

Abstract
This study describes a practical system that allows high-throughput genotyping of single nucleotide polymorphisms (SNPs) and detection of mutations by allele-specific extension on primer arrays. The method relies on the sequence-specific extension of two immobilized allele-specific primers that differ at their 3′-nucleotide defining the alleles, by a reverse transcriptase (RT) enzyme at optimized reaction conditions. We show the potential of this simple one-step procedure performed on spotted primer arrays of low redundancy by generating over 8000 genotypes for 40 mutations or SNPs. The genotypes formed three easily identifiable clusters and all known genotypes were assigned correctly. Higher degrees of multiplexing will be possible with this system as the power of discrimination between genotypes remained unaltered in the presence of over 100 amplicons in a single reaction. The enzyme-assisted reaction provides highly specific allele distinction, evidenced by its ability to detect minority sequence variants present in 5% of a sample at multiple sites. The assay format based on miniaturized reaction chambers at standard 384-well spacing on microscope slides carrying arrays with two primers per SNP for 80 samples results in low consumption of reagents and makes parallel analysis of a large number of samples convenient. In the assay one or two fluorescent nucleotide analogs are used as labels, and thus the genotyping results can be interpreted with presently available array scanners and software. The general accessibility, simple set-up, and the robust procedure of the array-based genotyping system described here will offer an easy way to increase the throughput of SNP typing in any molecular biology laboratory.
Presently, there are major efforts in progress to discover random SNPs covering the entire human genome, as well as SNPs located close to or within functionally important candidate genes. The SNPs are planned to be used as markers in large-scale searches for genes underlying common, multifactorial diseases by linkage disequilibrium mapping or genetic association studies (Schafer and Hawkins 1998; Collins et al. 1999). Functional SNPs in genes encoding drug-metabolizing enzymes, drug transporters, and receptors may form the basis for future design and development of new therapeutics. SNPs represent the most common form of human genetic variation, and functional SNPs encompass the majority of the mutant alleles actually causing or predisposing to human diseases. Although SNP markers are the most obvious targets for high-throughput genotyping today, diagnostics and carrier screening in classical Mendelian disorders would also benefit largely from technology that allows efficient, reproducible multiplex genotyping of sequence variations at the single nucleotide level at low cost. Technology for multiplex mutation detection would be particularly beneficial in disorders with pronounced allelic heterogeneity (Estivill et al. 1997; the Cystic Fibrosis Genetic Analysis Consortium, http://www.genet.sickkids.on.ca/cftr-cgi-bin/FullTable) and for analyzing panels of commonly recurring mutations (Bertina et al. 1994; Feder et al. 1996; Denoyelle et al. 1997; Peltonen et al. 1999).
To use microarrays with oligonucleotide reagents immobilized on small surfaces in miniaturized assays is a frequently proposed approach for multiplex genotyping of SNPs and large-scale mutation screening (reviewed by Southern 1996; Hacia 1999a). Microarray-based technology for analyzing the expression levels of thousands of genes per single experiment is well established (Iyer et al. 1999; reviewed by Brown and Botstein 1999). In contrast, the progress in the analyses of known sequence variants on microarrays has been much less spectacular. Only a handful of studies describing the analysis of predefined human SNPs or disease-causing mutations on microarrays have appeared in the literature. Even in the largest of these studies, either the number of analyzed samples (Wang et al. 1998; Hacia et al. 1998a, 1999b) or the number of analyzed SNPs (Pastinen et al. 1998a, 1998b) were small, and, consequently, only a limited number of genotypes were produced in each study.
There are two major hurdles for highly parallel screening of SNPs on microarrays. The first is the necessity of amplifying the DNA regions spanning the mutations or SNPs by the PCR to achieve sufficient sensitivity and specificity of detecting single-base variation in the complexity of the human genome limits the capacity of genotyping assays. Efforts to perform multiplex PCR reactions with tens or hundreds of fragments in a reproducible way have failed (Wang et al. 1998; Hacia et al. 1999b; Cho et al. 1999). The second is the detection reaction itself. A key requirement for a scoring method for genomic SNPs is that it be able to distinguish unequivocally between homozygous and heterozygous allelic variants in the diploid human genome. Differential hybridization with allele-specific oligonucleotide (ASO) probes is the most commonly used reaction principle in microarray format. The specificity of genotyping by ASO hybridization depends strongly on the nucleotide sequence context of the SNPs and on the reaction conditions (Conner et al. 1983; Southern et al. 1992). Therefore, multiplex ASO hybridization reactions in microarray formats are performed using arrays carrying a highly redundant set of probes (Cronin et al. 1996; Hacia et al. 1998a, 1999b; Sapolsky et al. 1999). The power of genotyping SNPs using high-density ASO arrays was shown to be limited in recent mapping studies, in which 400–500 SNPs were analyzed and the correct genotype could only be assigned at 60%–70% of the analyzed sites (Cho et al. 1999; Wang et al. 1998; Hacia et al. 1999b).
