

Abstract
Superoxide production via NADPH-oxidase (NOX) has been shown to play a role in variety of neurological disorders including Alzheimer’s disease (AD). To improve our understanding of the NOX system and cognitive impairment, we studied the various protein components of the phagocytic isoform (NOX2) in the frontal and temporal cortex of age and postmortem matched samples. Individuals underwent antemortem cognitive testing and postmortem histopathologic assessment to determine disease progression and assignment to one of the following groups: no cognitive impairment (NCI), preclinical AD (PCAD), mild cognitive impairment (MCI), early AD (eAD) and mild to moderate AD (mAD). Biochemical methods were used to determine overall NOX activity as well as levels of the different subunits (gp91phox, p67phox, p47phox, p40phox, and p22phox ). Overall enzyme activity was significantly elevated in the MCI cohort in both cortical regions when compared to the NCI cohort. This activity level remained elevated in the AD groups. Only the NOX cytosolic subunit proteins (p67phox, p47phox, and p40phox ) were significantly elevated with disease progression, while the membrane bound subunits (gp91phox and p22phox ) remained stable. In addition, there was a robust correlation between NOX activity and the individual’s cognitive status such that as the enzyme activity increased, cognitive performance decreased. Collectively, these data show that NADPH-oxidase upregulated in frontal and temporal cortex suggest that increases in NOX associated redox pathways might participate in early pathogenesis and contribute to AD progression.
Keywords: NADPH-oxidase, preclinical AD, mild cognitive impairment, Alzheimer’s disease
INTRODUCTION
Alzheimer’s disease (AD) is a progressive dementing disorder characterized clinically by impairment in memory, cognition and behavior. The neuropathology of the disease features increased numbers of neuritic plaques, neurofibrillary tangles, and the loss of synaptic connectivity in key neocortical regions [1, 2]. A major research emphasis in AD progression has recently been placed on early evaluation, with the hope of identifying the earliest clinical manifestations of the disease onset. One of the presumed early transition stages is the clinical diagnosis of mild cognitive impairment (MCI), defined as an individual expressing consistent, measurable memory impairment without dementia or significant disability related to activities of daily living [3, 4]. Many AD subjects often are first seen clinically with amnestic MCI, and likely represent an early opportunity for possible pharmacologic interventions. The drive to identify the earliest manifestations of AD has led to the concept of preclinical AD (PCAD), also called presymptomatic AD or prodromal AD. PCAD is assigned to cognitively normal individuals who at autopsy manifest sufficient AD pathologic hallmarks to meet at least one set of neuropathology guidelines for diagnosis of AD [5–7].
Oxidative stress, defined as a marked imbalance between reactive oxygen species (ROS) and its removal by antioxidant systems, has been implicated in many neurodegenerative diseases including AD [8–10]. There are multiple potential sources for ROS (e.g. mitochondrial respiration) that can contribute to the different disease processes. Including all sources of free radical production, the enzyme NADPH-oxidase (nicotinamide adenosine dinucleotide phosphate oxidase; NOX) is notable, as it is dedicated to the specific and deliberate production of superoxide (O2•−). The NOX family consists of at least seven different isoforms that have unique distribution patterns throughout the body. Some of the NOX enzymes are expressed throughout the CNS with areas such as the cortex and hippocampus demonstrating particularly high amounts in neurons, astrocytes and glia [11]. These enzyme complexes have multiple biological functions including cellular differentiation, apoptosis, and neuronal signaling during development. They have been implicated in NGF signaling and may play an important role in the nervous system’s plasticity response including learning and memory [12]. Other well known functions include a host defense coupled with inflammation and increased oxidative stress where it appears to contribute to neurodegenerative diseases such as AD [11, 13].
Brain phagocytes have a NOX complex that is composed of primarily five different subunits [14]. A catalytic subunit gp91phox (also called NOX2), along with subunit p22phox are embedded in the cell membrane. This flavocytochrome structure is regulated by the association of three cytosolic subunits p40phox, p47phox, and p67phox. In the presence of an appropriate stimulus, the cytosolic subunits translocate to the membrane and interact with the flavocytochrome to form an active complex that generates ROS. Previous studies have implicated changes in NOX activity to the progression of AD suggesting that increased activity may be one of the early events in the transition from normal cognition to dementia [15–17]. The present set of investigations was undertaken to more fully explore the relationship of NOX activity and AD with particular attention paid to the levels of the different subunits and the individuals cognitive test scores.
