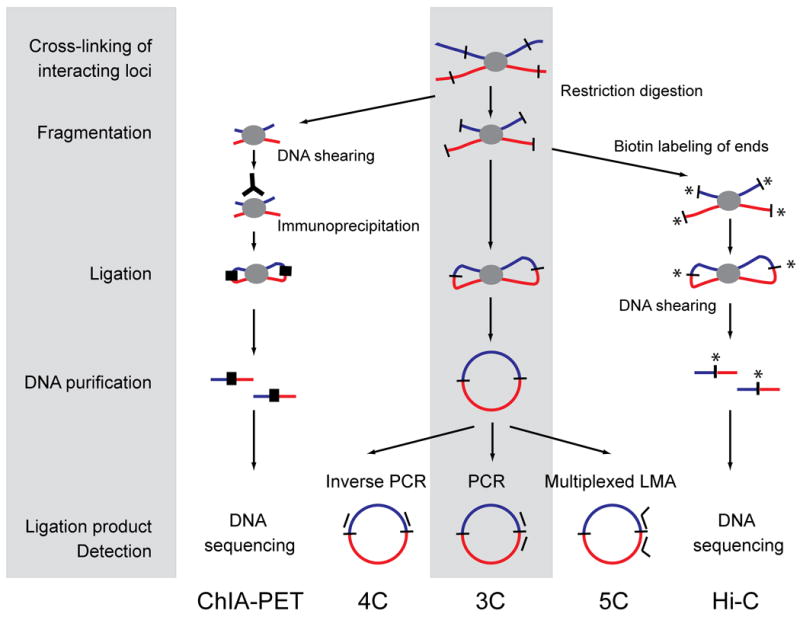
All 3C-based methods use the same principle of chromatin interaction detection (indicated in grey): cross-linking of interacting loci with formaldehyde, followed by DNA fragmentation, DNA ligation, DNA purification and finally ligation product detection. 3C employs restriction enzymes to fragment DNA, and regular PCR to detect ligation products, while 4C employs inverse PCR to detect all fragments ligated to a locus of choice. 5C uses multiplexed ligation mediated amplification (LMA) to detect large numbers of interactions simultaneously using pools of primers for thousands of loci of interest. CHiA-PET employs sonication to fragment cross-linked chromatin followed by an immunoprecipitation step prior to DNA ligation to enrich for loci bound by a protein of interest. Linkers are then ligated (black thick lines) and DNA is analyzed by direct deep sequencing. Finally, Hi-C employs restriction enzymes to fragment chromatin followed by filling in of the staggered ends using biotinylated nucleotides prior to DNA ligation. DNA is sheared and DNA fragments containing ligation junctions are purified using streptavidin-coated beads. DNA is then directly deep sequenced.
Comprehensive chromatin interaction datasets can be used to gain insights in the overall three-dimensional (3D) structure of chromatin. Here we will review recently developed approaches that integrate comprehensive chromatin interaction mapping technologies, cell imaging and 3D modeling to derive insights into chromosome folding [2, 6, 8, 14, 15].
Using long-range interaction data to build 3D models of chromatin
The notion that sets of chromatin interaction data could be used to estimate the overall 3D folding of chromatin has been around since the development of the 3C technology [2]. The idea is based on the realization that the interaction frequency of a pair of loci, as determined using any 3C-based assay, is inversely related to the average spatial distance between them. Indeed, imaging analyses have confirmed that this assumption is largely correct [8, 16], although the exact form of such relationship is unknown and likely to vary between different cell types or organisms. Nevertheless, when a sufficiently dense matrix of interaction frequencies is measured among a series of chromatin segments, e.g. representing a chromosome or a chromosomal domain, one can construct 3D models that place these chromatin segments in relative spatial positions that optimally satisfy the distance constraints derived from the interaction data. High-resolution 3D modeling of chromosomal domains is currently limited to small fractions of genomes (up to Mb domains) given that: (i) chromosomes are higly dynamic in nature and thus at larger scales we are expected to observe increasingly large variability between cells, and (ii) to identify deterministic solutions of larger genomic domains (e.g. whole human chromosomes and genomes), it is necessary to measure very large numbers of long-range interactions (restraints) that would allow for an efficient computational sampling of the 3D architecture of chromosomes.
