

Abstract
Background & Aims
Cholesterol gallstone disease (CGD) results from a biochemical imbalance of lipids and bile salts in the gallbladder bile. We investigated whether the xenobiotic receptor pregnane X receptor (PXR) has a role in pathogenesis of CGD.
Methods
Wild-type, PXR-null (PXR-/-), and CGD-sensitive C57L mice were placed on a lithogenic diet and then analyzed for CGD at the biochemical, histological, and gene-regulation levels.
Results
Loss of PXR sensitized mice to lithogenic diet-induced CGD, characterized by decreases in biliary concentrations of bile salts and phospholipids and an increases in the cholesterol saturation index (CSI) and formation of cholesterol crystals. The decreased bile acid pool size in PXR-/- mice that received lithogenic diets was associated with reduced expression of Cyp7a1, the rate-limiting enzyme of cholesterol catabolism and bile acid formation. The reduced expression of Cyp7a1 most likely resulted from activation of FXR and induction of fibroblast growth factor 15 in the intestine. In C57L mice given the PXR agonist pregnenolone-16α-carbonitrile or the herbal medicine St. John's wort, cholesterol precipitation was prevented, by increases in concentrations of biliary bile salt and a reduced CSI. PXR prevented CGD via its coordinate regulation of the biosynthesis and transport of bile salts in the liver and intestine.
Conclusions
PXR maintains biliary bile acid homeostasis and might be developed as a therapeutic target for CGD.
Keywords: Fgf15, PCN, nuclear receptor, gene regulation, transgenic mice
Introduction
Cholesterol gallstone disease (CGD) is one of the most common gastrointestinal diseases. Precipitation of excess cholesterol in the bile as solid crystals is a prerequisite for cholesterol gallstone formation. Cholesterol is only slightly soluble in aqueous media, but is made soluble in bile through mixed micelles composed of bile salts and phospholipids 1. Cholesterol precipitation can occur as a result of excessive cholesterol, deficiency of bile salts or phospholipids, or a combination of these factors 1. If unmanaged, the cholesterol crystals will aggregate, fuse, and ultimately form pathologic gallstones.
Biliary secretion of bile salts and lipids is regulated by an elaborate network of transporters. Cholesterol secretion is mediated by the ABC binding cassette (ABC) transporters Abcg5 and Abcg8 2. The secretion of phospholipids is controlled by Abcb4, a P-glycoprotein member of the multi-drug resistance gene family 3. Biliary bile salts consist of those newly synthesized in the liver and those that have undergone enterohepatic cycling. Cholesterol 7α-hydroxylase (Cyp7a1) is the rate-limiting enzyme of bile acid biosynthesis, catalyzing the conversion of cholesterol to bile acids 4. The majority of bile acids, once emptied from the gallbladder to the intestine, are reabsorbed as facilitated by the apical sodium-dependent bile acid transporter (Asbt) and organic solute transporter α/β (Ostα/β) in the ileum, and then transported back to the liver through the hepatic uptake transporters sodium (Na)-taurocholate cotransport protein (Ntcp) and the organic anion transporting polypeptides (Oatps) 5. Both newly synthesized and reabsorbed bile acids are secreted into the bile by the bile acid export pump (Bsep) and multidrug resistance protein 2 (Mrp2) 5. Mrp3 and Mrp4 play a role in the sinusoid export of conjugated bile salts and promote their renal secretion 6.
Genetic background has a major impact on CGD susceptibility. In humans, gallstone prevalence is linked to ethnicity and displays familial clustering 7. In mice, when challenged with a lithogenic diet, certain strains, such as the C57L mice, formed gallbladder cholesterol crystals within a week; whereas some other strains were resistant to CGD. Quantitative trait locus (QTL) analysis identified genomic regions that harbored the causative genetic variations in susceptible strains 8, 9. In QTL analysis, several nuclear receptors, including PXR, were suggested as potential lithogenic (Lith) genes 9.
PXR, highly expressed in the liver and small intestine, was initially identified as a xenobiotic receptor regulating the expression of drug metabolizing enzymes and transporters 10. Subsequent studies suggested that PXR is also implicated in bile acid metabolism 11, 12. PXR can be activated by bile acids and their precursors, and activation of PXR plays an essential role in the detoxification of bile acids and relief of cholestasis 11, 12. Knowing PXR has a role in preventing cholestasis, it remains to be determined whether this receptor plays a role in the pathogenesis of CGD.
In this report, we showed that loss of PXR sensitized mice to lithogenic diet-induced CGD, whereas a pharmacological activation of PXR prevented CGD. We conclude that PXR is an anti-lithogenic receptor.
Methods
Animals, lithogenic diet, and drug treatment
PXR-/- mice were previously described 13. PXR-/- and their WT control littermates were maintained on a mixed background of C57BL/6J and 129SvJ. C57L mice were purchased from the Jackson laboratory. The lithogenic diet (Harlan Teklad TD-02189) contains 21% butterfat, 1.5% cholesterol, 0.5% cholic acid, and 23% casein. Male PXR-/- and WT mice were fed with lithogenic diet for 4 weeks at which WT mice were largely resistant to CGD and the diet did not cause appreciable hepatotoxicity. Male C57L mice were fed with lithogenic diet for 1 week, because they are genetically sensitive to CGD 8. When necessary, daily i.p. injection of PCN (30 mg/kg) or oral gavage of SJW (300 mg/kg) was given for 4 days before the lithogenic diet, and the drug treatment was continued until the completion of the experiments. Mice were then sacrificed and gallbladders were removed by ligation of the cystic duct after overnight fasting. Gallbladder bile and its contents were gently squeezed into tubes after cholecystectomy, and were immediately examined by a polarized microscope for cholesterol crystals and gallstones. The remaining bile was used for biochemical analysis. The use of mice in this study complied with relevant federal guidelines and institutional policies.
Bile duct cannulation and collection of hepatic bile
Bile duct cannulation was performed as described 14. Briefly, the lower end of the common bile duct of mice fasted overnight was ligated and the common bile duct was cannulated with a PE-10 polyethylene catheter below the entrance of the cystic duct. The cystic duct was then doubly ligated and a cholecystectomy was performed. Hepatic bile was collected by gravity every 15 min for the first hour, and then every hour for 8 hrs.
