

Abstract
Caloric excess has been postulated to disrupt cardiac function via: i) the generation of toxic intermediates; ii) via protein glycosylation and iii) through the generation of reactive oxygen species. It is now increasingly being recognized the nutrient intermediates themselves may modulate metabolic pathways through the post-translational modifications of metabolic enzymes. In light of the high energy demand of the heart, these nutrient mediated modulations in metabolic pathway functioning may play an important role in cardiac function and in the capacity of the heart to adapt to biomechanical stressors. In this review the role of protein acetylation and deacetylation in the control of metabolic programs are explored. Although, not extensively investigated directly in the heart, the emerging data support that these nutrient mediated post-translational regulatory events modulate: i) cardiac metabolic pathways; ii) integrate nutrient flux mediated post-translational effects with cardiac function and iii) may be important in the development of cardiac pathology. Areas of investigation that need to be explored are highlighted.
Keywords: sirtuins, acetyltransferase, NAD+, Acetyl-CoA, Cardiac contraction, Cardiac hypertrophy
1. Introduction
The modern scourges of obesity and type 2 diabetes mellitus (T2DM) have multiple disease consequences including the potential to reverse decades of medical therapeutics and interventions that have diminished cardiac morbidity and mortality [1]. The cardiac health effects of nutrient excess is epitomized by the strong correlation between body mass index and the development of cardiac hypertrophy [2, 3] and heart failure [4]. However, whether caloric excess directly perturbs myocardial contractile function and hypertrophy, independent of the systemic effects of obesity and diabetes such as endothelial dysfunction, hypertension and hyperlipidemia, is fiercely contested, and has been difficult to delineate in epidemiologic studies.
If caloric excess does perturbs myocardial growth and function, sequelae would be expected to manifest as direct perturbations in cellular metabolic pathway biology in parallel with alterations in cardiac pump functioning and/or in the cardiac growth program. A reductionist approach can be employed to investigate the role of nutrient perturbations on metabolic pathways and to link these to effects to cardiomyocyte function. The focus of this review is on the effects of nutrient intermediates NAD+/NADH and acetyl-CoA and their role on post-translational modifications in linking metabolism, mitochondrial biology and myocardial function. The hypothesis explored contends that these intermediates indirectly and directly modulate metabolism and mitochondrial function, which in turn, may orchestrate changes in cardiac muscle function and growth. The objective of this manuscript is to review the current knowledge pertaining to: i) the modulation in cardiac function in response to changes in caloric load; ii) the role of NAD+-linked biology to changes in metabolic and mitochondrial function; iii) the role of acetyl-CoA as a signaling intermediate to modulate metabolic biology and iv) an overview of evidence how these metabolic pathway mediated regulatory effects alter cardiac hypertrophy and function. An additional important energetic-dependent system that modulates cardiac biology is the AMPK signaling system. The role of AMPK in nutrient sensing and as a signaling modulator has recently been extensively reviewed [5] and is not a primary focus in this manuscript. Additional mechanisms that orchestrating ‘nutrient’ mediated perturbations in cardiac and skeletal muscle metabolism and mitochondrial function include lipotoxicity [6, 7], the consequences of protein glycosylation [8], and via caloric-excess mediated augmentation of reactive oxygen species (ROS) [9, 10] with subsequent detrimental sequelae. These mechanisms are similarly not the focus of this review.
2. Modulation in cardiac function with changes in dietary load
In order to establish whether ‘nutrients’ themselves modulate cardiac contractile function, proof of concept studies have been performed at the extreme limits of nutrient levels. Experimental and human studies are discussed here to show that at the macronutrient level, substrate alterations can modify numerous aspects of cardiac metabolism and function.
Changes in fuel substrate levels appear to initially affect cardiac relaxation or diastole. Diastolic dysfunction is defined as abnormalities in the relaxation phase of the cardiac mechanical cycle, that delays ventricular filling and can result in symptoms and signs of heart failure [11]. Although diastolic dysfunction was initially proposed to occur independently of systolic contractile dysfunction it is now recognized that a relative degree of contractile dysfunction accompanies filling/relaxation deficits [11]. The effects of nutrient biology on the continuum of diastolic and systolic function and cardiac hypertrophy are reviewed.
2.1. Caloric restriction
The acute restriction in caloric intake provokes adipose tissue lipolysis resulting in elevated levels of circulating non-esterified fatty acids [12]. This metabolic intervention results in increased cardiac exposure to fatty acids where incorporation of triglyceride content into the myocardium can be measured by 1H-magnetic resonance spectroscopy (1H-MRS). Human studies have been performed using 1H-MRS in parallel with cardiac functional imaging to correlate changes in cardiac triglyceride content and ventricular pump function. Fasting up to 3 days, or restriction of calories over the same period, result in progressive uptake of triglycerides into the myocardium with a triglyceride ‘dose-dependent’ associated attenuation in diastolic relaxation [13, 14]. Interestingly this acute triglyceride-level dependent attenuation in cardiac relaxation appears to be independent of alterations in high-energy phosphate levels [13].
Longer term caloric restriction results in a reduction in circulating free fatty acid and triglyceride levels [15, 16]. This dietary intervention results in both a reduction in cardiac triglyceride content and an improvement in diastolic function in obese type 2 diabetic subjects [16]. In otherwise healthy individuals long-term caloric restriction additionally protects against age-associated decline in diastolic function [17]. Unfortunately in this latter study, myocardial triglyceride content was not measured and the contribution of changes in triglyceride levels to this ameliorative phenotype was not shown. Systemic changes including a reduction in blood pressure may have also contributed to the preservation of cardiac relaxation in this study. The validity of change in intracardiac triglyceride and the modification in cardiac function is further supported by a human study showing a strong correlation between the age-associated increase in myocardial triglyceride content and impaired diastolic relaxation [18].