Materials & Methods
Frozen frontal and temporal cortex samples were obtained from the University of Kentucky Rapid Autopsy Program of the Alzheimer’s Disease Clinical Center. Individuals were classified as no cognitive impairment (NCI), PCAD, MCI, early AD (eAD) or mild to moderate AD (mAD) based upon cognitive testing and histopathologic analysis. The diagnosis of probable AD was made according to criteria developed by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association [18]. Classification of MCI was based only upon cognitive testing [3]. Individuals included in these studies, were followed longitudinally for approximately 8 years, agreed to an annual clinical evaluation and to brain donation at the time of death. A total of 19 NCI subjects were without history of dementia or other neurological disorders and underwent annual mental status testing and semiannual physical and neurological exams as part of the UK ADC normal volunteer longitudinal aging study. In addition to the NCI cohort, 11 PCAD, 11 MCI, 8 eAD and 11 mAD subjects were used to quantify NOX activity and protein subunits in the isolated membrane fractions. The diagnosis of MCI, PCAD eAD, mAD and NCI were defined by consensus conference. All NCI and PCAD subjects had neuropsychological scores in the normal range with no evidence of cognitive impairments. Histological examination of NCI subjects showed only age-related changes and Braak stage score of 0-II, meeting the National Institute of Aging-Reagan Institute (NIA-RI) low-likelihood criteria for the histopathologic diagnosis of AD. PCAD subjects were distinguished as those individuals with antemortem psychometric test scores in the normal range, but sufficient AD pathology to meet intermediate or high NIA-RI criteria (Braak scores of II-V, with moderate or frequent neuritic plaque score according to Consortium to Establish a Registry for AD (CERAD). The clinical criteria for diagnosis of amnestic MCI included (1) memory complaints, (2) intact activities of daily living, (3) objective memory impairment for age and education, (4) failure to meet criteria for dementia, and (5) a clinical dementia rating (CDR) scale score of 0.5. The Braak stage scores had a range of I–V. Clinical progression to AD was diagnostically characterized by (1) a decline in cognitive functions from a previous higher level, (2) decline in one or more areas of cognition in addition to memory, (3) impaired activities of daily living, (4) a CDR score between 0.5–1, and (5) a clinical evaluation that excludes other causes of dementia. The criteria for eAD subjects included the above clinical progression plus a histopathologic diagnosis that included a Braak stage score of II–VI. For a mAD categorization, subjects demonstrated progressive intellectual decline as described above, a mini-mental status examination score (MMSE) less than that of the eAD cohort, and a Braak scores of V–VI. None of the mAD subjects were considered to be at the end-stage of the disease progression.
Estimation of NADPH-oxidase Activity
NADPH-oxidase activity was measured according to the method described earlier [19]. Briefly, homogenates of all samples were centrifuged at 13,000g for 10 min at 4°C, 100 μl supernatant were added in 900 μl HEPES–buffer containing L-NAME (1.0 mM), triethylenetetramine (1.0 mM), SDS (100 μM), lucigenin 20 μM, and the samples were aliquoted in 96-well plate. After addition of NADPH (0.2 mmol/L), enzyme activity was measured for 15 minutes in the luminometer (Synergy HT, BIO-TEK). Blanks were subtracted from sample-added wells and chemiluminescence data after blank subtraction are reported as percent counts/min/mg protein. Samples were also assayed in the presence of reaction inhibitors diphenyliodonium (100 μM) and quinacrine (1.0 mM).
Western-blots for NADPH-oxidase Protein Subunits
The key component proteins (gp91Phox, p67Phox, p47Phox, p40Phox and p22Phox) of NOX were studied by Western-blot method as described previously[19]. Briefly, collected supernatants were centrifuged at 100,000g for 1 h at 4°C to obtain membrane fractions. The 50μg protein was loaded with the appropriate marker (Bio-Rad) on a gradient gel (4–20% Tris-HCl), followed by transfer to polyvinylidene fluoride membrane using a semi-dry transfer for 2 hours at 15 volt. After 1 h blocking with 5% dry milk, primary antibodies for gp91Phox, p67Phox, p47Phox, p40Phox, p22Phox, and Na+/K+-ATPase, were added and incubated overnight at 4°C. The blot was washed and incubated for 1 hour with alkaline phosphatase conjugated secondary antibodies. The membrane was again washed and developed in Sigma Fast tablets (BCIP/NBT substrate). Blots were dried, scanned with Adobe Photoshop, and quantified with Scion Image (PC NIH Image). Total protein concentrations were measured using the Pierce BCA method (Sigma, St. Louis, MO). Because the function of NOX depends upon the complex formation, consisting of both cytosolic and membrane subunits, the present experiments focused on analyzing the translocated amount of cytosolic subunits into the plasma membrane based flavocytochrome. While the subunits are analyzed in both the cytosolic and membrane factions, only the values observed from the membrane factions are shown in the results section.