DNA-modifying enzymes, such as DNA polymerases (Shumaker et al. 1996; Pastinen et al. 1997; Head et al. 1997; Dubiley et al. 1999) and DNA ligases (Gunderson et al. 1998; Gerry et al. 1999) may provide better genotyping power than microarray-based ASO hybridization. In a direct experimental comparison we showed earlier that DNA polymerase-assisted, single nucleotide primer extension discriminates between genotypes more than tenfold better than hybridization with ASO probes in the same microarray format (Pastinen et al. 1997). Because the discrimination between sequence variants by the DNA polymerases or ligases is based on the high-sequence specificity of the enzyme, highly redundant oligonucleotide arrays are not required.
This paper describes a novel enzyme-assisted method that allows efficient high-throughput genotyping of multiplex amplified genomic SNPs and mutations in a microarray format. The method is based on the extension of two allele-specific primers that differ at their 3′-nucleotide, defining the allelic variants of the SNPs by a reverse transcriptase (RT). Multiplex PCR products are analyzed without purification steps directly on the microarray by a concurrent in vitro transcription and allele-specific extension reaction, in which multiple RNA targets are produced from each PCR product and labeled nucleotides are incorporated in the allele-specific primer extension reactions. The method is highly sensitive, ensuring robust genotyping despite the simple single-step procedure. We demonstrate the potential for accurate multiplex genotyping by the allele-specific primer extension method by applying it to 40 mutations or SNPs and producing >8000 genotypes with a success rate approaching 100%. The specificity of the method is further evidenced by genotyping several mutations without reduction in genotype discrimination power in the presence of a template complexity corresponding to 100 SNPs per reaction and by successful detection of 5% of a minority sequence variant at several sites. All the equipment and reagents required for performing the assay are generally available, and our miniaturized assay format allows simultaneous genotyping of 80 samples at minimized reagent costs.
RESULTS AND DISCUSSION
Reaction Principle and Assay Procedure
Figure 1 shows the principle and procedure of the novel allele-specific primer extension method for genotyping single nucleotide variations in a microarray format. Initially, we compared allele-specific primer extension to single nucleotide primer extension, with DNA as the template (Pastinen et al. 1997) and DNA polymerases in the reactions on primer arrays. We found that the discrimination between genotypes was similar for the two reaction principles, as recently also suggested by Dubiley et al. (1999). In the allele-specific extension method, only a single-detection reaction with a single fluorophore is required per sample, in contrast to four fluorophores or four detection reactions with the minisequencing method. Because only two immobilized primers per SNP are needed, a simple spotting procedure (Shalon et al. 1996) of presynthesized oligonucleotides can be used to construct the primer arrays, instead of manufacturing high-density arrays by sophisticated combinatorial chemistry (Pease et al. 1994), as is required for ASO hybridization.
Figure 1.

Steps and principle of allele-specific extension on primer arrays. Multiplex PCRs yielding amplicons tailed with 5′-T7 RNA polymerase promoter sequence are performed. The PCR products are added directly to the primer array, along with the reaction mixture, which contains both T7 RNA polymerase and rNTPs to generate RNA targets from the PCR products and RT and labeled dNTPs for the actual allele-specific genotyping reaction, which is illustrated in the inset on the right column. For genotyping a pair of allele-specific detection primers for each mutant or polymorphic site immobilized through their 5′ end are used. The two primers differ at their 3′ end, which is complementary to either of the variant alleles. The RT extends the immobilized detection primers with labeled dNTPs in a template-dependent manner. After the reaction, fluorescence scanning of the arrays and quantitation of the fluorescent signals are carried out using a commercial confocal scanner and software. The fluorescent signals from each primer pair are compared, and the signal ratios fall into distinct categories defining the genotype at each site. The timescale for the reaction steps is illustrated beside the left column of the figure.