The first low resolution 3D model of a complete chromosome was determined for yeast chromosome III [2]. In that case the 3C interaction data could be modeled using well-established polymer models that describe the chromatin fiber as a flexible freely jointed chain. Given that for such a polymer the relationship between interaction probability and average distance is precisely known, the 3C data could be directly translated into 3D distances. The set of distances then allowed a strictly mathematical calculation of a single 3D model of the chromosome that represents its average folding. The chromosome appeared to form a contorted ring, with the two telomeres closely juxtaposed.
Recently, several new approaches have been developed that use a more deterministic approach to 3D modeling of genomes and genomic domains [6, 14, 17, 18]. The commonality of such approaches is the integration of diverse experiments and computation to build 3D structures that maximally satisfy the experimental observations. These approaches are particularly useful for 3D modeling of chromosomal regions that are not readily described as a simple polymer due to the presence of prominent and abundant long-range interactions between specific distant DNA elements. This is particularly relevant for analysis of chromatin domains in complex genomes, such as those of human and mouse, where long-range looping interactions involved in gene regulation, e.g. between promoters, enhancer and insulators, are wide-spread [13, 19, 20]. For example, Jhunjhunwala and colleagues used a combination of FISH experiments and computation to determine a low-resolution architecture of the immunoglobulin heavy-chain (Igh) locus [17]. By computationally triangulating a set of distances between 12 fluorescent probes spanning the Igh locus, they proposed that the Igh locus is organized into compartments containing clusters of loops separated by linkers. More recently, the structure of the HoxA cluster was determined at low-resolution by minimizing its root mean square deviation to the inter-loci distances theoretically inferred from 5C interaction frequencies [18]. Duan and colleagues used a conceptually similar approach to build a 3D model of the entire yeast genome based on a genome-wide chromatin interaction dataset [6]. The resulting model is highly consistent with previous cytological observations. For instance, the previously described phenomenon of centromere clustering [21] was accurately reproduced. Finally, a higher resolution structure of the human α-globin domain was built using a comprehensive 5C dataset and the Integrative Modeling Platform (IMP, http:///www.integrativemodeling.org), and validated by microscopy [14]. IMP was originally developed as a general platform for integrating diverse types of structural information available for an object of interest and has been used to determine the architecture of the Nuclear Pore Complex [22, 23]. IMP translates input experimental data into points and restraints in 3D space. Then, IMP searches using a Monte Carlo protocol a set of optimal 3D structures that satisfy all imposed spatial restrains. The resulting 3D models represent the ensemble of solutions consistent with the diverse input data. In summary, for the examples outlined above, these methods allow the building of large ensembles of 3D models that all reasonably fit the chromatin interaction data. This is important because chromatin interaction data sets may represent the average of several dominant conformations that occur in the cell population, and the ensemble of 3D models could potentially identify these different conformations.
One important caveat of all these methods is that chromatin is highly dynamic. Thus, these static 3D models reflect overall tendencies for chromatin folding, and the specific path of the chromatin fiber in a given cell can differ substantially. For small highly constrained chromatin segments, e.g. the α-globin domain, these models represent accurate and biologically relevant structures, but one can expect that for larger genomic segments, i.e. whole human chromosomes that display a large degree of cell to cell variability, these approaches will not provide accurate structures. Clearly, there is significant room for improvement of modeling approaches that include the dynamic aspects of chromosomes.
The chromatin globule
In the human genome, genes and their regulatory elements, such as enhancers, are often spread out of over several hundred kilobases. Therefore, it is of significant interest to understand chromatin conformation of complex genomes at that length scale. 5C technology is particularly suited for obtaining comprehensive and high-resolution and quantitative interaction maps of chromatin domains up to several Mb [7].
5C and the IMP were combined to determine the first 3D models of a 500 Kb chromosomal domain on human chromosome 16 containing the α-globin locus and several house keeping genes [14]. 5C analysis of this gene rich region (Enm008) yielded a high resolution pairwise map of interactions between the genes and other elements. The 5C dataset was then converted into a set of average 3D distances and harmonic constraints that combined were used to generate an ensemble of optimal 3D models. In cells that do not express the α-globin genes (the lymphoblastoid cell line GM12878), but in which most other genes in the 500 Kb region were expressed, all models were highly similar to each other (Fig. 2A). The ensemble revealed an overall highly compacted chromatin domain, dubbed a “chromatin globule”. When the same analysis was repeated with K562 cells that express the α-globin genes, the domain appeared be more extended, but globular domains were again found.