2.2. The cardiac clock and metabolic and functional changes with diurnal rhythm
A continuous flux in substrate availability to the heart naturally occurs during the feeding and fasting states associated with the sleep-wake cycle and circadian rhythm. A link between the circadian clock, cardiac metabolism and contractile function has recently begun to be explored [19]. Here, it is intriguing to note, that in rats cardiac contractile function, glucose oxidation and oxygen consumption are significantly higher in the middle of the dark cycle compared to the middle of the light cycle [20]. This period coincides with the feeding period and correlates with the lowest circadian levels of circulating non-esterified fatty acid levels [20].
2.3. Caloric overload
Chronic nutrient overload results in the development of obesity and predisposes to insulin resistance and/or T2DM. These clinical conditions predispose to cardiac dysfunction and animal models have been interrogated to facilitate the investigation of the temporal effects of obesity and T2DM on myocardial functioning and metabolism [9].
The leptin deficient ob/ob mouse exhibits insulin resistance and diabetes in association with profound obesity. At 11 weeks of age, ob/ob mice demonstrate increase neutral lipid accumulation in the heart with echocardiographic parameters consistent with diastolic dysfunction [21] Furthermore, these mice have impaired mitochondrial respiratory capacity in association with diminished pyruvate dehydrogenase activity and a restricted glucose-dependent oxidative capacity response to increased workload [22]. In response to the direct addition of fat as the fuel substrate, oxygen consumption is enhanced, but at the expense of cardiac efficiency suggesting that the cardiac mitochondria in ob/ob mice are susceptible to lipid-mediated mitochondrial uncoupling [22]. Indeed, direct measurement of mitochondrial proton leak in the db/db (leptin receptor loss-of function mutation) mice supports the existence of fatty acid induced mitochondrial uncoupling with perturbed leptin signaling [23]. Further characterization of the db/db mice using an isolated working heart model system shows classical features of diastolic dysfunction as measured by an increased in the left ventricular end diastolic pressure and increased myocardial stiffness [24]. These perturbations are proposed to result from the diminution in myocardial efficiency as measured by the ratio between cardiac output (work) and oxygen consumed (MVO2). This excess in MVO2 was found to be exacerbated with higher levels of fat as the substrate for cardiac metabolism. db/db mice additionally show diminished glucose uptake and an early thickening of the myocardium in young mice, with progressive systolic dysfunction with aging [25]. The metabolic underpinning of these deficits is supported in that overexpression of the glucose transporter GLUT4 with concomitant increase in glucose oxidation rescues cardiac contractile function in db/db mice [26, 27].
Additional animal models that support caloric overload mediated disruption of cardiac function include the Otsuka Long-Evans Tokushima Fatty rats which show disrupted left ventricular diastolic filling [28] and evidence that cardiac contractile dysfunction is accelerated and mortality increased in hypertensive rats when they are subject to a high carbohydrate-diet [29]. Moreover, high-fat feeding of Wistar rats also evokes cardiac contractile dysfunction, which appears to result from enhanced fatty acid uptake through CD36 and intramyocellular triacylglycerol accumulation [30]. This observation is further supported by a study showing that obese Zucker rats fed a high fat ‘western diet’ exhibit increased cardiac triglyceride content within a week of this dietary intervention in parallel with reduced cardiac contractile functioning. Longer term exposure to this diet promotes fatty acid oxidation which correlates with the prevention of further deterioration in cardiac function or in progressive cardiac triglyceride accumulation [31]. Interestingly, a role for genetic susceptibility to developing this dietary response is inferred in that the Zucker lean control rats show a greater resistance to cardiac triglyceride accumulation and no adverse effects on the maintenance of cardiac power on this same diet [31].
Human subjects with diabetes or glucose intolerance similarly have increased cardiac steatosis as evident by 1H-MRS [32]. Furthermore, the measurement of high-energy phosphates in diabetic and non-diabetic subjects without clinical coronary artery disease and normal echocardiographic studies indirectly suggest that diabetic subjects can develop diminished cardiac energetics prior to evidence of measureable cardiac dysfunction [33]. Moreover, obesity is associated with evidence of cardiac hypertrophy and chamber dilatation, although dissecting out the relative contribution of nutrient excess from obesity associated effects on blood pressure and leptin homeostasis is difficult to delineate [34].
2.4. Caloric flux and modification in protein lysine-residue acetylation
The studies discussed above show that the modulation in cardiac substrate exposure can have measureable effects on cardiac function and structure. Additionally, calorie excess associated disease processes associated with obesity and T2DM can similarly affect cardiac function, although it should be reiterated that the relative contribution of ‘nutrient’ mediated effects in this biology is complicated by the comorbidities associated with these systemic disorders.
Nutrient-intermediate mediated changes in metabolic pathway biology are actively being explored and appear to function, in part, via post-translational modification of protein lysine-residues by acetylation [35]. The majority of investigation into this arena has occurred in the liver, where changes in protein acetylation status have been explored comparing fasting versus fed conditions. These studies demonstrate that the lysine-residues of enzymes controlling the spectrum of metabolic pathways employed to catabolize nutrients for energy production are modified by acetylation [35, 36] in a carbon source dependent fashion [35]. Interestingly, increasing glucose and fatty acid levels resulted in the increased acetylation in mitochondrial metabolic pathway enzymes [35], and conversely fasting results in the global deacetylation of mitochondrial proteins [36].