We tested the allele-specific primer extension reactions using RNA as the template and RT enzymes for the extension. We found that the excellent performance of the MMLV RT enzyme for allele discrimination at an elevated reaction temperature and the presence of trehalose to enhance the enzyme activity (see Methods) enabled us to design an extremely robust reaction procedure (Fig. 1). The multiplex PCR products are analyzed on the microarrays without purification, concentration, labeling, or fragmentation in a one-step reaction with all the required enzymes, nucleotide analogs, and other reagents. Less than 1% of the multiplex PCR products are sufficient for the reaction because the RNA polymerase amplifies the template up to 2000-fold (Eberwine et al. 1992) and several labeled nucleotide analogs are incorporated per RNA template in the allele-specific extension reaction. Use of DNA targets was abandoned as the single-stranded, amplified RNA target yielded higher signal-to-noise ratios. Little hands-on-time is needed for performing the reactions; the whole procedure can be completed in a single working day.
Mutation Panel and Multiplex PCR
The major known mutations underlying the diseases belonging to the Finnish disease heritage (Peltonen et al. 1999)—a set of recessive disorders more common in Finland than elsewhere in the world and a number of other diseases predisposing or causing mutations in the Finnish population—were included in the mutation panel to be analyzed using the allele-specific primer extension on microarrays (Fig. 2A). The mutation panel included 24 nucleotide substitutions, of which nine were transversions and 15 were transitions. Furthermore, five small deletions of 1–3-bp, and one was a large deletion of 1 kb, were included in the panel.
Figure 2.



Strategy and result from high-throughput genotyping on allele-specific primer extension arrays. (A) The design of an array is illustrated schematically. Eighty separate reaction chambers have been formed on each microscope glass slide, using a 384-well formatted silicon rubber grid. Each of these reaction chambers contain an identical array of 72 primers for detection of 36 mutations or SNPs. The vertical columns (A–D) contain pairs of allele-specific primers for each site ordered into nine horizontal rows (1–9). The acronyms corresponding to the diseases and the mutant or variant nucleotides analyzed on the array are given in the lower part of A. INCL = infantile neuronal ceroid lipofuscinosis (Vesa et al. 1995); AGU = aspartylglucosaminuria (Ikonen et al. 1991); FV = Factor V Leiden (Bertina et al. 1994); CCD = congenital chloride diarrhea (Höglund et al. 1996); APECED = autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (the Finnish-German APECED Consortium 1997); HTI = hereditary tyrosinemia type I (St-Louis et al. 1994); HFI = hereditary fructose intolerance (Cross et al. 1990); OAT1 & 2 = gyrate atrophy of the choroid and retina (Mitchell et al.1989); Batten = Batten disease (the International Batten Disease Consortium 1995); vLINCL = variant late infantile neuronal ceroid lipofuscinosis (Savukoski et al. 1998); NKH = nonketotic hyperglycinemia (Kure et al. 1992); CF Δ508/ΔTT394 = cystic fibrosis (Kere et al. 1994); A1AT = α1-antitrypsin deficiency Z-mutation (Cox et al. 1988); HH = hereditary hemochromatosis C282Y (Feder et al. 1996); DFNB=nonsyndromic deafness, connexin 26 35insG (Denoyelle et al. 1997); ODG = hypergonadotropic ovarian dysgenesis (Aittomaki et al. 1995); DTD = diastrophic dysplasia (Hästbacka et al. submitted); CNFmaj/min = congenital nephrosis (Kestilä et al. 1998); PKU R408W = phenylketonuria (Guldberg et al. 1995); MCAD = medium-chain acyl-CoA dehydrogenase deficiency (Matsubara et al. 1990); LCHAD = long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (Ijist et al. 1996); PT = prothrombin G20210A (Poort et al. 1996); MPS = mucopolysaccharidosis type I (Bunge et al. 1994); RS1/2 = X-linked juvenile retinoschisis (Huopaniemi et al. 1999); LPI = lysinuric protein intolerance (Torrents et al. 1999); Salla = Salla disease (Verheijen et al. 1999); SNP1 = WIAF-11062, SNP2 = WIAF-11091, SNP6 = WIAF-10964 (Cargill et al. 1999); SNP7 = HLA-H IVS2 (Beutler et al. 1996); SNP8 = HLA-H 5569 (Jeffrey et al. 1999) ; EPMR = progressive epilepsy with mental retardation (Ranta et al. 1999). (B) A fluorescence image of the array described in A obtained when 40 DNA samples were genotyped in duplicate (>2400 genotypes) by allele-specific primer extension. The genotype assignments for a larger number of samples based on numeric ratios from arrays are shown in Figures 3 and 4. (C) Enlargements of 16 individual arrays (a–p in B). The genotyping results from samples of mutation carriers and patients are indicated by the disease acronym and genotype below the array images. Both allele-specific primers give similar signal intensities in heterozygous samples, while only one of the primers gives a signal for homozygotes.