Biochemical analysis of bile, serum, urine, and feces
Biliary lipids were extracted as described 15. Phospholipid, cholesterol, and triglyceride levels were measured with assay kits from Stanbio (Boerne, TX). Total bile acids were quantified with an assay kit from Bioquanti (San Diego, CA). The cholesterol saturation index (CSI) was calculated according to Carey's critical tables 16. Urine and feces were collected with metabolic cages. Serum ALT and AST activities were determined with assay kits from Stanbio
Gas chromatography-mass spectrometry (GC-MS) analysis of biliary bile acids
These were performed as we have previously described 17 with minor modifications in sample preparation. Briefly, 10∼20 μl gallbladder bile was treated with 0.6 ml of acetone/methanol/6M HCl (36:4:0.4, v:v:v) at 37 °C for 14 hrs to remove the conjugated sulfonyl group, and then with 15% (w/v) NaOH at 120 °C for 2 hrs in an autoclave to deconjugate taurine or glycine moiety. Samples were then extracted with hexane twice to remove cholesterol, and then acidified with 2 M HCl. Deconjugated bile acids were extracted with diethyl ether and converted to methyl esters with trimethylsilyldiazomethane and to trimethylsilyl derivatives with N, O-bis(trimethylsilyl)trifluoroacetamide plus trimethylchlorosilane. Gas-chromatographic separation was performed as described 17. The calculation of bile salt hydrophobicity was according to a published method 18.
Western blot analysis
This was performed as previously described with minor modifications 19. The primary antibodies used include those against Fgf15 (sc-27177), Bsep (sc-74500) and Cyp7a1 (sc-25536) from Santa Cruz (Santa Cruz, CA). The Asbt, Ostα and Ostβ antibodies were generous gifts from Dr. Paul Dawson 19.
Quantitative real-time RT-PCR
Total RNAs were extracted with the TRIZOL reagent. Real-time PCR using SYBR Green-based assay was performed with the ABI 7300 real-time PCR system. Primer sequences are listed in Supplementary Table 1.
Statistical analysis
Data are expressed as the means ± S.E. One-way ANOVA Tukey's test was performed for statistical analysis using GraphPad Prism version 4.0. P values of less than 0.05 were considered statistically significant.
Results
Loss of PXR sensitized mice to CGD
To determine the effect of loss of PXR on lithogenesis, we challenged age-matched wild type (WT) and PXR-/- mice with a lithogenic diet for 4 weeks. As shown in Fig 1A, 92% of PXR-/- mice developed gallstones, whereas the penetrance in WT mice was 18%. Compared to those of WT mice, the gallbladders of PXR-/- mice appeared turbid and full of precipitates and stones (Fig. 1B). Microscopic examination of the gallbladder bile revealed numerous cholesterol crystals in PXR-/- mice, whereas WT mice were largely free of cholesterol precipitates, exhibiting only occasional aggregated vesicles in most animals (Fig. 1C). Histological analysis of the gallbladder of PXR-/- mice revealed signs of inflammation, including thickening of gallbladder wall, infiltration of granulocytes in the stromal layer, and submucosal vasodilatation (Fig. 1D). Neither WT nor PXR-/- mice showed signs of CGD when maintained under a chow diet (Supplementary Fig. 1).
Figure 1. Loss of PXR sensitized mice to CGD.
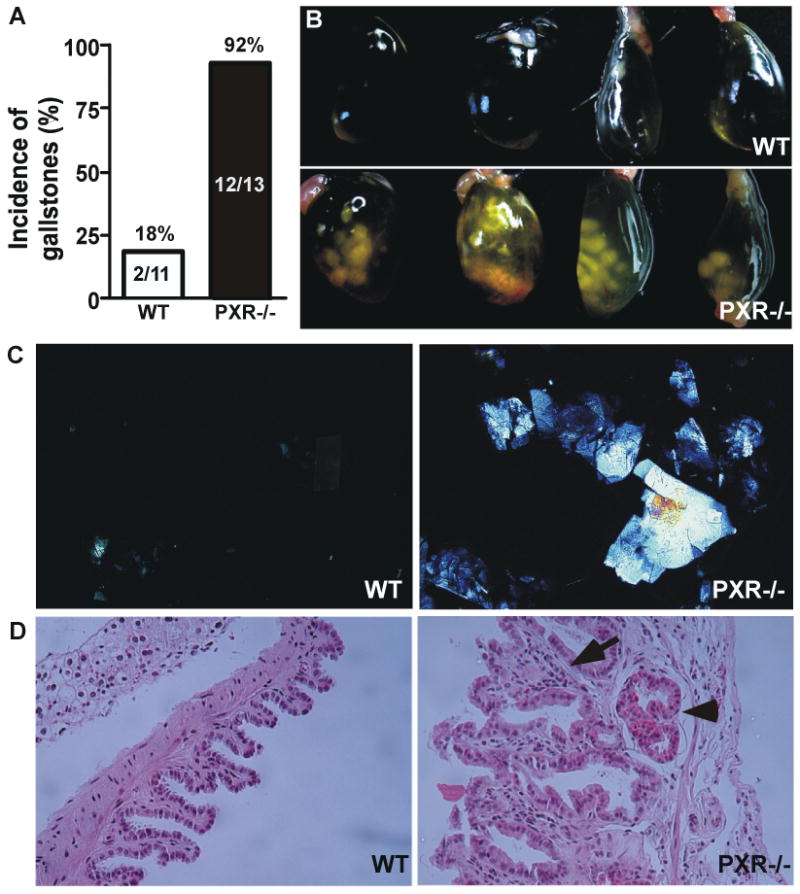
Male WT and PXR-/- mice were subjected to lithogenic diet for 4 weeks. (A) Incidence of gallstones with the numbers of mice labeled. (B) Gross appearance of representative gallbladders. (C) Polarizing light microscopic examination of cholesterol crystals. (D) Histological examination of the gallbladder by H&E staining. The arrow indicates the thickness of the gallbladder wall and stromal granulocyte infiltration. The arrowhead indicates submucosal vasodilatation.
Loss of PXR altered the biochemical composition of the gallbladder bile, and affected bile flow and biliary lipid outputs
On chow diet, PXR-/- mice had biliary bile acid, phospholipid, and cholesterol levels similar to their WT counterparts (Supplementary Fig. 1). However, upon a 4-week lithogenic diet treatment, the biliary concentrations of bile acids (Fig. 2A, left panel) and phospholipids (Fig. 2A, right panel) were significantly decreased in PXR-/- mice compared to their WT counterparts. The biliary concentration of cholesterol was unchanged (Fig. 2B, left panel). The combined effect of biochemical alterations led to an increased CSI in PXR-/- mice (Fig. 2B, right panel), providing a biochemical mechanism for the lithogenic phenotype of PXR-/- mice.