As the activities of cytosolic and mitochondrial metabolic enzymes are altered by the change in acetylation status [35, 37], the regulatory enzymes orchestrating these post-translational modifications may be important in modulating nutrient handling and cardiac function. Interestingly, the most characterized ‘nutrient’ dependent alteration in this program is caloric restriction induced protein lysine-residue deacetylation via the activation of the sirtuin family of deacetylase enzymes. The biochemistry and biology of this enzymatic system has recently been extensively reviewed [38, 39] but will be briefly discussed in the context of cardiac metabolism, growth and contractile function.
3. Sirtuin enzyme mediated metabolic protein deacetylation and cardiac functional consequences
3.1. The sirtuin family of enzymes
In contrast to the class I, II and IV histone deacetylases, which are zinc dependent, the sirtuins are designated as class III deacetylases and are NAD+-dependent enzymes. The founding member of these enzymes is yeast Sir2, which was found to silences chromatin via deacetylation of histones [40]. Sir2 enzymes have been shown to mediate lifespan extension in yeast, worms and flies and are postulated to function, in part, via the modulation of mitochondrial function [41]. Mammals have 7 sirtuin enzymes designated as SIRT1 through SIRT7. The mammalian sirtuins are phylogenetically divided into five subclasses based on the homology of their 250 amino acid core domain [42]. The mitochondrial enriched SIRT3 clusters with SIRT1 and SIRT2 in subclass I. These three enzymes show closest homology to yeast Sir2 and exhibit the most robust deacetylase activity. The additional mitochondrial enriched sirtuins SIRT4 and SIRT5 are assigned to subclasses II and III, and exhibit predominant ADP-ribosyltransferase and weak deacetylase activity respectively [43, 44] and SIRT6 and SIRT7 are classified as subclass IV enzymes [42].
Furthermore, the sirtuin isoforms have distinct tissue distributions and subcellular localizations [45]. With the exception of SIRT4 and SIRT7, all other family members are robustly expressed in the heart [46]. The subcellular localization of the sirtuins is probably a central feature in dictating their biological targets. Furthermore, their locations are not exclusive and may be dynamic under specific conditions. For example, SIRT1 is exclusively nuclear during cardiac embryogenesis and then displays both nuclear and cytoplasm postnatal localization [47]. Similarly, the subcellular localization of SIRT3 is predominantly in the mitochondrial matrix [48–50], although some studies suggest that SIRT3 is exclusively mitochondrial [51] while others show nuclear and cytosolic locations in whole tissue preparations [52] and following overexpression [46, 53, 54]. Whether changes in the subcellular localization of SIRT3 are associated with biological stressors, are tissue specific [50, 52] and/or result from the genetic manipulation studies is not completely resolved [55]. SIRT2, appears to be exclusively cytoplasmic, SIRT5 in the inner mitochondrial membrane or matrix and SIRT6 and SIRT7 in the nucleus [44, 45, 56].
3.2. NAD biochemistry
Sirtuin activation is directly linked to the energetic and redox status of the cell as measured by the ratio of NAD+:NADH, by the absolute levels of NAD and by the NAD+ catabolite nicotinamide [57–59]. NAD+ is a cofactor in the deacetylation reaction whereby the nicotinamide ribose is cleaved at its glycosidic bond to yield nicotinamide and the ribose accepts the acetyl group from the sirtuin substrate to produce O-acetyl-ADP-ribose. The biochemistry of this metabolic intermediate is not the focus of this review, but is beginning to be explored [60]. Interestingly, the nicotinamide generated in the deacetylation reaction itself inhibits sirtuin activity and nicotinamide-depletion during NAD+ biosynthesis inversely activates sirtuins [61].
In vertebrates de novo NAD+ biosynthesis is the minor NAD+ generation pathway, and uses tryptophan and nicotinic acid as metabolic precursors. This pathway does, however, appear to be induced by exercise and following the administration of peroxisome proliferator activated receptor alpha (PPARα) agonists [62]. The major pathway to generate NAD+ involves the salvage of NAD+ using nicotinamide as the precursor. In mammals there are two intermediary steps in NAD+ salvage, initiated by the conversion of nicotinamide to nicotinamide mononucleotide (NMN) via the nicotinamide phosphoribosyltransferase (NAMPT) enzyme. Nicotinamide/nicotinic acid mononucleotide adenylyltransferase (NMNAT) then converts NMN to NAD+. These biochemical pathways are most well characterized in the nucleus, and are pivotal for the activity of SIRT1 [63]. Moreover, NAMPT has been identified as the rate-controlling step in NAD+ biosynthesis in that overexpression of Nampt but not Nmnat increased cellular NAD+ levels [63].
The investigation into the biology of NAD+ in the mitochondria has begun to be explored, and the identification of a mitochondrial-enriched NMNAT isoform implicates that subcellular compartment specific functioning of NAD biosynthesis may be operational [64]. This is further supported in that the metabolic stress of fasting increases mitochondrial NAMPT with the concomitantly increase in mitochondrial NAD+ levels [65].
The regulation and role of this metabolic pathway in controlling cardiac metabolism and contractile function have not been extensively explored, although the transcript and protein levels of NAMPT are downregulated in the murine heart in response to pressure overload-induced cardiac hypertrophy and the overexpression of NAMPT in cardiomyocytes increased cellular NAD+ and ATP levels [66]. Taken together these data would suggest an ameliorative role of this regulatory pathway and of sirtuin activation in cardiac metabolism and in the adaptation to pathologic hypertrophic growth. Further support of this concept is that the administration of NAD+ to mice or primary cardiomyocytes exposed to hypertrophic agonist simulation, ameliorates the hypertrophic phenotype in parallel with the maintenance of cellular ATP levels [67]. The activation of this NAD+-mediated anti-hypertrophic program appears to function via SIRT3 mediated activation of the AMPK program, and the attenuation of redox stress [67]. The direct metabolic consequences of this and their effect, if any, on the hypertrophic program have not been ascertained. No data to date appears to have explored the manipulation of the NAD+ metabolic pathway and its effects on cardiac contractile functioning.