A multiplex PCR procedure for amplifying 30 DNA fragments spanning the 31 mutant sites was optimized. Although primers for as-short-as-possible amplicons with similar PCR-annealing temperatures were selected, no more than eight fragments could be amplified per PCR mixture reproducibly. The number of fragments that previously have been successfully and reproducibly amplified by multiplex PCR followed by accurate genotyping on microarrays, range from four to ten fragments per PCR reaction (Hacia et al. 1998a; Pastinen et al. 1998a), whereas multiplex PCRs of more than 40 fragments can be performed at the cost of significantly reduced accuracy and success in the genotype assignment (Wang et al. 1998; Hacia et al. 1999b; Cho et al. 1999). The multiplex PCR amplification remains a rate-limiting factor for developing truly high-throughput genotyping systems both using high-density ASO chips and using our allele-specific primer extension reaction.
Array Design and Genotyping Capacity
The two allele-specific detection primers per mutation or SNP have 3′-ends that are complementary to either of the variant alleles (Fig. 1). Up to three discriminative bases were used for detection of deletions, such as the cystic fibrosis CFTRΔF508 mutation. For detecting the Batten disease mutation, which is a 1 kb deletion (International Batten Disease Consortium 1995), we used separate PCR primers for the normal and mutant alleles, and detection primers spanning the 5′ deletion breakpoint end with three discriminative bases. At the optimized conditions three detection primers out of the 62 used gave significant template-independent signals, which were most likely due to the formation of intramolecular hairpin-loop structures, allowing self-primed extension. This phenomenon is known to occur from previous single nucleotide primer extension assays (Nikiforov et al. 1994). For the 10% of mutations that had detection primers forming hairpin loops, the primers were redesigned to detect the opposite DNA strand, thus solving the problem. By initially designing arrays that interrogate both DNA strands would obviate the need to reverse the orientation of unfavorably performing detection primers.
To increase the capacity of the multiplex genotyping procedure, we devised a simple system for preparing silicon rubber grids, forming separate reaction chambers for 80 samples on each microscope slide. Figure 2A shows the design of an array for analyzing 80 individual samples for 36 mutations or SNPs. Figure 2B shows a fluorescence image obtained after genotyping 40 samples in duplicate on this array, and Figure 2C shows the result from 16 individual samples in greater detail. With our current printing density of 230 μm spacing between spots, 4800 genotypes can be extracted from a single microscope slide. Using printing pins for smaller spot sizes, up to 600 spots would fit into each reaction chamber at 100 μm spacing, increasing the throughput of the system to >20,000 genotypes per slide.
Genotype Discrimination
The allele-specific primer extension method was evaluated in more than 400 assays, using the arrays for screening 31 disease mutations and arrays for a panel of SNPs in the factor V and HLA-H genes. Samples from known disease carriers (90 carriers, 74 samples), along with the available homozygote patient samples (n = 10) and anonymous Finnish samples of unknown genotype (n = 49), were analyzed for the mutations. Duplicate PCR and genotyping reactions were performed for the homozygous patient and unknown samples. The results of these 192 assays are presented numerically as scatter plots in Figure 3, which shows a clear clustering of signal ratios corresponding to the three genotypes. The difference in the ratios between the signals from the normal and mutant allele-specific reactions varied from 3-fold to over 100-fold depending on the mutation; the average of all sites was 22-fold. 5740 out of 5952 genotyped sites (96%) yielded signal intensities over the predefined threshold value of 2-fold over background. In this assay all the carrier and patient genotypes were assigned correctly (84 samples, 100 variants). Furthermore, 14 carriers were identified among the 49 unknown samples; these genotypes were confirmed by the reference methods. Only eight ambiguous genotypes (0.1%) due to signal ratios falling outside the three distinct clusters were observed.