Figure 2. Loss of PXR altered the biochemical composition of the gallbladder bile, and affected bile flow and biliary lipid outputs. (A and B).

The concentrations of bile acids (A, left panel), phospholipids (A, right panel), and cholesterol (B, left panel) in the gallbladder bile; as well as the cholesterol saturation index (B, right panel). Mice were the same as those used in Fig. 1. (C-F) Biliary bile acid output (C), phospholipid output (D), bile flow rate (E), and cholesterol output (F), as measured bile duct cannulation. Insert in (C) is the bile acid pool size. N=7 for each group. * P<0.05; **P<0.01.
We then performed bile duct cannulation to understand the dynamics of bile flow and fresh biliary lipid outputs. The biliary bile acid (Fig. 2C) and phospholipid (Fig. 2D) outputs were substantially lower in PXR-/- mice, consistent with the lipid profile of gallbladder bile. By using the area under the bile acid secretion curve (AUC) during the first 4 hrs as the surrogate estimate of bile salt pool size 14, we found the pool size was significantly decreased in PXR-/- mice (Fig. 2C, insert). Decreased bile salt pool size is known to predispose patients to CGD 1. The bile flow was increased in PXR-/- mice (Fig. 2E), suggesting a compensatory mechanism to correct the decreased pool size. The biliary output of bilirubin was increased especially at the late time point (Supplementary Fig. 2). This observation was consistent with the reported increase of bilirubin detoxification and clearance in PXR-/- mice 20, and the increased bilirubin output may have contributed as a driving force for the increased bile flow. Interestingly, the cholesterol output was higher in PXR-/- mice (Fig. 2F). The higher cholesterol output and unchanged cholesterol concentration in the gallbladder bile of PXR-/- mice suggested differential rates of cholesterol absorption in the gallbladders of WT and PXR-/- mice.
When the individual bile acid species in the gallbladder bile was analyzed by GC-MS, we found the concentration of cholic acid (CA) was decreased in PXR-/- mice. Interestingly, the concentrations of both the hydrophilic muricholate (MCA) and the hydrophobic deoxycholate (DCA) were decreased in PXR-/- mice, leading to decreased bile salt hydrophobicity in this genotype (Supplementary Fig. 3). We also analyzed the hepatic lipid profile. As expected, treatment of WT mice with lithogenic diet increased hepatic levels of cholesterol, triglycerides, phospholipids, and bile acids (Supplementary Table 2), a profile consistent with the high-fat, high-cholesterol, and high-bile acid nature of the lithogenic diet. Interestingly, under the lithogenic diet, PXR-/- mice had a hepatic cholesterol level similar to WT mice, despite having a higher biliary cholesterol output. Instead, PXR-/- mice had higher hepatic concentrations of phospholipids and bile acids, suggesting an impaired efflux of bile acids and phospholipids. The serum biochemistry is summarized in Supplementary Table 3. Lithogenic diet-fed PXR-/- mice had increased serum concentration of bile acids and decreased serum concentration of phospholipids, whereas the circulating concentration of cholesterol was unchanged. The lower serum concentration of phospholipids was consistent with a previous report that cholic acid feeding decreased circulating phospholipid level in PXR-/- mice 21. The lithogenic diet did not cause appreciable hepatotoxicity in either genotype, as confirmed by the measurement of serum levels of total bilirubin, alanine aminotransferase (ALT), and aspartate aminotransferase (AST) (Supplementary Table 3). The loss of function effect on lithogenesis was specific for the xenobiotic receptor PXR, because loss of CAR, a sister xenobiotic receptor of PXR, had little effect on CGD susceptibility (Supplementary Fig. 4). The lithogenic diet had little effect on the expression of PXR and its target genes Cyp3a11 and Oatp2 in WT mice (Supplementary Fig. 5).
Loss of PXR altered the basal and lithogenic diet responsive gene expression in the liver and intestine
To understand the mechanism by which loss of PXR sensitized mice to CGD, we profiled the expression of genes whose products can impact the biliary homeostasis. Among bile acid transporters, Ntcp and Oatps are hepatic uptake transporters, whereas Bsep and Mrp2 are responsible for canalicular efflux 5. Mrp3 and Mrp4 play a role in the sinusoid export and subsequent renal secretion of conjugated bile acids 6. In the intestine, Asbt and Ostα/β are responsible for the uptake and efflux of bile acids, respectively. Cholesterol efflux is facilitated by Abcg5 and Abcg8. Abcb4 is responsible for canalicular efflux of phospholipids, whereas Abca1 is a basolateral transporter that effluxes both cholesterol and phospholipids.
When the expression of hepatic bile acid transporters was profiled, we found that on a chow diet, the expression of Oatp2 and Mrp3 was decreased and increased in PXR-/- mice, respectively. When challenged with the lithogenic diet, the expression of Oatp2 and Ntcp was significantly reduced in PXR-/- mice (Fig. 3A). Among canalicular efflux transporters, the expression of Bsep was markedly suppressed in PXR-/- mice, whereas the expression of Mrp2 was not affected. The expression of Mrp4, but not Mrp3, was induced in PXR-/- mice (Fig. 3A). Consistent with the induction of Mrp4, the urinary (Fig. 3B) and serum (Supplementary Table 3) bile acid levels were increased in lithogenic diet-fed PXR-/- mice.
Figure 3. Loss of PXR altered the basal and lithogenic diet responsive expression of bile acid transporters.