Data Analysis
Differences in NOX activity and its protein components are reported as mean ± standard deviation. Differences between group means were evaluated with a one-way ANOVA coupled with a Student Newman-Keuls post-hoc test (StatView 5.0, SAS Institute). Association between cognitive scores and biochemical markers were examined using a Spearman correlation. For significant differences, alpha was set at 0.05.
Results
The demographic parameters of each cohort are provided in Table 1. Statistical analysis revealed that groups were well matched in terms of age, education, post mortem interval, and brain weight. By design there was a significant difference in the MMSE scores [F (4,55) = 50.769, p < 0.0001] with the NCI and PCAD groups having scores significantly higher (p < 0.05) than the other cohorts but not differing from each other. A chi-square analysis failed to detect any significant gender differences between groups. The NCI cohorts had significantly lower Braak scores than all other groups. The PCAD group was not significantly different from the MCI and eAD cohorts. The mAD cohort had significantly higher Braak scores than all other groups.
TABLE 1.
Mean Demographic Data for All Groups.
| NCI (n=19) | PCAD (N=11) | MCI (n=11) | eAD (n=8) | mAD (n=11) | |
|---|---|---|---|---|---|
| Age (yr) | 83.2 ± 7.7 | 86.6 ± 6.3 | 91.3 ± 4.5 | 88.1 ± 6.1 | 82.5 ± 7.4 |
| Education (yr) | 16.1 ± 2.2 | 16.4 ± 1.8 | 16.8 ± 1.7 | 13.6 ± 2.5 | 15.1 ± 4.2 |
| Sex (M/F) | 11/8 | 4/7 | 4/7 | 4/4 | 6/5 |
| MMSE | 28.0 ± 2.0 | 28.7 ± 1.6 | 24.2 ± 3.5 * | 23.6 ± 3.0 * | 14.4 ± 4.7 * |
| PMI (h) | 3.0 ± .6 | 2.9 ± .8 | 3.0 ± .6 | 3.0 ± .9 | 3.4 ± 1.2 |
| Brain Wt. (g) | 1170 ± 116 | 1172 ± 167 | 1155 ± 139 | 1167 ± 60 | 1141 ± 112 |
Standard deviations following the ± sign
p < 0.05 compared to normal
NCI, no cognitive impairment, PCAD, preclinical AD, MCI, mild cognitive impairment, eAD, early AD, mAD, mild-moderate AD, MMSE, Mini Mental State Examination; PMI, postmortem interval
NADPH-oxidase Activity
Frontal Cortex
The overall NOX activity in the frontal cortex was analyzed as a function of disease progression. A one-way ANOVA demonstrated a significant main effect [F (4,55) = 12.309, p < 0.0001] for overall NOX activity (Fig. 1A). Post hoc analysis revealed a significant increase in the MCI cohort compared to the NCI group. The NOX activity remained elevated throughout the course of the disease progression. While there was a trend toward an increase in the PCAD group, it did not reach significance. The NOX complex is activated when the cytosolic subunits are phosphorylated resulting in translocation to the membrane domains. Analysis of the various subunits revealed that the membrane components did not significantly change while the cytosolic components significantly increased with the disease progression (Fig. 2A). A one-way ANOVA showed a significant progression effect for p67Phox [F(4,55) = 22.807, p < 0.0001], p47Phox [F(4,55) = 9.713, p < 0.0001], and p40Phox [F(4,55) = 11.121, p < 0.0001] such that all of the cytosolic subunits increased (Fig. 3A,C,E). The AD cohorts manifested the highest levels for all three cytosolic subunits with the mAD cohort significantly different from all other groups. Although there was a trend toward an increase in the MCI group, it did not reach significance. An analysis also determined a significant correlation between the subject’s Braak score and overall NOX activity in the frontal cortex (r = 0.553, p < 0.0001).