Figure 2. 3D model of the α-globin gene domain in lymphoblastoid cells reveals a globular domain.
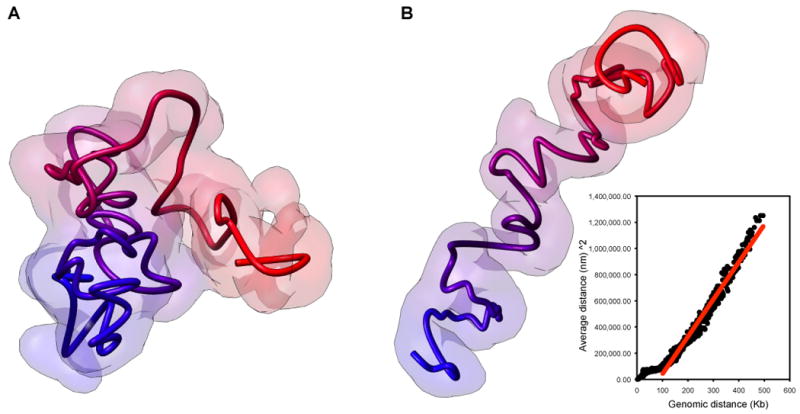
A. Chromatin globule formed by the 500 Kb α-globin gene containing domain in lymphoblastoid cells (From [14]). Long-range interactions between genes and elements such as enhancers and insulators lead to a collapse of the fiber to form a compact chromatin globule. B. 3D model of the α-globin gene containing domain in the absence of looping interactions. A theoretical 5C interaction map was calculated assuming a freely jointed chain, and a 3D model was calculated exactly as in A and as described in [14]. The model accurately reproduces the expected relationship between genomic distance between loci and spatial distance for a freely jointed chain [(spatial distance)2 ∼ genomic distance] (inset: Black dots: observed data; Red line: expected relationship) [11]. Deviations from this expected relationship are observed for smaller genomic distances, consistent with the fact that the feely jointed chain model is only valid for long-range interactions between sites separated by several times the persistence length of the fiber.
The 3D models correctly reproduced the known looping interactions [24, 25] between the HS40 enhancer and the α-globin genes in K562 cells specifically [14]. Interestingly, the core of the globules was found to be enriched in genes that are actively transcribed while non-transcribed chromatin was located at the periphery of the globules. This suggests that formation of compact chromatin globules is most likely driven by the existence of multiple long-range interactions between genes and between genes and putative regulatory elements. Besides the previously described looping interactions, the 5C analysis identified several novel frequent long-range looping contacts between genes and functional elements [14]. Although the functional relevance of these interactions is currently not known, they may be involved in regulation of various genes in the region. We hypothesize that these looping interactions may be directly responsible for formation of a compact chromatin globule. Here we provide some support for this idea by modeling the same 500 Kb region as an unconstrained random walk polymer. We generated a theoretical 5C dataset for the same set of loci (restriction fragments) throughout the 500 Kb region (Supplemental File 1) using the well-established relationship for a linear polymer between genomic distance to contact probability ([11] and Supplemental Material). This expected 5C dataset was then used to calculate an ensemble of 3D models in exactly the same way as was used to model the experimental 5C data of the region ([14] and Supplemental Material). The resulting models reveal a significantly more extended conformation than the 3D models built with the experimental 5C data (Fig. 2B). Importantly, when we analyzed the distances between the loci in the models they showed the expected relationship with the contact probabilities of the elements, as predicted by polymer theory (Fig. 2B, and Supplemental File 2) [2, 26]. This analysis shows that looping interactions are required for the otherwise extended chromatin fiber to fold into a compact globule. In addition, the fact that the random walk models accurately predict the average distances between the loci provides strong support for the general empirical approach for converting contact probability into constraints used for modeling.