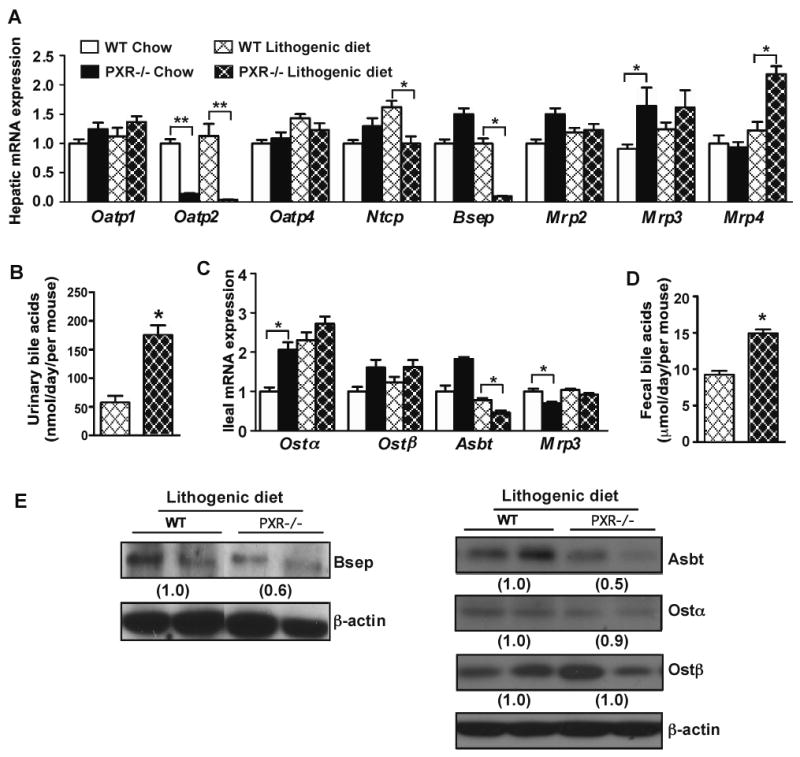
(A) Hepatic mRNA expression of bile acid transporters as measured by real-time PCR. (B) Urinary levels of bile acids. (C) Ileal mRNA expression of bile acid transporters. (D) Fecal output of bile acids. N=7 for each group. * P<0.05; **P<0.01. (E) Expression of hepatic (left panel) and ileal (right panel) bile acid transporter proteins was measured by Western blot analysis. The relative average protein level was determined by densitometry and labeled underneath the bands.
Among intestinal bile acid transporters, the expression of Ostα and Mrp3 was increased and decreased in chow diet-fed PXR-/- mice (Fig. 3C), respectively. When challenged with the lithogenic diet, PXR-/- mice showed a decreased expression of Asbt, whereas the expression of Ostα/β and Mrp3 was not affected (Fig. 3C). PXR-/- mice showed an increased fecal secretion of bile acids (Fig. 3D), consistent with the notion that Asbt is the primary transporter responsible for the re-absorption and enterohepatic circulation of bile acids 22. The down-regulation of Bsep and Asbt protein expression in lithogenic diet-fed PXR-/- mice was confirmed by Western blot analysis (Fig. 3E).
When the expression of hepatic phospholipid transporters was profiled, we found the expression of Abcb4 was modestly but significantly decreased in lithogenic diet-fed PXR-/- mice (Fig. 4A), consistent with the lower biliary phospholipid output (Fig. 2D). Upon lithogenic diet feeding, the expression of Abca1 was indiscriminately increased in both WT and PXR-/- mice (Fig. 4A). Among cholesterol transporters, the basal expression of Abcg5 and Abcg8 in the liver, but not intestine, was increased in PXR-/- mice (Fig. 4B). Upon lithogenic diet treatment, the hepatic expression of Abcg8 and intestinal expression of both Abcg5 and Abcg8 in PXR-/- mice was higher than WT mice (Fig. 4B). The expression of Npc1l1, an intestinal cholesterol transporter important for dietary cholesterol absorption, was also higher in lithogenic diet-fed PXR-/- mice (Fig. 4B).
Figure 4. Effect of loss of PXR on the expression of cholesterol and phospholipid transporters and genes involved in bile acid synthesis. (A and B).
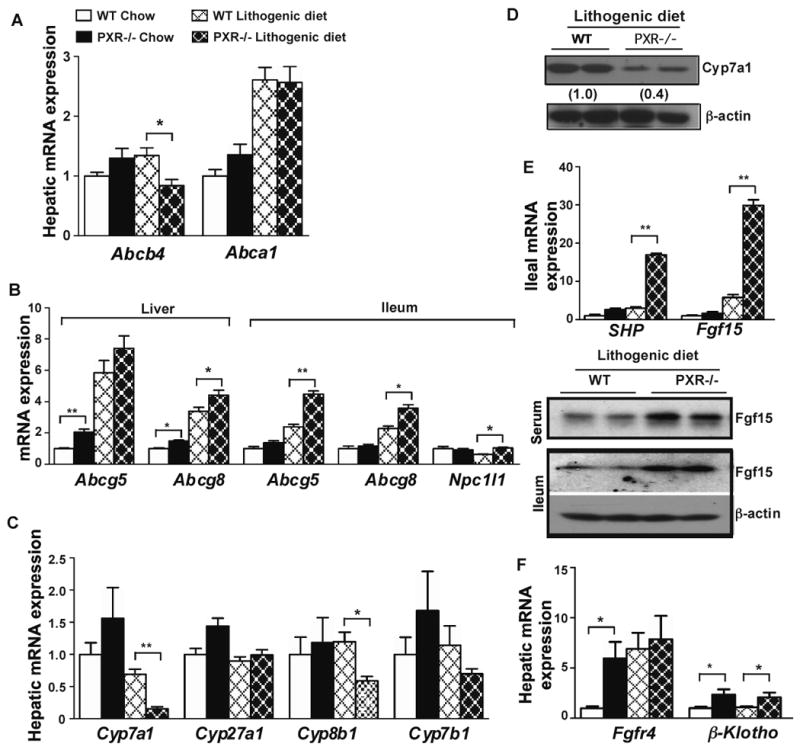
Hepatic expression of phospholipid transporters (A), and hepatic and ileum expression of cholesterol transporters (B) as measured by real-time PCR. (C) Hepatic expression bile acid synthesis enzyme genes. (D) Expression of Cyp7a1 protein was measured by Western blot analysis. (E) SHP and Fgf15 mRNA expression in the ileum (top panel), and Fgf15 protein expression in the serum and ileum (bottom panel). (F) Hepatic mRNA expression of Fgfr4 and β-Klotho. N=5 for each group. * P<0.05; **P<0.01.