FIG. 1.

Levels of NADPH-oxidase (NOX) activity analyzed in human (A) frontal and (C) temporal cortex from individuals classified as NCI, PCAD, MCI, eAD, and mAD. Bars represent group mean ± SD. *p < 0.01 versus NCI, #p < 0.01 versus PCAD. Scatter plots show the association between NOX activity and the subject’s score on the Mini Mental Status Exam (B) FC and (D) TC. Line used to represent the direction of the association and does not indicate a line of regression. *** p < 0.001 Spearman’s rho. In both FC and TC, individuals with increased NADPH-oxidase activity had lower MMSE scores.
FIG. 2.
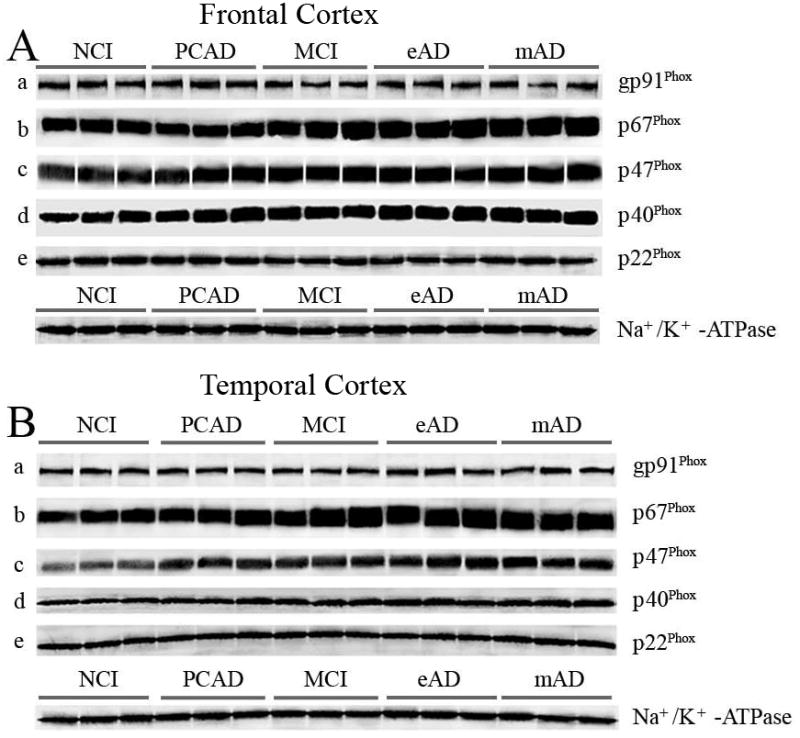
Representative Western-blots demonstrating changes in NADPH-oxidase protein subunits in the isolated membrane fractions from NCI, PCAD, MCI, eAD, and mAD subjects in frontal cortex (A), and temporal cortex (B) as a function of disease progression. The values were normalized with standard membrane marker Na+/K+ -ATPase. Isolated membrane fractions from all subjects were processed for immunobloting followed by Western-blot.
FIG. 3.

Levels of cytosolic protein subunits of NADPH-oxidase (p67Phox, p47Phox, p40Phox) (A,C,E) analyzed in membrane fractions of the frontal cortex from individuals classified as NCI, PCAD, MCI, eAD, and mAD. Values are plotted as percent change from the levels observed in the NCI cohort. Bars represent group mean ± SD. *p < 0.01 versus NCI, #p < 0.01 versus PCAD, $ p < 0.01 versus MCI. Scatter plots show the relationship between the individual’s score on the MMSE and the levels of the different cytosolic subunits (B,D,E). Each point represents an individual subject. Line used to represent the direction of the association and does not indicate a line of regression. *** p < 0.001 Spearman’s rho
Temporal Cortex
The overall NOX activity in the temporal cortex mirrored that of the frontal region. A one-way ANOVA demonstrated a significant main effect for disease progression [F(4,53) = 16.589, p < 0.0001] (Fig. 1C). Post hoc analysis revealed a significant increase in the three cognitively impaired groups compared to the NCI cohort. As with the frontal region, there was a non-significant trend for an increase in the PCAD group possibly indicating an early change in the activity. Examination of the specific subunits revealed a significant change in each of the cytosolic proteins and no significant change in those that are inserted in the membrane (Fig. 2B). A one-way ANOVA showed a significant progression effect for p67Phox [F(4,53) = 20.655, p < 0.0001], p47Phox [F(4,53) = 20.886, p < 0.0001], and p40Phox [F(4,53) = 12.655, p < 0.0001] such that all of the cytosolic subunits increased (Fig. 4A,C,E). Post hoc analysis revealed that early significant increases were observed in the MCI group for both the p67Phox and p47Phox subunits. This increase remained significantly elevated for both AD groups as well. For the p40Phox subunit, only the mAD cohort demonstrated a statistically significant increase. An analysis also determined a significant correlation between the subject’s Braak score and overall NOX activity in the temporal cortex (r = 0.553, p < 0.0001).