3.3. Sirtuin control of substrate metabolism and cardiac muscle function
It has been well established that cardiac substrate utilization is altered with the development of cardiac hypertrophy and the transition to heart failure [68, 69]. This metabolic remodeling results in the diminution in fatty acid oxidation as a reducing equivalent source and an augmentation in the use of glucose to generate energy. The role of this metabolic remodeling in this progressive pathophysiology is uncertain. Interestingly, the supplementation of fatty acids as substrates can delay the onset of pressure-overload induced cardiac hypertrophy and possibly delay the progression to heart failure [70, 71], in parallel with the maintenance of fatty acid oxidation [72]. Whether sirtuin biology regulates these programs is not known, although a role of sirtuins in regulating substrate utilization is increasingly becoming apparent.
Although, the role of SIRT1 in regulating fat metabolism has been quite extensively investigated, it has not directly been studied in the heart. In skeletal muscle, under low nutrient conditions SIRT1 increases fatty acid oxidation and leads to a metabolic switch from glucose oxidation to fatty acid oxidation [73]. In this nutrient limited environment AMPK enhances SIRT1 activity by increasing cellular NAD+ level, resulting in the deacetylation and modulation of cognate substrates including PGC-1α, FOXO1 and FOXO3a to regulate skeletal muscle energy metabolism [74]. In an apparent perpetuating cycle, the deacetylation of AMPK kinase LKB1 by SIRT1 concordantly increases AMPK activity [75, 76]. Additionally, in a caloric excess environment SIRT1 is downregulated in skeletal muscle in parallel with the suppression of mitochondrial respiratory function [77]. In the postnatal heart fatty acids make up the major carbon source for ATP production [69]. From a nutrient perspective it is interesting to note that SIRT1 overexpression protects cardiomyocytes from serum starvation induced cell death [78]. Furthermore, the modest induction of SIRT1 in the heart in transgenic mice prevents aging associated cardiac dysfunction and protects against paraquat-induced oxidative stress [79]. This ameliorative effect is ‘gene-dose dependent’ as a very high copy number of the SIRT1 transgene induces cardiomyopathy and increases oxidative stress [79]. Whether the ameliorative effects of SIRT1 in the heart function, in part, through the modulation of fat metabolism has not been determined.
SIRT3 has recently been reported to promote mitochondrial fatty-acid oxidation, in the heart and other tissues, through the deacetylation and activation of long chain acyl-CoA dehydrogenase (LCAD) [80]. A direct link between SIRT3 controlled fatty acid oxidation, cardiac hypertrophy and heart failure has not been established, however, mice with genetic depletion of SIRT3, possess greater susceptibility to angiotensin II induced cardiac hypertrophy compared to wildtype mice and an anti-hypertrophic effect of SIRT3 overexpression functions, in part, via its reactive oxygen species modulatory effects [81].
Although not directly related to cardiac metabolism or cardiac contractile function several mitochondrial proteins have been identified as SIRT3 targets which link SIRT3 to various metabolic pathways. Acetyl-CoA synthetase 2 (AceCS2) was the first mitochondrial proteins found to be deacetylated and activated by SIRT3 [82, 83]. AceCS2 converts free acetate, which is generated from endogenous cellular reactions or absorbed from the gut, into an active metabolite acetyl-CoA for energy production through the TCA cycle. Interestingly, AceCS2 activation plays an important role in acetate conversion under ketogenic conditions such as diabetes. A second enzyme regulating ketone body production, i.e. mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 is similarly activated by SIRT3 to facilitate energy production under fasting conditions [84]. In addition, other two mitochondrial matrix proteins, GDH, an enzyme important for amino acid metabolism, and isocitrate dehydrogenase 2 (IDH2), a key regulation enzyme for TCA cycle are also deacetylated and activated by SIRT3 [50, 56]. Interestingly, several components of the electron transfer chain (ETC) have found to be regulated by SIRT3. For example, NDUFA9, a subunit for ETC complex I [85], succinate dehydrogenase flavoprotein (SdhA), the complex II subunit [86], and ATP synthase alpha and beta subunits [87, 88] were shown to interact with SIRT3. Interaction with SIRT3 increases both complex I and complex II activities [85, 86], implying that SIRT3 is a central player in energy metabolism. Consistent with this, SIRT3 is found to regulate and maintain tissue basal ATP levels in vivo [85]. SIRT3 activation additionally modulates reactive oxygen species levels by activation of MnSOD [89] and by increasing levels of reduced glutathione [90]. These effects on reactive oxygen species level modulation, may, hypothetically have cardioprotective effects, although this too has not been directly explored. One laboratory has shown that the absence of SIRT3 predisposes to the spontaneous development of cardiac hypertrophy [81]. However, in general SIRT3 knockout mice have shown no obvious basal metabolic abnormalities [50], suggesting that the regulatory role of SIRT3 is most likely operational under certain ‘nutrient ‘stress conditions, such as during caloric restriction and in response to caloric excess. The mitochondrial metabolic pathways regulated by SIRT3 are schematized in figure 1.
Fig 1.