The biosynthesis of bile acids in the liver is controlled by multiple CYP enzymes. We found the mRNA expression of Cyp7a1 and Cyp8b1, the two key enzymes in bile acid synthesis 4, was markedly suppressed in lithogenic diet-fed PXR-/- mice (Fig. 4C). The suppression of Cyp7a1 protein expression was confirmed by Western blot analysis (Fig. 4D). The expression of Cyp27a1 and Cyp7b1 was not affected. A decreased expression of Cyp7a1 has been linked to a heightened gallstone susceptibility 23. To understand the mechanism of Cyp7a1 suppression, we evaluated the expression of small heterodimerization partner (SHP) and fibroblast growth factor 15 (Fgf15), two FXR target genes that play an important role in the negative feedback control of Cyp7a1 transcription 24-26. Loss of PXR had little effect on the basal expression of SHP and Fgf15. However, upon lithogenic diet treatment, the mRNA expression of intestinal SHP and Fgf15 was robustly induced in PXR-/- mice (Fig. 4E, top panel). The induction of Fgf15 protein in the serum and ileum was confirmed by Western blot analysis (Fig. 4E, bottom panel). Fgf15 is secreted from the intestine into the circulation, binds to the hepatic Fgf receptor 4 (Fgfr4), and subsequently inhibits Cyp7a1 expression 26. The basal expression of Fgfr4 was increased in PXR-/- mice. Upon lithogenic diet treatment, the expression of Fgfr4 in WT mice increased, but there was no further induction in PXR-/- mice. The expression of β-klotho, the co-receptor of Fgfr4 27, 28, was induced in PXR-/- mice regardless of the diet (Fig. 4F). These results suggested that the activation of FXR and induction of Fgf15 in the intestine might be responsible for the suppression of Cyp7a1 in the liver.
When the gene expression was profiled in CAR-/- mice, we found that the majority of lithogenic diet-responsive hepatic and intestinal gene regulation observed in PXR-/- mice was absent in CAR-/- mice. These included the suppression of Oatp2, Ntcp, Bsep, Asbt, Abcb4, Cyp7a1 and Cyp8b1, as well as the induction of Mrp4, Abcg5 and Abcg8. The ileal induction of Fgf15 was absent. The induction of SHP was observed in CAR-/- mice, but the induction was not as dramatic as their PXR-/- counterparts (Supplementary Fig. 6).
Pharmacological activation of PXR prevented CGD in C57L mice
Having demonstrated that loss of PXR sensitized mice to CGD, we went on to determine whether a pharmacological activation of PXR conferred resistance to CGD. In this experiment, the CGD-sensitive C57L mice 8 were subjected to lithogenic diet for one week, in the presence or absence of the PXR agonists pregnenolone-16α-carbonitrile (PCN) or St. John's wort (SJW) 29, before being analyzed for CGD. The incidence of cholesterol crystals is summarized in Fig. 5A. As expected, all vehicle-treated mice showed large aggregates and cholesterol crystals, whereas little sequelae of CGD were observed in PCN- or SJW-treated mice (Fig. 5B). Treatment with PCN resulted in an increased biliary concentration of bile acids and decreased concentration of cholesterol (Fig. 5C). PCN had little effect on the phospholipid level (Fig. 5D). The overall effect of PCN was a significantly decreased CSI (Fig. 5E). Compared to the vehicle-treated mice, PCN-treated mice showed decreased thickness of gallbladder wall and lack of signs of inflammation (Fig 5F). This regimen of PCN did not cause appreciable hepatotoxicity, as supported by the unchanged serum levels of ALT and AST (Supplementary Fig. 7). A similar profile of SJW-responsive biliary biochemistry is shown in Supplementary Fig. 8.
Figure 5. Pharmacological activation of PXR prevented CGD in C57L mice.
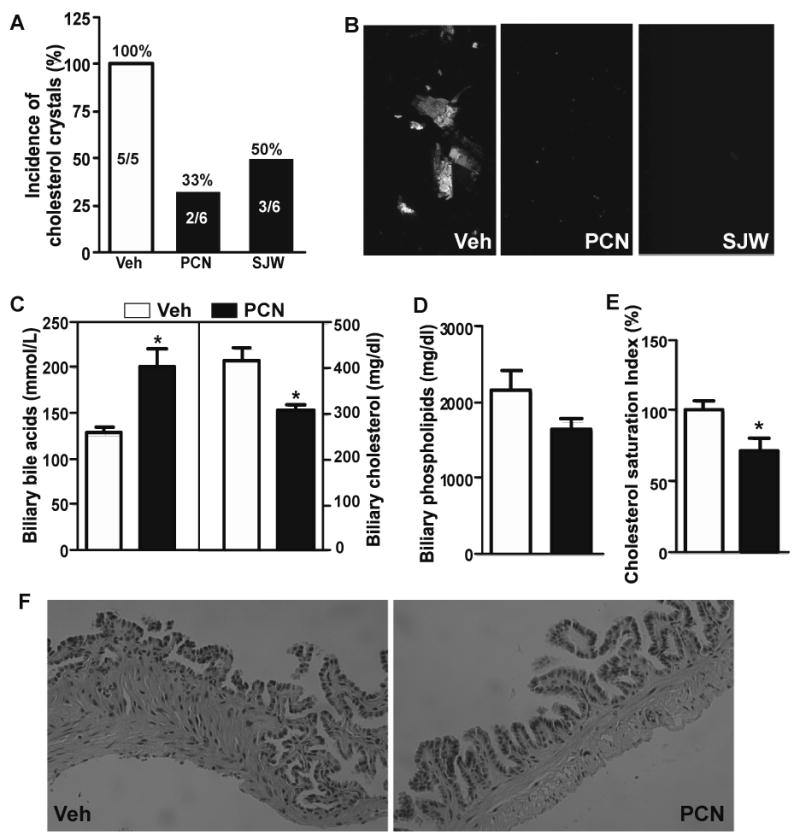
(A) Incidence of gallstones in male C57L mice treated with vehicle (Veh), pregnenolone-16α-carbonitrile (PCN), or St. John's wort (SJW). All mice were fed with lithogenic diet for one week. The numbers of mice are labeled. (B) Microscopic examination of cholesterol crystals. (C and D) Biliary concentrations of bile acids (C, left panel), cholesterol (C, right panel), and phospholipids (D). (E) Calculated cholesterol saturation index. (F) Histological examination of the gallbladder by H&E staining. * P<0.05.
The litho-preventive effect of PCN was PXR dependent, because treatment of PXR-/- mice with PCN had little effect in preventing CGD and altering the biliary biochemistry (Supplementary Fig. 9). Ketoconazole has been reported as a PXR antagonist 30. Interestingly, treatment with ketoconazole (50 mg/kg) for two weeks showed a modest but significant effect in sensitizing WT mice to CGD, which was supported by the biliary biochemistry (Supplementary Fig. 10).