FIG. 4.

Levels of cytosolic protein subunits of NADPH-oxidase (p67Phox, p47Phox, p40Phox) (A,C,E) analyzed in membrane fractions of the temporal cortex from individuals classified as NCI, PCAD, MCI, eAD, and mAD. Values are plotted as percent change from the levels observed in the NCI cohort. Bars represent group mean ± SD. *p < 0.01 versus NCI, #p < 0.01 versus PCAD, $ p < 0.01 versus MCI. Scatter plots show the relationship between the individual’s score on the MMSE and the levels of the different cytosolic subunits (B,D,E). Each point represents an individual subject. Line used to represent the direction of the association and does not indicate a line of regression. *** p < 0.001 Spearman’s rho
NOX Activity and Cognitive Testing
All the samples were normalized with membrane specific marker protein Na+/K+-ATPase (Fig. 2). The NOX activity and expression of the different subunits were correlated with the MMSE scores observed within 12 months of each subject’s death. Figures 1B and 1D show the association between overall NOX activity in the frontal and temporal cortex and the results of cognitive testing. A correlation showed a very strong association between the subject’s most recent MMSE score and overall NOX activity in both the frontal (r = 0.526, p < 0.0001) and temporal (r = 0.461, p < 0.0005) cortex. Membrane-bound protein subunits (gp91Phox, p22Phox) failed to demonstrate a significant association ( p > 0.1) for either the frontal or temporal cortex (Fig. 5), but did reveal a significant association (p < 0.0001) for the cytosolic proteins (p67Phox, p47Phox, p40Phox) in the frontal cortex (Fig. 3B,D,F) and the temporal cortex (Fig. 4B,D,F). The relationship between the subjects’ CDR scores and the overall NOX activity was significant for both the frontal (r = 0.702, p < 0.0001) and temporal (r = 0.703, p < 0.0001) regions.
FIG. 5.

Levels of membrane subunit proteins of NADPH-oxidase (gp91Phox, and p22Phox) in the frontal (A,C) and temporal (E,G) cortex from individuals classified as NCI, PCAD, MCI, eAD, and mAD. Values are plotted as percent change from the levels observed in the NCI cohort. Bars represent group mean ± SD. Scatter plots show the relationship between the individual’s score on the MMSE and the levels of the different membrane subunits (B,D,F,H). Each point represents an individual subject. Line used to represent the direction of the association and does not indicate a line of regression.
Discussion
The present results support and extend previous findings of enhanced NOX activity in MCI suggesting an increase early in the progression of the disease. Our data, obtained from human brain autopsy samples, also evaluated individuals that have recently transitioned from MCI to AD. Enzyme activity in the MCI cohort was significantly increased and remained elevated in individuals with moderate AD. One of the strong features of the present investigation was the use of two different neocortical areas (frontal, temporal) known to be intimately involved in the disease progression [2, 8, 20–22].
NOX is a unique enzyme with the primary purpose of producing ROS. This production of ROS has been highly conserved in mammalian systems and differs from mitochondrial ROS production occurring as part of normal cellular respiration in the production of ATP. Certain levels of ROS are extremely important for a variety of normal biological functions including growth factor regulation, calcium signaling, and phagocytosis/inflammation [23–25]. Excessive amounts of ROS can be extremely detrimental resulting in elevated levels of oxidative stress that appear to play an important role in numerous degenerative diseases including AD [8, 26, 27]. The NOX enzyme complex is highly expressed in phagocytes and may play a critical role in the neuroinflammatory response associated with AD [28, 29] where ROS production to occurs as part of a phagocyte-mediated host defense.