Figure 3.

Scatter plots showing the genotype assignment at 31 mutant sites for 192 samples from known mutation carriers, homozygous patients, and unknown individuals. The acronyms for the diseases above each scatter-plot are as explained in the Figure 2A legend. The ratios between the signal intensities from the normal and mutant allele-specific reactions are given on the y-axis. The ratios are near the value 1 in heterozygotes, elevated in normal homozygotes, and reduced in mutant homozygotes. The x-axis shows the signal intensities from each sample after correction for the average local background signal from the arrays. The red asterisks in the heterozygote clusters indicate that synthetic templates were analyzed because no carrier samples were available.
In a recent study, a map comprising 412 SNPs in Arabidopsis thaliana was analyzed using high-density, ASO-based microarrays carrying 72,500 probes, corresponding to 176 probes per SNP, and the correct genotype was assigned for as little as 57% of the analyzed SNPs (Cho et al. 1999). Similarly, a success rate of 71% was achieved for genotyping 558 human SNPs using a microarray carrying over 62,000 probes (Wang et al. 1998; Hacia et al. 1999b). Such low success rates as these obtained with random SNP markers could be a problem for analysis of defined sets of disease-causing mutations or functional SNPs.
We also evaluated our allele-specific primer extension method in a second array design, with seven SNPs and the FVLeiden mutation in the coagulation factor V gene and two SNPs and the hemochromatosis HHC282Y mutation in the HLA-H gene. The reaction conditions were identical to the optimized conditions applied in this first assay design. Over 200 anonymous samples of known genotype with respect to the two mutations were typed. At each of the 11 variant sites, the signal ratios clustered distinctly according to the genotypes (Fig. 4). The tighter clustering of the genotypes as compared to the mutation panel (Fig. 3) is mainly attributable to triplicate primers on the arrays reducing S/N variation due to nonuniform spotting. Also the PCR procedure was easier to optimize because of a lower degree of multiplexing. The first set of the allelespecific detection primers was used without redesign. The discrimination between genotypes was unequivocal; the average difference in signal ratios was 18.0-fold over all sites, and the smallest average discrimination was 7.9-fold. In all samples the genotypes for the FVLeiden and HHC282Y mutations were correctly assigned (n = 462). Three out of the seven analyzed SNPs in the FV gene were not polymorphic in the samples analyzed here. Two of these SNPs were previously reported to have frequencies below 5% for the rare allele (Cargill et al. 1999). The allele frequencies for the other six SNPs are similar to those estimated previously in other Caucasian populations (Cargill et al. 1999; Beutler et al. 1997; Jeffrey et al. 1999). The genotypes for the SNPs are in Hardy-Weinberg equilibrium in the samples negative for the FVLeiden and the HHC282Y mutations. These results from determining >2000 genotypes at 11 SNPs, along with the 6000 genotypes determined on the mutation screening array with an accuracy of nearly 100%, show the excellent power and general applicability of the allele-specific primer extension method for different types of mutations and SNPs.
Figure 4.

Scatter plots showing the results from genotyping a panel of variant nucleotides in the factor V and the HLA-H genes. The SNPs from the FV gene had been selected from those described by Cargill et al. (1999). The polymorphism WIAF-10964 lies in exon 10 as the FVLeiden mutation (Bertina et al. 1994), while the other SNPs are located in exon 13 of the gene. The polymorphism IVS2 lies in intron 2 (Beutler et al. 1997), the HHC282Y mutation in exon 4 (Feder et al. 1996) and 5569A/G in intron 4 (Jeffrey et al. 1999) of the HLA-H gene. The six FV and two HLA-H gene SNPs, along with FVLeiden and HHC282Y mutations, were analyzed in 53 known FVLeiden mutation and in 84 known HHC282Y mutation carriers or homozygotes, and in 94 individuals known to be of normal genotype at both the mutant sites. The signal intensity ratios are given on the y-axis and the signal intensities on the x-axis.
Effect of Template Complexity
The effect of the complexity of the template on the allele-specific primer extension reactions was tested by analyzing a 7-plex PCR product for seven homozygous mutations separately and after increasing the number of amplicons present during the reaction 4-fold and 16-fold, respectively (Fig. 5). Decreased signal intensity is evident with increased template complexity. However, the specificity of the assay expressed as the signal ratios between correct primer extension over misincorporation is not markedly affected. This result shows that higher degrees of multiplexing of the allele-specific primer extension reaction will be possible without loss of genotype discrimination power.