Mitochondrial pathway targets of SIRT3. The metabolic pathways, protein complexes and individual proteins deacetylated by SIRT3 are highlighted by asterisks. In general, the deacetylation of these proteins results in increased protein/pathway activation. The exception is the deacetylation of cyclophilin D (Cyp D), as this modification inhibits this protein to attenuate susceptibility to increased mitochondrial permeability. Additional abbreviations: TCA – tricarboxylic acid; PDH – pyruvate dehydrogenase; I – IV – represent the complex of the election transfer chain; Fo and F1 are components of ATP synthase; ANT – adenine nucleotide translocase, UCP – uncoupling proteins, VDAC – voltage dependent anion channel.
Again, although the metabolic component of their function has not been directly ascertained, a role of SIRT2 and SIRT7 appear to be operational in the cardiac derived cells and the heart respectively. Although not directly explored in the intact heart, the cytosolic sirtuin SIRT2 modulates mitochondrial function via the modulation of mitochondrial pro-apoptotic proteins and via mitochondrial antioxidant enzyme regulation [91, 92]. SIRT7 resides in the nucleoli and following its genetic depletion mice develop and die from cardiac hypertrophy and an inflammatory cardiomyopathy [93]. The absence of SIRT7 in primary murine cardiomyocytes enhances p53 acetylation, additionally leading to increased apoptosis and increased susceptibility to oxidative and genotoxic stressors [93].
4. Metabolic protein acetylation
A counter-regulatory program to counterbalance sirtuin deacetylase function is less well established. However, in much the same way as histone acetyltransferases (HAT’s) counterbalance the histone deacetylases (HDAC’s), it is expected that sirtuin acetyltransferases should exist. With respect to HAT’s and sirtuin acetyltransferase enzymes, acetyl-CoA would be expected to be the essential cofactor for protein acetylation [94]. Calorie mediated control of this process has been succinctly demonstrated where excess glucose availability has been shown to be result in the ATP-citrate lyase dependent generation of nuclear acetyl-CoA from citrate with the subsequent acetylation of histones and the induction of genes encoding for glucose uptake and metabolic enzymes [95].
The first functional characterization of a sirtuin deacetylase demonstrated that acetyl transferase general control of amino-acid synthesis (GCN5) acetylates PGC-1β to inhibit its activity to counter the deacetylation function of SIRT1 [96]. The inhibition of the predominant cardiac PGC isoform, PGC-1α has similarly been shown to be acetylated and inhibited by the nuclear transcriptional coactivator and acetyltransferase p300 [97]. Although not directly studied in the heart, one could propose that these or other acetyltransferases could function to dampen the PGC regulated mitochondrial biogenesis and β-oxidation enzymes programs central to cardiac contractile functioning and stress resilience [98, 99].
As acetyl-CoA is a predominant intermediate of both pyruvate dehydrogenase and the mitochondrial fatty acid β-oxidation cycle it would not be unreasonable that this metabolite may function within mitochondria as a ‘sensor’ of caloric excess. Indeed, dynamic regulation of mitochondrial acetyl-CoA levels are suggested by its accumulation in liver mitochondria following high-fat feeding and diabetes [100], and excess glucose and fatty acids have been shown to increase the acetylation of mitochondrial metabolic cycle proteins with a concomitant modulation of enzyme activities [35]. However, to date no mitochondrial enriched acetyl transferase has been identified to employ acetyl-CoA for protein acetylation and/or to counterregulate the deacetylase actions of SIRT3. These proteins need to be identified and characterized before this putative nutrient regulated program could be investigated in the regulation mitochondrial function in general and more specifically in the heart in response to nutrient load mediated alterations in cardiac growth and function.
5. Conclusions
There is increasing recognition that ‘nutrient’ intermediates can play an important role in regulating metabolic pathways via the modulation of the acetylation status of metabolic enzymes. This review highlights new areas of research and as importantly, significant deficits in our knowledge of the role of caloric-load biology in the modulation of cardiac substrate utilization and as to whether these regulatory programs play a role in cardiac adaptation and maladaptation. The schematic figure 2 shows proposed functioning of these programs in the modulation of cardiac contractile function and in response to hypertrophic stressors. The calorie excess scenario in this schematic additionally highlights the consequences of excess nutrient availability as an innate cardiac ‘toxin’. It is also important to recognize that the metabolic regulatory effects and the resulting cardiac consequences of the change in metabolic enzyme acetylation status probably functions to fine-tune these biomechanical and bioenergetic interactions, versus operating in a binary fashion displaying ‘all or nothing’ effects. This is best illustrated where the knockout of SIRT3 has a subtle phenotype with metabolic effects most apparent under maximal activation of the deacetylase program [80]. Nevertheless, chronic alternations in ‘fine-tuning’ of metabolism that may be operational in caloric excess conditions such as obesity and T2DM probably still have profound effects on the high-energy demand system orchestrating cardiac contractile function and it’s adaptation to extra-cardiac biomechanical stressors. We look forward to exciting advances in the understanding of this regulation as additional targets of sirtuin biology are delineated. Finally, the identification and characterization of the sirtuin counter-regulatory programs, especially in the modulation of metabolic enzymes remains enigmatic and may be pivotal in our understanding of nutrient mediated modulation in cardiac function.
Fig 2.