Pharmacological activation of PXR resulted in a pattern of gene expression favorable for anti-lithogenesis
Among bile acids transporters, the hepatic expression of Oatp2 and Mrps 2, 3 and 4 increased in PCN-treated C57L mice (Fig. 6A). The intestinal expression of Asbt and Mrp3 was also increased in PCN-treated C57L (Fig. 6B). The induction of Oatp2 and Asbt was consistent with the suppression of the same genes in PXR-/- mice (Fig. 3A and 3C). Mrp3/4 and Asbt promote the sinusoid efflux and intestinal bile acid re-absorption, respectively. The induction of Mrp3/4 and Asbt may have contributed to a higher serum level of bile acids in PCN-treated mice (Fig. 6C). PCN had little effect on the expression of phospholipid and cholesterol transporters (data not shown).
Figure 6. Pharmacological activation of PXR resulted in a pattern of gene expression favorable for anti-lithogenesis. (A and B).

The mRNA expression of bile acid transporters in the liver (A) and ileum (B) as measured by real-time PCR. Mice were the same as those used in Fig. 5. (C) Serum bile acid levels. (D) The mRNA expression bile acid synthesis enzyme genes. (E) Ileal mRNA expression of SHP and Fgf15. N=5 for each group. * P<0.05; **P<0.01.
The expression of Cyp7a1 was induced by PCN (Fig. 6D), consistent with the suppression of the same gene in PXR-/- mice (Fig. 4C). The expression of Cyp7b1was modestly reduced, whereas the expression of Cyp8b1 and Cyp27a1 was unchanged (Fig. 6D). PCN inhibited the ileal expression of SHP and Fgf15 (Fig 6E), consistent with the induction of the same genes in lithogenic diet-fed PXR-/- mice (Fig. 4E). As a positive control, this regimen of PCN was efficient in activating the PXR target gene Cyp3a11 (Supplementary Fig. 11).
Discussion
In this report, we have revealed a novel function for PXR in preventing lithogenesis. The sensitization of PXR-/- mice to CGD was due to the decreased concentrations of bile acids and phospholipids in the gallbladder bile and consequently increased CSI. Major genes whose expression were affected in PXR-/- mice and may have contributed to the lithogenic phenotype are summarized in Fig. 7. Conversely, a pharmacological activation of PXR by PCN or SJW inhibited lithogenesis in C57L mice, which was associated with an increased biliary concentration of bile acids. It is interesting to note that SJW, a widely used herbal medicine 29, has been recommended as a home remedy for gallstones (http://www.gallstonesremoval.com/home-remedy-for-gallstones/).
Figure 7. Summary of the major gene regulation in PXR-/- mice that may have contributed to the lithogenic phenotype.

Shaded and open ovals indicated genes that were down-regulated and up-regulated, respectively in lithogenic diet-fed PXR-/- mice. Open rectangles indicate genes whose expression was not affected. The gene regulation, in conjunction with the lithogenic diet, led to decreased biliary concentrations of bile acids and phospholipids, increased CSI, and sensitization to CGD.
Previous reports showed that PXR was essential for bile acid detoxification 11, 12, 31, suggesting that PXR might be critical for bile acid homeostasis. Indeed, altered bile acid metabolism appeared to be the primary mechanism by which PXR affected lithogenesis. In the gallbladder bile, the cholesterol solubility is maintained by the balance among cholesterol, bile acids, and phospholipids 32. The decreased biliary concentration of bile acids in PXR-/- mice resulted in cholesterol super-saturation and precipitation. In contrast, an increased biliary concentration of bile acids in PCN- or SJW-treated C57L mice was effective in preventing CGD. The effect of PXR on biliary bile acids was consistent with several incidental reports. Treatment of patients with rifampicin, an antibiotic and potent PXR agonist, increased biliary bile acid concentration 33. In contrast, treatment with ketoconazole, an antifungal drug and PXR antagonist 30, decreased bile acid output in rodents 34.
As to the mechanism for the decreased bile acid pool size in lithogenic diet-fed PXR-/- mice, one interesting finding is the suppression of Cyp7a1, the rate-limiting enzyme of bile acid synthesis 4. The expression of Cyp7a1 can be regulated by several negative feedback loops. In the liver, SHP binds to LRH-1 and decreases the recruitment of coactivators to the Cyp7a1 gene promoter 24, 25. In the intestine, Fgf15 is secreted into the portal circulation, binds to Fgfr4 in the liver, and activates the JNK signaling pathway, causing loss of coactivator binding to the Cyp7a1 gene promoter 26. Both SHP and Fgf15 are FXR target genes. We showed that the intestinal expression of SHP and Fgf15 was dramatically induced in lithogenic diet-fed PXR-/- mice, which might have accounted for the suppression of Cyp7a1. In contrast, the anti-CGD effect of PCN in C57L mice was associated with an increased biliary concentration of bile acids, an increased expression of Cyp7a1, and a decreased expression of intestinal SHP and Fgf15. Interestingly, the PXR effect on the expression of SHP and Fgf15 appeared to be tissue-specific, because the hepatic expression of these two genes was not affected (data not shown). PXR has been reported to be a FXR target gene 35. The expression of FXR in our PXR models was not affected (data not shown). Although the mechanism by which PXR regulates the expression of SHP and Fgf15 remains to be determined, our results highly suggest a functional crosstalk between PXR and FXR.
The effects of nuclear receptors on lithogenesis have been reported for LXR and FXR. The anti-lithogenic effect of PXR is opposite to the lithogenic effect of LXR 15. The lithogenic effect of LXR was due to the combined effect of increased biliary concentrations of cholesterol and phospholipids, and decreased biliary concentration of bile salt. The expression of LXR target gene Srebp-1c, but not LXRα itself, was increased in both WT and PXR-/- mice upon lithogenic diet treatment, likely due to the high concentration of cholesterol in this diet. However, the expression of LXRα and Srebp-1c was not different between these two genotypes regardless of the diet (Supplementary Fig. 12), suggesting that activation LXR cannot account for the sensitization of PXR-/- mice. Loss of FXR was also shown to sensitize mice to CGD 36. There are several differences between the PXR and FXR models. Loss of PXR had a major effect on the expression of bile acid transporters, whereas FXR-/- mice showed profound dysregulation of phospholipid transporters. The regulation of Asbt, which we believe plays an important role in the PXR model, was not evaluated in the FXR model 36. Another major difference is that under the lithogenic diet, the biliary bile salt hydrophobicity index was increased in FXR-/- mice, but it was decreased in PXR-/- mice. The discrepancy may be due to the unexpected decrease of hydrophobic DCA in PXR-/- mice, which was not observed in FXR-/- mice 36. We reason the benefit of lower hydrophobicity in PXR-/- mice may have been overshadowed by the decreased bile acid pool size and increased CSI, and thus PXR-/- mice showed an overall phenotype of increased sensitivity to CGD. Finally, the bile acid synthesis was suppressed in lithogenic diet-fed PXR-/- mice, which was opposite to the increased bile acid synthesis in lithogenic diet-fed FXR-/- mice. Androgen receptor (AR) and estrogen receptor α (ERα) have also been reported to be involved in CGD 37, 38. We found the mRNA expression of AR and ERα was decreased and increased, respectively and regardless of the diet. However, the expression of their responsive genes, such as Igf-1, Hmgcs and Hmgcr, was unchanged (Supplementary Fig. 13).