Figure 5.
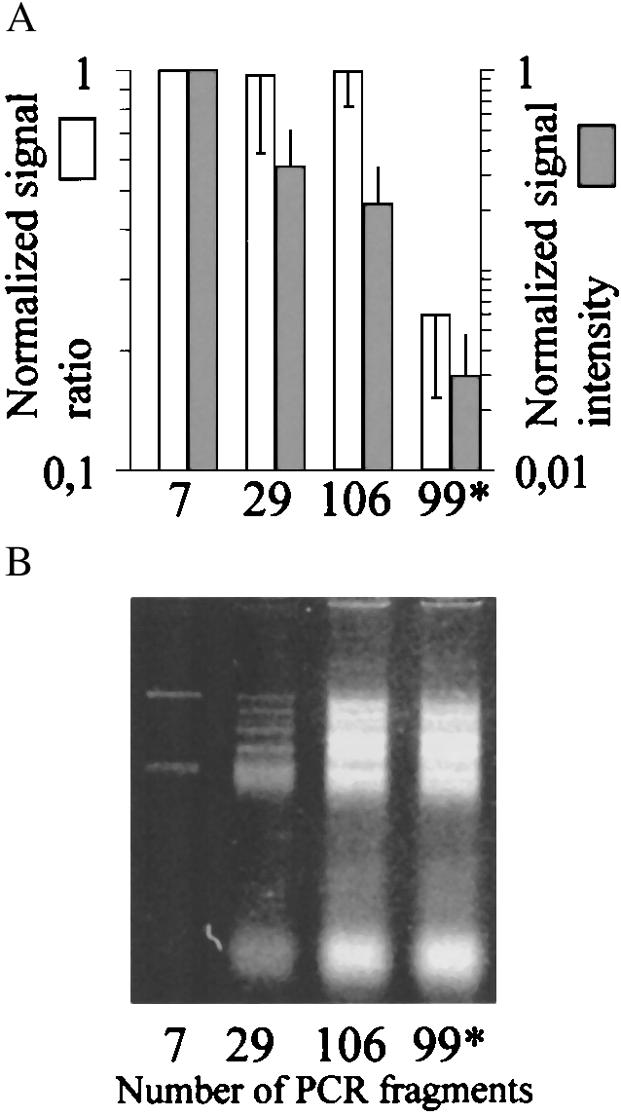
Effect of template complexity on the allele-specific extension reaction on primer arrays. (A)Average signal intensity ratios and signal intensities, obtained when seven mutations amplified in a single 7-plex PCR (indicated by 7), were genotyped directly and at increased complexity in the presence of 22 additional (indicated as 7 + 22 = 29) and 99 additional (106) PCR fragments in similar molar amounts. A mixture containing all the other PCR fragments (99*), except the 7-plex PCR product, was included as control for the template-independent extension or cross reaction with nonspecific targets. The average signal intensity ratio (reflecting specific vs. misincorporation of CY5-dNTPs) and signal intensity from quadruplicate genotyping reactions were given the value 1 for the 7-plex PCR product. The signal intensity ratios and signal intensities for the complex mixtures are given in relation to this value. The standard deviations were calculated for the overall change in signal ratio and signal intensity for the seven analyzed mutant sites. (B) Agarose gel image of the mixtures of PCR products analyzed in the experiment described in A.
Detection of Minority Mutations
Finally, we evaluated how well the allele-specific primer extension method performs in multiplex detection of mutations present as a minority in a mixture with an excess of the normal sequence. For this analysis a dual-color labeling strategy was devised to normalize the signal ratios with respect to differences in signal intensity at different positions on the array (see Methods). According to this analysis at eight sites, as little as 5% of the mutant sequence can be distinguished from the normal sequence (Fig. 6). This ability of multiplex detection of mutant-sequence variants in the presence of an excess of normal sequence shows the robustness of the allele-specific primer extension reactions on the microarrays. This result suggests that heterozygous mutations could be detected in pooled samples containing DNA from ten individuals. Such a pooling strategy would be extremely valuable for reducing the number of required genotyping assays in large-scale, population-directed screening for carriers of multiple rare recessive mutations. Analogously, the allele-specific primer extension method on microarrays could be applied to multiplex screening of somatic mutations present as a minority of the cells in tissue samples.