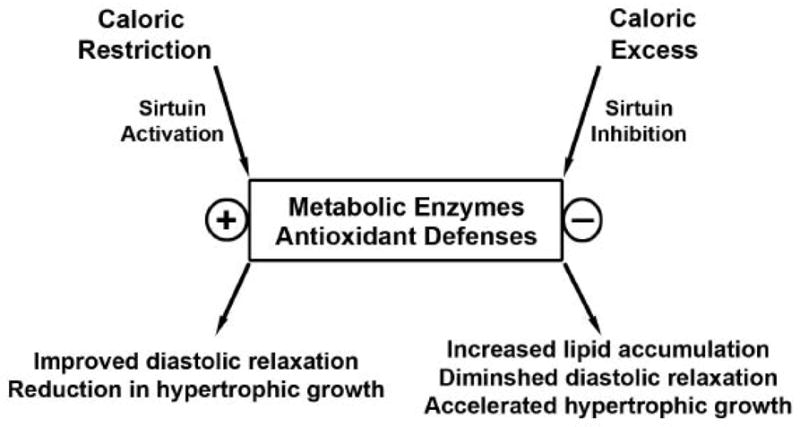
Schematic of sirtuin mediated regulatory events under ‘caloric load’ control and proposed effects on cardiac function. The plus sign represents protein activation and the minus sign, protein inhibition.
Acknowledgments
MNS is funded by the Division of Intramural Research of the National Heart Lung and Blood Institute of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Capewell S, Ford ES, Croft JB, Critchley JA, Greenlund KJ, Labarthe DR. Cardiovascular risk factor trends and potential for reducing coronary heart disease mortality in the United States of America. Bull World Health Organ. 88:120–130. doi: 10.2471/BLT.08.057885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rider OJ, Francis JM, Ali MK, Byrne J, Clarke K, Neubauer S, Petersen SE. Determinants of left ventricular mass in obesity; a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson. 2009;11:9. doi: 10.1186/1532-429X-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rider OJ, Petersen SE, Francis JM, Ali MK, Hudsmith LE, Robinson MR, Clarke K, Neubauer S. Ventricular hypertrophy and cavity dilatation in relation to body mass index in females with uncomplicated obesity. Heart. doi: 10.1136/hrt.2009.185009. [DOI] [PubMed] [Google Scholar]
- 4.Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–313. doi: 10.1056/NEJMoa020245. [DOI] [PubMed] [Google Scholar]
- 5.Kim AS, Miller EJ, Young LH. AMP-activated protein kinase: a core signalling pathway in the heart. Acta Physiol (Oxf) 2009;196:37–53. doi: 10.1111/j.1748-1716.2009.01978.x. [DOI] [PubMed] [Google Scholar]
- 6.Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta. 2010;1801:311–319. doi: 10.1016/j.bbalip.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brindley DN, Kok BP, Kienesberger PC, Lehner R, Dyck JR. Shedding light on the enigma of myocardial lipotoxicity: the involvement of known and putative regulators of fatty acid storage and mobilization. Am J Physiol Endocrinol Metab. 2010;298:E897–E908. doi: 10.1152/ajpendo.00509.2009. [DOI] [PubMed] [Google Scholar]
- 8.Smit AJ, Hartog JW, Voors AA, van Veldhuisen DJ. Advanced glycation endproducts in chronic heart failure. Ann N Y Acad Sci. 2008;1126:225–230. doi: 10.1196/annals.1433.038. [DOI] [PubMed] [Google Scholar]
- 9.Sack MN. Type 2 diabetes, mitochondrial biology and the heart. J Mol Cell Cardiol. 2009;46:842–849. doi: 10.1016/j.yjmcc.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pagel-Langenickel I, Bao J, Pang L, Sack MN. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev. 2010;31:25–51. doi: 10.1210/er.2009-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desai A, Fang JC. Heart failure with preserved ejection fraction: hypertension, diabetes, obesity/sleep apnea, and hypertrophic and infiltrative cardiomyopathy. Heart Fail Clin. 2008;4:87–97. doi: 10.1016/j.hfc.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Goldrick RB, Hirsch J. Serial studies on the metabolism of human adipose tissue. II. Effects of caloric restriction and refeeding on lipogenesis, and the uptake and release of free fatty acids in obese and nonobese individuals. J Clin Invest. 1964;43:1793–1804. doi: 10.1172/JCI105053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Meer RW, Hammer S, Smit JW, Frolich M, Bax JJ, Diamant M, Rijzewijk LJ, de RA, Romijn JA, Lamb HJ. Short-term caloric restriction induces accumulation of myocardial triglycerides and decreases left ventricular diastolic function in healthy subjects. Diabetes. 2007;56:2849–2853. doi: 10.2337/db07-0768. [DOI] [PubMed] [Google Scholar]
- 14.Hammer S, van der Meer RW, Lamb HJ, Schar M, de RA, Smit JW, Romijn JA. Progressive caloric restriction induces dose-dependent changes in myocardial triglyceride content and diastolic function in healthy men. J Clin Endocrinol Metab. 2008;93:497–503. doi: 10.1210/jc.2007-2015. [DOI] [PubMed] [Google Scholar]
- 15.Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci USA. 2004;101:6659–6663. doi: 10.1073/pnas.0308291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammer S, Snel M, Lamb HJ, Jazet IM, van der Meer RW, Pijl H, Meinders EA, Romijn JA, de RA, Smit JW. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function. J Am Coll Cardiol. 2008;52:1006–1012. doi: 10.1016/j.jacc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- 17.Meyer TE, Kovacs SJ, Ehsani AA, Klein S, Holloszy JO, Fontana L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. J Am Coll Cardiol. 2006;47:398–402. doi: 10.1016/j.jacc.2005.08.069. [DOI] [PubMed] [Google Scholar]
- 18.van der Meer RW, Rijzewijk LJ, Diamant M, Hammer S, Schar M, Bax JJ, Smit JW, Romijn JA, de RA, Lamb HJ. The ageing male heart: myocardial triglyceride content as independent predictor of diastolic function. Eur Heart J. 2008;29:1516–1522. doi: 10.1093/eurheartj/ehn207. [DOI] [PubMed] [Google Scholar]
- 19.Durgan DJ, Young ME. The cardiomyocyte circadian clock: emerging roles in health and disease. Circ Res. 2010;106:647–658. doi: 10.1161/CIRCRESAHA.109.209957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young ME, Razeghi P, Taegtmeyer H. Clock genes in the heart: characterization and attenuation with hypertrophy. Circ Res. 2001;88:1142–1150. doi: 10.1161/hh1101.091190. [DOI] [PubMed] [Google Scholar]
- 21.Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144:3483–3490. doi: 10.1210/en.2003-0242. [DOI] [PubMed] [Google Scholar]