We propose that the compact and looped chromatin globules are related, but also different from the previously described active “chromatin hub” [27, 28]. Analysis of the mouse β-globin locus led to the identification of a set of looping interactions between the β-globin genes, their distal enhancer (the Locus Control Region) and distal CTCF-bound elements. 3C studies suggested that all these elements cluster together in a single complex. As described above chromatin globule formation also required long-range looping interactions, but the resulting structure is distinct from an active chromatin hub because the elements do not all cluster in one complex, and the structure is not dedicated to regulation of a single set of related genes. Instead, the chromatin globule is a “collapsed” chromatin fiber driven by a network of interactions between nearby genes and gene regulatory elements.
We believe that chromatin globule formation is related to the previously described clustering of active genes, e.g. at so-called transcription factories. Both cytological and 3C-based studies found that active genes tend to associate with other active genes [3, 29, 30]. Given that long-range interactions are most frequent for loci located near each other along the same chromosome, contiguous active chromosomal domains would most easily fold into compact globules.
Globule formation: a general property of active chromatin domains?
Analysis of a large set of loci in a number of studies suggests that long-range interactions between genes and distal elements are common throughout the genome, and these interactions ar directly related to gene regulation. For instance, several studies identified long-rang looping interactions among CTCF-bound elements, gene promoters and enhancers [20, 31-34]. This would predict that many or all gene-rich regions would be highly looped and to fold into higher order structures that may be similar to compact chromatin globules as observed for the α-globin region. Indeed, we recently analyzed the HoxA locus, and found that groups of active HoxA genes cluster together in spatial domains [35]. If this is generally true, chromosomes can be viewed as a series of cell type specific compact globular domains that each represents groups of active genes and associated regulatory elements. In support of this, cytological observations of active chromatin domains revealed series of globular domains [36].
Long-range interactions, and globule formation, may not be limited to associations between active loci: long-range contacts have also been detected between silenced loci in yeast [16], and between widely spaced polycomb-bound Hox loci in flies [37]. In the case of the Hox loci these interactions were shown to contribute to their appropriate regulation.
Beyond the chromatin globule
Chromatin globules are driven by the frequent looping interactions between genes and regulatory elements that are located relatively close along the length of the chromosome. On top of these strong (frequent) looping interactions 4C and Hi-C experiments have detected weaker (less frequent) interactions between groups of active genes separated by large genomic distances (tens of Mb) [3, 5, 8]. We speculate that those weaker long-range interactions bring distal globules together in space resulting in larger aggregates of active chromatin domains (Fig. 3). This model proposes a hierarchical chromosome structure in which the local chromatin globule forms the first building block (Fig. 3). As a result of association of groups of chromatin globules a compartmentalized chromosome organization in which active chromatin is spatially separated from inactive chromatin would naturally emerge, consistent with results obtained with Hi-C [8]. The functional relevance of these very long-range interactions between active loci is currently far from clear. Possibly their main role is simply related to organizing chromosomes, which is an important phenomenon by itself, with only indirect and possibly non-specific effects on gene expression.
Figure 3. Chromatin globules as a first building block of chromosome conformation.