This study documented changes not only in the overall NOX activity but also in different protein subunits known to regulate gp91phox. Results show that overall NOX activity increased in both the frontal and temporal cortex early in the disease progression. These findings support two previous studies indicating a significant increase in frontal cortex NOX activity in AD [16] and temporal cortex in MCI [15]. The latter study is notable in that NOX activity did not remain elevated in the AD group. This disparity with the present findings may be due to the fact that AD subjects in this study were possibly earlier in the disease progression and didn’t fall into the “late stage” category. Our findings for both frontal and temporal regions demonstrate a significant increase in NOX activity throughout the disease progression most notably in the more advanced stages of the disease. We used tissue from individuals that were both early in the conversion from MCI to AD (eAD) and individuals with mild to moderate AD (mAD). Our analysis also included individuals with possible PCAD, characterized as cognitively normal individuals with abundant pathology at autopsy. Similar to previously published data [15] we did not observe a significant increase in NOX activity, although the variance within this subgroup of subjects was larger than the NCI group. There did appear to be a trend in the data possibly suggesting a group in transition, although this is only speculation. In addition to overall activity, we also monitored levels of each of the primary subunits involved in the NOX complex. Our results again support the previous reports in AD[16] and MCI [15] and extend those findings to include possible changes in other subunits. In our study, the flavocytochrome proteins remained stable throughout the disease progression while the cytosolic subunits were all significantly elevated. Not only the cytosolic subunits(p47phox, p67phox ) are of particular importance in the activation of the flavocytochrome oxidase activity [30], but also membrane bound gp91phox isoform itself. It is the direct contact of p67phox with gp91phox that modulates the transfer of electrons from NADPH, whereas p47phox initiates the assembly of the enzyme complex and serves as an adapter protein for p67phox subunit [31]. Because two different types of assays were used in assessing overall NOX activity and the subunits, the activity of both does not always match. The biochemical studies show the actual level of NOX activity while the Western blots give an indication of the subunit expression, which isn’t always a one to one relationship with activity. The overall expression of p67phox and p47phox in the temporal cortex is almost identical with the overall temporal NOX activity even with a trend in the PCAD group. While the case isn’t as clear in the frontal cortex, it is certainly clear that there is a progressive increase in the subunit activity. Our data show that NOX function is increased by translocation and assembly of the subunits. Isoform pg91 is the catalytic center of the NOX complex, such that any change in the levels of gp91 can alter NOX activity. We didn’t observe any significant change in pg91phox in either frontal or temporal regions throughout the progression of the disease. Somewhat surprising was the finding that the temporal cortex appeared to be more involved in the early stages of the disease than the frontal cortex. This differential response may indicate that the temporal association areas may have a greater predisposition than frontal association areas in the early stages of the disease.
Although NOX has been proposed to participate in several neurodegenerative syndromes, the detrimental actions of this enzyme complex appear to be most strongly associated with age-related chronic disease processes, such as Parkinson’s disease, atherosclerosis, and AD. Based on these observations, the term “antagonistic pleiotropy” has been coined to explain a potential role of NOX in the brain [24]. In this scenario, the physiologic production of ROS gathers an advantage in early life where it is essential in molecular process underlying a variety of biological functions such as signal transduction, synaptic plasticity, and memory [32–37]. Sustained activation of NOX ends in harmful effects with aging essentially by increasing oxidative stress. The present findings indicate that increased NOX activity is associated with the early expression of dementia most likely as a result of the increased ROS and subsequent oxidative stress. We have previously shown that early in the disease progression there is a significant accumulation of protein modification as a result of ROS in synaptosomes isolated from the frontal cortex that correlated very strongly with the MMSE [8]. The overall progression of NOX activity in the frontal cortex shows a striking similarity with our previous results showing the progression of oxidative stress in the post-mitochondrial and synaptosomal fractions [8]. The centrifugation techniques used in the present study essentially removed the mitochondrial fraction.