- 22.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- 23.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 24.How OJ, Aasum E, Severson DL, Chan WY, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes. 2006;55:466–473. doi: 10.2337/diabetes.55.02.06.db05-1164. [DOI] [PubMed] [Google Scholar]
- 25.Yue P, Arai T, Terashima M, Sheikh AY, Cao F, Charo D, Hoyt G, Robbins RC, Ashley EA, Wu J, Yang PC, Tsao PS. Magnetic resonance imaging of progressive cardiomyopathic changes in the db/db mouse. Am J Physiol Heart Circ Physiol. 2007;292:H2106–2118. doi: 10.1152/ajpheart.00856.2006. [DOI] [PubMed] [Google Scholar]
- 26.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 27.Semeniuk LM, Kryski AJ, Severson DL. Echocardiographic assessment of cardiac function in diabetic db/db and transgenic db/db-hGLUT4 mice. Am J Physiol Heart Circ Physiol. 2002;283:H976–982. doi: 10.1152/ajpheart.00088.2002. [DOI] [PubMed] [Google Scholar]
- 28.Mizushige K, Yao L, Noma T, Kiyomoto H, Yu Y, Hosomi N, Ohmori K, Matsuo H. Alteration in left ventricular diastolic filling and accumulation of myocardial collagen at insulin-resistant prediabetic stage of a type II diabetic rat model. Circulation. 2000;101:899–907. doi: 10.1161/01.cir.101.8.899. [DOI] [PubMed] [Google Scholar]
- 29.Sharma N, Okere IC, Barrows BR, Lei B, Duda MK, Yuan CL, Previs SF, Sharov VG, Azimzadeh AM, Ernsberger P, Hoit BD, Sabbah H, Stanley WC. High-sugar diets increase cardiac dysfunction and mortality in hypertension compared to low-carbohydrate or high-starch diets. J Hypertens. 2008;26:1402–1410. doi: 10.1097/HJH.0b013e3283007dda. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ouwens DM, Diamant M, Fodor M, Habets DD, Pelsers MM, El HM, Dang ZC, van den Brom CE, Vlasblom R, Rietdijk A, Boer C, Coort SL, Glatz JF, Luiken JJ. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia. 2007;50:1938–1948. doi: 10.1007/s00125-007-0735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burgmaier M, Sen S, Philip F, Wilson CR, Miller CC, III , Young ME, Taegtmeyer H. “Western” Diet, Obesity(Silver Spring) 2010. Metabolic Adaptation Follows Contractile Dysfunction in the Heart of Obese Zucker Rats Fed a High-Fat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. 2007;116:1170–1175. doi: 10.1161/CIRCULATIONAHA.106.645614. [DOI] [PubMed] [Google Scholar]
- 33.Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, Radda GK, Neubauer S, Clarke K. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. 2003;107:3040–3046. doi: 10.1161/01.CIR.0000072789.89096.10. [DOI] [PubMed] [Google Scholar]
- 34.de las FL, Waggoner AD, Mohammed BS, Stein RI, Miller BV, III, Foster GD, Wyatt HR, Klein S, Davila-Roman VG. Effect of moderate diet-induced weight loss and weight regain on cardiovascular structure and function. J Am Coll Cardiol. 2009;54:2376–2381. doi: 10.1016/j.jacc.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 37.Lin YY, Lu JY, Zhang J, Walter W, Dang W, Wan J, Tao SC, Qian J, Zhao Y, Boeke JD, Berger SL, Zhu H. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009;136:1073–1084. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu Z, Scott I, Webster BR, Sack MN. The emerging characterization of lysine residue deacetylation on the modulation of mitochondrial function and cardiovascular biology. Circ Res. 2009;105:830–841. doi: 10.1161/CIRCRESAHA.109.204974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 41.Guarente L. Mitochondria--a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 43.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 44.Nakagawa T, Lomb DJ, Haigis MC, Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell. 2009;137:560–570. doi: 10.1016/j.cell.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 46.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 47.Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007;282:6823–6832. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- 48.Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci USA. 2002;99:13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr, Weissman S, Verdin E, Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cooper HM, Spelbrink JN. The human SIRT3 protein deacetylase is exclusively mitochondrial. Biochem J. 2008;411:279–285. doi: 10.1042/BJ20071624. [DOI] [PubMed] [Google Scholar]
- 52.Sundaresan NR, Samant SA, Pillai VB, Rajamohan SB, Gupta MP. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol Cell Biol. 2008;28:6384–6401. doi: 10.1128/MCB.00426-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakamura Y, Ogura M, Tanaka D, Inagaki N. Localization of mouse mitochondrial SIRT proteins: shift of SIRT3 to nucleus by co-expression with SIRT5. Biochem Biophys Res Commun. 2008;366:174–179. doi: 10.1016/j.bbrc.2007.11.122. [DOI] [PubMed] [Google Scholar]
- 54.Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007;21:920–928. doi: 10.1101/gad.1527307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bao J, Lu Z, Joseph JJ, Carabenciov D, Dimond CC, Pang L, Samsel L, McCoy JP, Jr, Leclerc J, Nguyen P, Gius D, Sack MN. Characterization of the murine SIRT3 mitochondrial localization sequence and comparison of mitochondrial enrichment and deacetylase activity of long and short SIRT3 isoforms. J Cell Biochem. 2010;110:238–247. doi: 10.1002/jcb.22531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol. 2008;382:790–801. doi: 10.1016/j.jmb.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 57.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 58.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Cohen H, Lin SS, Manchester JK, Gordon JI, Sinclair DA. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J Biol Chem. 2002;277:18881–18890. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- 59.Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, Garcia JA. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009;324:1289–1293. doi: 10.1126/science.1169956. [DOI] [PubMed] [Google Scholar]