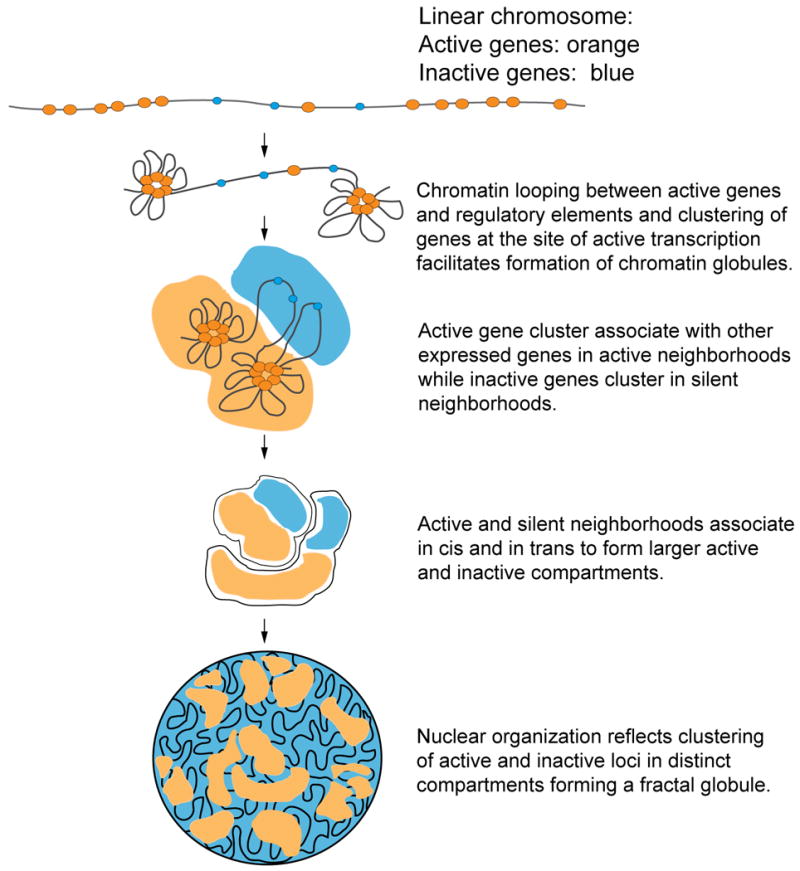
Hierarchical model of chromosome organization, driven by globule formation, and interactions between globules.
Currently, this model is highly speculative. Future chromatin interaction mapping, 3D modeling and further development of theoretical polymer models such as the Dynamic Looping model and the Fractal Globule model [8, 12, 38] that may describe these experimentally observed collapsed chromatin states, may provide insights into dynamic folding properties of chromatin domains, and ultimately whole chromosomes.
Supplementary Material
Acknowledgments
J.D. and A.S. are supported by a grant from the NIH (HG003143) and the Keck Foundation. MAM-R and D.B. are supported by the Spanish Ministerio de Ciencia e Innovación (BIO2007/66670).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of special interest (*) or outstanding interest (**)
- 1.van Steensel B, Dekker J. Genomics tools for unraveling chromosome architecture. Nat Biotechnol. 2010;28(10):1089–1095. doi: 10.1038/nbt.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dekker J, et al. Capturing Chromosome Conformation. Science. 2002;295(5558):1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 3.Simonis M, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38(11):1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 4.Zhao Z, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38(11):1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 5.Schoenfelder S, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010 Jan;42(1):53–61. doi: 10.1038/ng.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6**.Duan Z, et al. A three-dimensional model of the yeast genome. Nature. 2010;465(7296):363–367. doi: 10.1038/nature08973. This paper presents a genome-wide interaction map of the yeast genome. This data set is then used to build a three-dimensional model of the genome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dostie J, et al. Chromosome Conformation Capture Carbon Copy (5C): A Massively Parallel Solution for Mapping Interactions between Genomic Elements. Genome Res. 2006;16(10):1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8**.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. doi: 10.1126/science.1181369. This paper described a 3C-based method (Hi-C) for genome-wide mapping of long-range interactions. Hi-C is used to generate an interaction map for the human genome, which reveals that chromosomes are compartmentalized and that chromatin fold in a new polymer state called the fractal globule. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Fullwood MJ, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462(7269):58–64. doi: 10.1038/nature08497. This work described a methdo that combines chromatin immunoprecipitation and 3C-based proximity ligation. The method is used to detect interactions between sites bound by the estrogen receptor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sachs RK, et al. A random-walk/giant-loop model for interphase chromosomes. Proc Natl Acad Sci USA. 1995;92(7):2710–2714. doi: 10.1073/pnas.92.7.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rippe K. Making contacts on a nucleic acid polymer. Trends Biochem Sci. 2001;26(12):733–740. doi: 10.1016/s0968-0004(01)01978-8. [DOI] [PubMed] [Google Scholar]
- 12.Bohn M, Heermann DW. Diffusion-driven looping provides a consistent framework for chromatin organization. PLoS One. 2010;5(8):e12218. doi: 10.1371/journal.pone.0012218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miele A, Dekker J. Long-range chromosomal interactions and gene regulation. Mol BioSyst. 2008;4(11):1046–57. doi: 10.1039/b803580f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14**.Baù D, et al. The three-dimensional folding of the alpha-globin gene domain reveals formation of chromatin globules. Nat Struct Mol Biol. 2011;18(1):107–114. doi: 10.1038/nsmb.1936. This paper presents a method for building three-dimensional models of chromatin domains using the Integrative Modeling Platform and comprehensive 5C interaction datasets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baù D, Marti-Renom MA. Structure determination of genomic domains by satisfaction of spatial restraints. Chromosome Res. 2011;19(1):p25–35. doi: 10.1007/s10577-010-9167-2. [DOI] [PubMed] [Google Scholar]