A major finding in the present study was the strong association between the NOX activity and the individual’s cognitive scores. As the levels of NOX activity increased the MMSE scores declined in a linear fashion. Because of the extremely limited range of cognitive scores in the NCI, PCAD and MCI groups, it was not possible to carry out a correlation analysis on these individual cohorts separately. There was very close agreement between the two cortical regions on measures of overall suggesting NOX activity that the increase was not limited to a specific area or system. Analysis of the individual subunits also showed a close association with the cognitive testing for the cytosolic but not the membrane bound proteins. The CDR scores, a purely clinical assessment of cognitive competency, also showed a very robust correlation with the overall NOX activity in both cortical regions. Since one of the major criterions for differentiating PCAD from NCI was the presence of increased tau pathology, it was important to assess the possible association between the Braak scores and the NOX activity. The presence of neurofibrillary tangles has been recently reported to show a strong relationship with the progression of the disease [1, 38]. In the present study there was a very strong association between the NOX activity and Braak score. Although there was no statistical difference between NCI and PCAD group in terms of NOX activity, the data clearly indicate a trend in that direction and may be signaling the earliest state of the transition toward MCI.
In AD, activation of microglia by A beta results in increased NOX activity and subsequent elevated levels of oxidative stress, significantly contributing to the pathophysiology of the disease process [39–42]. Microglia activation may be the predominant source of AD-related oxidative stress suggesting that targeting microglia and neuroinflammation may provide a key in the therapeutic approach to modifying the disease progression. A recent study using an in vivo AD model has shown that using a nonsteroidal anti-inflammatory drug can significantly alter oxidative damage. It is believed to be working through the disruption of important NOX signaling cascades utilized by microglia [43].
Acknowledgments
This work was supported by the National Institutes of Health: AG27219, PO1 AG14449, AG028383. The authors declare that they have no competing financial interests.
ABBREVIATIONS
- AD
Alzheimer’s disease
- eAD
early Alzheimer’s disease
- mAD
mild/moderate Alzheimer’s disease
- CDR
clinical dementia rating
- MCI
mild cognitive impairment
- MMSE
mini-mental status examination score
- NCI
no cognitive impairment
- NIA-RI
National institute of health-Reagan institute
- NOX
NADPH-oxidase
- NOX2
NADPH-oxidase subunit gp91phox
- PCAD
preclinical Alzheimer’s disease
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheff SW, Price DA. Alzheimer’s disease-related alterations in synaptic density: neocortex and hippocampus. J Alzheimers Dis. 2006;9:101–115. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- 3.Petersen RC, Morris JC. Mild cognitive impairment as a clinical entity and treatment target. Arch Neurol. 2005;62:1160–1163. doi: 10.1001/archneur.62.7.1160. discussion 1167. [DOI] [PubMed] [Google Scholar]
- 4.Winblad B, Palmer K, Kivipelto M, Jelic V, Fratiglioni L, Wahlund LO, Nordberg A, Backman L, Albert M, Almkvist O, Arai H, Basun H, Blennow K, de Leon M, DeCarli C, Erkinjuntti T, Giacobini E, Graff C, Hardy J, Jack C, Jorm A, Ritchie K, van Duijn C, Visser P, Petersen RC. Mild cognitive impairment--beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med. 2004;256:240–246. doi: 10.1111/j.1365-2796.2004.01380.x. [DOI] [PubMed] [Google Scholar]
- 5.Morris JC, Storandt M, McKeel SW, Rubin eH, Price JL, Grant EA, Berg L. Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurology. 1996;46:707–719. doi: 10.1212/wnl.46.3.707. [DOI] [PubMed] [Google Scholar]
- 6.Galvin JE, Powlishta KK, Wilkins K, McKeel DW, Jr, Xiong C, Grant E, Storandt M, Morris JC. Predictors of preclinical Alzheimer disease and dementia: a clinicopathologic study. Arch Neurol. 2005;62:758–765. doi: 10.1001/archneur.62.5.758. [DOI] [PubMed] [Google Scholar]