- 60.Tong L, Denu JM. Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbapap.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 62.Ito Y, Yonekura R, Maruta K, Koike T, Nakagami Y, Shibata K, Saito K, Nagamura Y. Tryptophan metabolism was accelerated by exercise in rat. Adv Exp Med Biol. 2003;527:531–535. doi: 10.1007/978-1-4615-0135-0_61. [DOI] [PubMed] [Google Scholar]
- 63.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 64.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 65.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de CR, Sauve AA, Sinclair DA. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu CP, Hariharan N, Alcendor RR, Oka S, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through autophagy in cardiomyocytes. Autophagy. 2009;5:1229–1231. doi: 10.4161/auto.5.8.10275. [DOI] [PubMed] [Google Scholar]
- 67.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMPK pathway. J Biol Chem. 2009 doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 69.Sack MN, Kelly DP. The energy substrate switch during development of heart failure: gene regulatory mechanisms (Review) Int J Mol Med. 1998;1:17–24. doi: 10.3892/ijmm.1.1.17. [DOI] [PubMed] [Google Scholar]
- 70.Okere IC, Young ME, McElfresh TA, Chess DJ, Sharov VG, Sabbah HN, Hoit BD, Ernsberger P, Chandler MP, Stanley WC. Low carbohydrate/high-fat diet attenuates cardiac hypertrophy, remodeling, and altered gene expression in hypertension. Hypertension. 2006;48:1116–1123. doi: 10.1161/01.HYP.0000248430.26229.0f. [DOI] [PubMed] [Google Scholar]
- 71.Berthiaume JM, Bray MS, McElfresh TA, Chen X, Azam SM, Young ME, Hoit BD, Chandler MP. Myocardial contractile response to physiological stress improves with high saturated fat feeding in heart failure. Am J Physiol Heart Circ Physiol. doi: 10.1152/ajpheart.00270.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chess DJ, Khairallah RJ, O’Shea KM, Xu W, Stanley WC. A high-fat diet increases adiposity but maintains mitochondrial oxidative enzymes without affecting development of heart failure with pressure overload. Am J Physiol Heart Circ Physiol. 2009;297:H1585–H1593. doi: 10.1152/ajpheart.00599.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, Cohen RA, Zang M. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283:27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pagel-Langenickel I, Bao J, Joseph JJ, Schwartz DR, Mantell BS, Xu X, Raghavachari N, Sack MN. PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J Biol Chem. 2008;283:22464–22472. doi: 10.1074/jbc.M800842200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2alpha, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95:971–980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 79.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 80.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci USA. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci USA. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, Jacobson MP, Verdin E. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010;12:654–661. doi: 10.1016/j.cmet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cimen H, Han MJ, Yang Y, Tong Q, Koc H, Koc EC. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49:304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Law IK, Liu L, Xu A, Lam KS, Vanhoutte PM, Che CM, Leung PT, Wang Y. Identification and characterization of proteins interacting with SIRT1 and SIRT3: implications in the anti-aging and metabolic effects of sirtuins. Proteomics. 2009;9:2444–2456. doi: 10.1002/pmic.200800738. [DOI] [PubMed] [Google Scholar]
- 88.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic Biol Med. 2010;49:1230–1237. doi: 10.1016/j.freeradbiomed.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 90.Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–514. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 92.Lynn EG, McLeod CJ, Gordon JP, Bao J, Sack MN. SIRT2 is a negative regulator of anoxia-reoxygenation tolerance via regulation of 14-3-3 zeta and BAD in H9c2 cells. FEBS Lett. 2008;582:2857–2862. doi: 10.1016/j.febslet.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res. 2008;102:703–710. doi: 10.1161/CIRCRESAHA.107.164558. [DOI] [PubMed] [Google Scholar]
- 94.Ladurner AG. Chromatin places metabolism center stage. Cell. 2009;138:18–20. doi: 10.1016/j.cell.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 95.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kelly TJ, Lerin C, Haas W, Gygi SP, Puigserver P. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J Biol Chem. 2009 doi: 10.1074/jbc.M109.015164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 98.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 99.McLeod CJ, Pagel I, Sack MN. The mitochondrial biogenesis regulatory program in cardiac adaptation to ischemia--a putative target for therapeutic intervention. Trends Cardiovasc Med. 2005;15:118–123. doi: 10.1016/j.tcm.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 100.Horie S, Isobe M, Suga T. Changes in CoA pools in hepatic peroxisomes of the rat under various conditions. J Biochem. 1986;99:1345–1352. doi: 10.1093/oxfordjournals.jbchem.a135602. [DOI] [PubMed] [Google Scholar]