We used both the PXR ablation and activation models to demonstrate the role of PXR in the pathogenesis of CGD. The phenotypes in lithogenesis, biliary bile acid level, and several key gene regulations were largely consistent between these two models. However, we did notice some inconsistencies in the biliary lipid profile. Specifically, PXR-/- mice showed decreased biliary phospholipids, whereas the biliary phospholipid level was unchanged in PCN- or SJW-treated WT mice. In another example, PXR ablation did not change biliary cholesterol, whereas PXR activation decreased biliary cholesterol level. These results suggested that PXR ablation and activation could have overlapping, yet different, mechanism in regulating biliary lipid homeostasis. We cannot exclude the possibility that compensatory mechanisms were involved in the absence of PXR.
The current work has firmly established PXR as a lithogenic (Lith) gene. Previous QTL analysis on inbred mice identified Lith14 on the chromosome 16 9. A genome-wide scan on CGD patients suggested the linkage of CGD to three regions on the human chromosome 3q, two of which are highly homologous to the mouse chromosome 16 39. PXR colocalizes with Lith14 on the human chromosome 3q and mouse chromosome 16, and thus was postulated as a positional candidate gene for CGD.
The loss of function effect on lithogenesis is specific for the xenobiotic receptor PXR, because loss of CAR had little effect on CGD. CAR is a sister receptor of PXR that has also been implicated in bile acid detoxification 31, 40, 41. CAR and PXR share many of their target genes and pathophysiological functions. Interestingly, CAR was also suggested to be a candidate lithogenic gene 9. The differential effect of PXR and CAR on lithogenesis, although paradoxical, is consistent with the notion that these two receptors have overlapping, yet distinct, spectrums of target genes and physiological functions.
At present, removal of the gallbladder is the surgical treatment for gallstones, which is limited by its invasive nature, risk of side effects, and high cost 42. The non-surgical treatment of gallstones is the oral administration of ursodeoxycholate, a hydrophilic bile salt that promotes the dissolution of gallstones. The clinic benefit of ursodeoxycholate is limited due to the difficulty in dose optimization 43. Our results suggest that pharmacological or neutraceutical activation of PXR may represent a novel approach for the treatment of CGD.
Supplementary Material
Acknowledgments
We than Dr. Paul Dawson (Wake Forest University) for his generous gifts of the Asbt, Ostα and Ostβ antibodies.
Grant Support: This work was supported in part by the NIH grant DK083952 to W.X., and an AHA Postdoctoral Fellowship 09POST2280546 to J.H.
Abbreviations
- Abc
ATP binding cassette
- Asbt
apical sodium-dependent s transporter
- Bsep
bile salt export pump
- CGD
cholesterol gallstone disease
- CSI
cholesterol saturation index
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- LXR
liver X receptor
- Mrp
multidrug resistance protein
- Ntcp
Na+-taurocholate cotransport proteins
- Oatp
organic anion transporting polypeptides
- Ost
organic solute transporter
- PCN
pregnenolone-16α-carbonitrile
- PXR
pregnane X receptor
- QTL
quantitative trait locus
- SHP
small heterodimerization partner
- SJW
St. John's wort
- WT
wild type
Footnotes
Author conflict of interest disclosures: No conflicts of interest exist.
Author contributions: Study concept and design (JH, WX); acquisition of data (JH, SN, MX); analysis and interpretation of data (JH, SN, MM, WX); drafting of the manuscript (JH, WX); critical revision of the manuscript for important intellectual content (MM); statistical analysis (JH); obtained funding (WX).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Portincasa P, Moschetta A, Palasciano G. Cholesterol gallstone disease. Lancet. 2006;368:230–9. doi: 10.1016/S0140-6736(06)69044-2. [DOI] [PubMed] [Google Scholar]
- 2.Yu L, Gupta S, Xu F, et al. Expression of ABCG5 and ABCG8 is required for regulation of biliary cholesterol secretion. J Biol Chem. 2005;280:8742–7. doi: 10.1074/jbc.M411080200. [DOI] [PubMed] [Google Scholar]
- 3.Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–62. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 4.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–74. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 5.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50:2340–57. doi: 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mennone A, Soroka CJ, Cai SY, et al. Mrp4-/- mice have an impaired cytoprotective response in obstructive cholestasis. Hepatology. 2006;43:1013–21. doi: 10.1002/hep.21158. [DOI] [PubMed] [Google Scholar]
- 7.Everhart JE, Khare M, Hill M, et al. Prevalence and ethnic differences in gallbladder disease in the United States. Gastroenterology. 1999;117:632–9. doi: 10.1016/s0016-5085(99)70456-7. [DOI] [PubMed] [Google Scholar]
- 8.Khanuja B, Cheah YC, Hunt M, et al. Lith1, a major gene affecting cholesterol gallstone formation among inbred strains of mice. Proc Natl Acad Sci U S A. 1995;92:7729–33. doi: 10.1073/pnas.92.17.7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyons MA, Wittenburg H. Cholesterol gallstone susceptibility loci: a mouse map, candidate gene evaluation, and guide to human LITH genes. Gastroenterology. 2006;131:1943–70. doi: 10.1053/j.gastro.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 10.Kliewer SA, Moore JT, Wade L, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 11.Xie W, Radominska-Pandya A, Shi Y, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A. 2001;98:3375–80. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Staudinger JL, Goodwin B, Jones SA, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–74. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xie W, Barwick JL, Downes M, et al. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435–9. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 14.Wang DQ, Lammert F, Paigen B, et al. Phenotypic characterization of lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice. Pathophysiology of biliary lipid secretion. J Lipid Res. 1999;40:2066–79. [PubMed] [Google Scholar]