- 7.Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55:370–376. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 8.Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mamelak M. Alzheimer’s disease, oxidative stress and gammahydroxybutyrate. Neurobiol Aging. 2007;28:1340–1360. doi: 10.1016/j.neurobiolaging.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 11.Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- 12.Tejada-Simon MV, Serrano F, Villasana LE, Kanterewicz BI, Wu GY, Quinn MT, Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci. 2005;29:97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkinson BL, Landreth GE. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J Neuroinflammation. 2006;3:30. doi: 10.1186/1742-2094-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 15.Bruce-Keller AJ, Gupta S, Parrino TE, Knight AG, Ebenezer PJ, Weidner AM, LeVine H, 3rd, Keller JN, Markesbery WR. NOX activity is increased in mild cognitive impairment. Antioxid Redox Signal. 2010;12:1371–1382. doi: 10.1089/ars.2009.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun. 2000;273:5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- 17.de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis. 2006;9:167–181. doi: 10.3233/jad-2006-9209. [DOI] [PubMed] [Google Scholar]
- 18.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 19.Ansari MA, Keller JN, Scheff SW. Protective effect of Pycnogenol in human neuroblastoma SH-SY5Y cells following acrolein-induced cytotoxicity. Free Radic Biol Med. 2008;45:1510–1519. doi: 10.1016/j.freeradbiomed.2008.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terry RD, Peck A, DeTeresa R, Schechter R, Horoupian DS. Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann Neurol. 1981;10:184–192. doi: 10.1002/ana.410100209. [DOI] [PubMed] [Google Scholar]
- 21.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 22.Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 23.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 24.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Chong ZZ, Li F, Maiese K. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog Neurobiol. 2005;75:207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 28.McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis. 2010;19:355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 29.McNaull BB, Todd S, McGuinness B, Passmore AP. Inflammation and anti-inflammatory strategies for Alzheimer’s disease--a mini-review. Gerontology. 2010;56:3–14. doi: 10.1159/000237873. [DOI] [PubMed] [Google Scholar]
- 30.Rotrosen D, Yeung CL, Leto TL, Malech HL, Kwong CH. Cytochrome b558: the flavin-binding component of the phagocyte NADPH oxidase. Science. 1992;256:1459–1462. doi: 10.1126/science.1318579. [DOI] [PubMed] [Google Scholar]
- 31.Dang PM, Cross AR, Quinn MT, Babior BM. Assembly of the neutrophil respiratory burst oxidase: a direct interaction between p67PHOX and cytochrome b558 II. Proc Natl Acad Sci U S A. 2002;99:4262–4265. doi: 10.1073/pnas.072345299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci. 2009;29:2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowan MJ, Klyubin I, Wang Q, Anwyl R. Mechanisms of the inhibitory effects of amyloid beta-protein on synaptic plasticity. Exp Gerontol. 2004;39:1661–1667. doi: 10.1016/j.exger.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 34.Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26:5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeLeo FR, Burritt JB, Yu L, Jesaitis AJ, Dinauer MC, Nauseef WM. Processing and maturation of flavocytochrome b558 include incorporation of heme as a prerequisite for heterodimer assembly. J Biol Chem. 2000;275:13986–13993. doi: 10.1074/jbc.275.18.13986. [DOI] [PubMed] [Google Scholar]
- 36.Lapouge K, Smith SJ, Groemping Y, Rittinger K. Architecture of the p40-p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox. J Biol Chem. 2002;277:10121–10128. doi: 10.1074/jbc.M112065200. [DOI] [PubMed] [Google Scholar]
- 37.Samhan-Arias AK, Duarte RO, Martin-Romero FJ, Moura JJ, Gutierrez-Merino C. Reduction of ascorbate free radical by the plasma membrane of synaptic terminals from rat brain. Arch Biochem Biophys. 2008;469:243–254. doi: 10.1016/j.abb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 39.Abramov AY, Canevari L, Duchen MR. Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta. 2004;1742:81–87. doi: 10.1016/j.bbamcr.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Block ML. NADPH oxidase as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008;9(Suppl 2):S8. doi: 10.1186/1471-2202-9-S2-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milton RH, Abeti R, Averaimo S, DeBiasi S, Vitellaro L, Jiang L, Curmi PM, Breit SN, Duchen MR, Mazzanti M. CLIC1 function is required for beta-amyloid-induced generation of reactive oxygen species by microglia. J Neurosci. 2008;28:11488–11499. doi: 10.1523/JNEUROSCI.2431-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park KW, Baik HH, Jin BK. IL-13-induced oxidative stress via microglial NADPH oxidase contributes to death of hippocampal neurons in vivo. J Immunol. 2009;183:4666–4674. doi: 10.4049/jimmunol.0803392. [DOI] [PubMed] [Google Scholar]
- 43.Wilkinson BL, Cramer PE, Varvel NH, Reed-Geaghan E, Jiang Q, Szabo A, Herrup K, Lamb BT, Landreth GE. Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.06.014. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]