- 16.Miele A, Bystricky K, Dekker J. Yeast silent mating type loci form heterochromatic clusters through silencer protein-dependent long-range interactions. PLoS Genet. 2009;5(5):e1000478. doi: 10.1371/journal.pgen.1000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17*.Jhunjhunwala S, et al. The 3D structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions. Cell. 2008;133(2):265–279. doi: 10.1016/j.cell.2008.03.024. This paper employs imaging and polymer modeling to gain insights into the three-dimensional folding of the immunoglobulin hevay chain locus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraser J, et al. Chromatin conformation signatures of cellular differentiation. Genome Biol. 2009;10(4):R37. doi: 10.1186/gb-2009-10-4-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dekker J. Gene regulation in the third dimension. Science. 2008;319(5871):1793–1794. doi: 10.1126/science.1152850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin QW, Fuchs J, Loidl J. Centromere clustering is a major determinant of yeast interphase nuclear organization. J Cell Sci. 2000;113:1903–1912. doi: 10.1242/jcs.113.11.1903. [DOI] [PubMed] [Google Scholar]
- 22.Alber F, et al. The molecular architecture of the nuclear pore complex. Nature. 2007;450(7170):695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- 23.Alber F, et al. Determining the architectures of macromolecular assemblies. Nature. 2007;450(7170):683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- 24.Zhou GL, et al. Active chromatin hub of the mouse alpha-globin locus forms in a transcription factory of clustered housekeeping genes. Mol Cell Biol. 2006;26(13):5096–5105. doi: 10.1128/MCB.02454-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vernimmen D, et al. Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J. 2007;26(8):2041–2051. doi: 10.1038/sj.emboj.7601654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dekker J. Mapping in vivo chromatin interactions in yeast suggests an extended chromatin fiber with regional variation in compaction. J Biol Chem. 2008;283(50):34532–34540. doi: 10.1074/jbc.M806479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tolhuis B, et al. Looping and Interaction between Hypersensitive Sites in the Active beta-globin Locus. Mol Cell. 2002;10(6):1453–1465. doi: 10.1016/s1097-2765(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 28.de Laat W, Grosveld F. Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003;11(5):447–459. doi: 10.1023/a:1024922626726. [DOI] [PubMed] [Google Scholar]
- 29.Osborne CS, et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36(10):1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 30.Cook PR. A model for all genomes: the role of transcription factories. J Mol Biol. 2010;395(1):1–10. doi: 10.1016/j.jmb.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 31.Splinter E, et al. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2004;20(17):2349–2354. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurukuti S, et al. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci U S A. 2006;103(28):10684–10689. doi: 10.1073/pnas.0600326103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nativio R, et al. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009;5(11):e1000739. doi: 10.1371/journal.pgen.1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hadjur S, et al. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460(7253):410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang KC, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene activation. Nature. 2011 doi: 10.1038/nature09819. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller WG, et al. Generic features of tertiary chromatin structure as detected in natural chromosomes. Mol Cell Biol. 2004;24(21):9359–9370. doi: 10.1128/MCB.24.21.9359-9370.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bantignies F, Roure V, Comet I, Leblanc B, Schuettengruber B, Bonnet J, Tixier V, Mas A, Cavalli G. Polycomb-dependent regulatory contacts between distant Hox loci in Drosophila. Cell. 144(2):214–226. doi: 10.1016/j.cell.2010.12.026. [DOI] [PubMed] [Google Scholar]
- 38.Mirny LA. The fractal globule as a model of chromatin architecture in the cell. Chromosome Res. 2011;19(1):37–51. doi: 10.1007/s10577-010-9177-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.