- 15.Uppal H, Zhai Y, Gangopadhyay A, et al. Activation of liver X receptor sensitizes mice to gallbladder cholesterol crystallization. Hepatology. 2008;47:1331–42. doi: 10.1002/hep.22175. [DOI] [PubMed] [Google Scholar]
- 16.Carey MC. Critical tables for calculating the cholesterol saturation of native bile. J Lipid Res. 1978;19:945–55. [PubMed] [Google Scholar]
- 17.Nishida S, Ozeki J, Makishima M. Modulation of bile acid metabolism by 1alpha-hydroxyvitamin D3 administration in mice. Drug Metab Dispos. 2009;37:2037–44. doi: 10.1124/dmd.109.027334. [DOI] [PubMed] [Google Scholar]
- 18.Heuman DM. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J Lipid Res. 1989;30:719–30. [PubMed] [Google Scholar]
- 19.Dawson PA, Hubbert M, Haywood J, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280:6960–8. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saini SP, Mu Y, Gong H, et al. Dual role of orphan nuclear receptor pregnane X receptor in bilirubin detoxification in mice. Hepatology. 2005;41:497–505. doi: 10.1002/hep.20570. [DOI] [PubMed] [Google Scholar]
- 21.Masson D, Lagrost L, Athias A, et al. Expression of the pregnane X receptor in mice antagonizes the cholic acid-mediated changes in plasma lipoprotein profile. Arterioscler Thromb Vasc Biol. 2005;25:2164–9. doi: 10.1161/01.ATV.0000183674.88817.fb. [DOI] [PubMed] [Google Scholar]
- 22.Dawson PA, Haywood J, Craddock AL, et al. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem. 2003;278:33920–7. doi: 10.1074/jbc.M306370200. [DOI] [PubMed] [Google Scholar]
- 23.Xie Y, Blanc V, Kerr TA, et al. Decreased expression of cholesterol 7alpha-hydroxylase and altered bile acid metabolism in Apobec-1-/- mice lead to increased gallstone susceptibility. J Biol Chem. 2009;284:16860–71. doi: 10.1074/jbc.M109.010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu TT, Makishima M, Repa JJ, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–15. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 25.Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–26. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 26.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–25. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Ge H, Gupte J, et al. Co-receptor requirements for fibroblast growth factor-19 signaling. J Biol Chem. 2007;282:29069–72. doi: 10.1074/jbc.C700130200. [DOI] [PubMed] [Google Scholar]
- 28.Lin BC, Wang M, Blackmore C, et al. Liver-specific activities of FGF19 require Klotho beta. J Biol Chem. 2007;282:27277–84. doi: 10.1074/jbc.M704244200. [DOI] [PubMed] [Google Scholar]
- 29.Moore LB, Goodwin B, Jones SA, et al. St. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci USA. 2000;97:7500–2. doi: 10.1073/pnas.130155097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang H, Wang H, Sinz M, et al. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene. 2007;26:258–68. doi: 10.1038/sj.onc.1209788. [DOI] [PubMed] [Google Scholar]
- 31.Uppal H, Toma D, Saini SP, et al. Combined loss of orphan receptors PXR and CAR heightens sensitivity to toxic bile acids in mice. Hepatology. 2005;41:168–76. doi: 10.1002/hep.20512. [DOI] [PubMed] [Google Scholar]
- 32.Wang DQ, Carey MC. Complete mapping of crystallization pathways during cholesterol precipitation from model bile: influence of physical-chemical variables of pathophysiologic relevance and identification of a stable liquid crystalline state in cold, dilute and hydrophilic bile salt-containing systems. J Lipid Res. 1996;37:606–30. [PubMed] [Google Scholar]
- 33.Marschall HU, Wagner M, Zollner G, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129:476–85. doi: 10.1016/j.gastro.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Princen HM, Huijsmans CM, Kuipers F, et al. Ketoconazole blocks bile acid synthesis in hepatocyte monolayer cultures and in vivo in rat by inhibiting cholesterol 7 alpha-hydroxylase. J Clin Invest. 1986;78:1064–71. doi: 10.1172/JCI112662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung D, Mangelsdorf DJ, Meyer UA. Pregnane X receptor is a target of farnesoid X receptor. J Biol Chem. 2006;281:19081–91. doi: 10.1074/jbc.M600116200. [DOI] [PubMed] [Google Scholar]
- 36.Moschetta A, Bookout AL, Mangelsdorf DJ. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nat Med. 2004;10:1352–8. doi: 10.1038/nm1138. [DOI] [PubMed] [Google Scholar]
- 37.Meyer TE, O'Brien TG, Andreotti G, et al. Androgen receptor CAG repeat length and risk of biliary tract cancer and stones. Cancer Epidemiol Biomarkers Prev. 2010;19:787–93. doi: 10.1158/1055-9965.EPI-09-0973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang HH, Afdhal NH, Wang DQ. Estrogen receptor alpha, but not beta, plays a major role in 17beta-estradiol-induced murine cholesterol gallstones. Gastroenterology. 2004;127:239–49. doi: 10.1053/j.gastro.2004.03.059. [DOI] [PubMed] [Google Scholar]
- 39.Puppala S, Dodd GD, Fowler S, et al. A genomewide search finds major susceptibility loci for gallbladder disease on chromosome 1 in Mexican Americans. Am J Hum Genet. 2006;78:377–92. doi: 10.1086/500274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wagner M, Halilbasic E, Marschall HU, et al. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology. 2005;42:420–30. doi: 10.1002/hep.20784. [DOI] [PubMed] [Google Scholar]
- 41.Stedman CA, Liddle C, Coulter SA, et al. Nuclear receptors constitutive androstane receptor and pregnane X receptor ameliorate cholestatic liver injury. Proc Natl Acad Sci U S A. 2005;102:2063–8. doi: 10.1073/pnas.0409794102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnston DE, Kaplan MM. Pathogenesis and treatment of gallstones. N Engl J Med. 1993;328:412–21. doi: 10.1056/NEJM199302113280608. [DOI] [PubMed] [Google Scholar]
- 43.Portincasa P, Di Ciaula A, Wang HH, et al. Medicinal treatments of cholesterol gallstones: old, current and new perspectives. Curr Med Chem. 2009;16:1531–42. doi: 10.2174/092986709787909631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.