Figure 6.

Detection of minority mutations in mixed samples with an excess of normal sequence by dual-color, allele-specific primer extension reactions. A control sample, which is a normal homozygote for all the tested mutations (0% mutant allele), was first analyzed on all the arrays by extension with CY3-labeled dNTPs. The same arrays were then subjected to a second round of extension, in which the mixed samples containing varying amounts of mutant allele were analyzed using CY5-labeled dNTPs in the allele-specific reactions. The acronyms for the mutations above each diagram are as in Figure 3. The proportion of mutant allele (%) in the samples is given below the diagrams. The CY-5 signal ratios between the mutant and normal alleles in the mixed samples normalized for the corresponding CY-3 ratios in the control sample and further normalized to 1 are given on the y-axis. The error bars show range of signal ratios in triplicate.
The signal ratios in Figure 6 indicate that the sensitivity of detecting mutations depends on the sequence context and that considerably <5% of the A1AT, INCL, and AGU mutations could be distinguished from the background. In earlier work we have shown that a minority of <1% of the AGU mutation can be detected in large-pooled DNA samples by individually performed minisequencing primer extension reactions (Syvänen et al. 1992). Similarly, a recent study showed that minority mutations could be detected using ligation-based zip-code arrays at the 1% level (Gerry et al. 1999), but in this study only one mutation was assayed in an individual reaction. Because enzyme-mediated detection of minority mutations compares favorably to ASO hybridization in individual reactions (Farr et al. 1988), it can be expected that this will be the case also in multiplex microarray-based analyses. This assumption is supported by a result from ASO hybridization performed in a low-redundancy array format with eight probes per mutation, in which the arrays failed to discriminate mutations present at <50% in the target (Gunthard et al. 1998). To date, none of the studies using high-density ASO chips have addressed the issue of detecting minority sequence variants.
Our study describes an enzyme-assisted method for genotyping on DNA microarrays and shows that the method is accurate in multiplex genotyping. Multiplex genotyping using ASO hybridization requires highly redundant probe sets and thus high-density arrays (Pease et al. 1994) which can only be fabricated with sophisticated instrumentation. In contrast, our method requires only two probes per mutation and thus can be set up with a simple spotting robot. The poor specificity of ASO-based microarray systems in certain sequence contexts is a well-recognized problem, and a variety of strategies have been employed to overcome this problem, such as an electronic stringency changer (Gilles et al. 1999), the use of modified bases in the targets and the inclusion of chaotropic agents in the buffer (Hacia et al. 1998b; Nguyen et al. 1999), and the monitoring of melting curves of the hybrids (Drobyshev et al. 1997). The high fidelity of the RT enzyme in our system ensures good genotype discrimination at a single set of reaction conditions. In comparison to other enzyme-assisted methods (Shumaker et al. 1996; Pastinen et al. 1997; Gerry et al. 1999; Head et al. 1997; Gunderson et al. 1998; Dubiley et al. 1999; Tang et al. 1999). the present method has the significant practical advantage that the post-PCR preparation and the genotyping reaction are combined into a single step, allowing efficient handling of multiple samples in parallel, even without automation. Furthermore, our reaction is performed using only one or two fluorophores, which is compatible with all available array scanners. We estimate the cost of the assay, including oligonucleotide synthesis, to be 0.1–0.3 US dollars per genotype depending on the degree of multiplexing and the number of samples analyzed. Because the complete system is fully accessible to any molecular biology laboratory thriving to scale up its current genotyping capacity, we believe that our novel microarray-based method will greatly benefit various large-scale SNP-typing or mutation-screening projects.
METHODS
DNA Samples
Blood samples from known patients and heterozygous carriers of most of the disease mutations were obtained from clinicians involved in investigating the diseases. (For a list of the diseases and analyzed mutations, see the Figure 2 legend.) A few samples from carriers of the recessive disease mutations and all the samples from carriers of the FVLeiden and hemochromatosis HHC282Y mutations were identified in a large-scale population study in Finland (Pastinen et al., in prep.). The samples of unknown genotype analyzed in our study were either provided by voluntary laboratory personnel or were anonymous population samples. Informed consent was obtained from all subjects. The DNA was extracted from whole blood by a standard method (Bell 1981). Altogether over 400 samples were genotyped using two array designs for 40 mutations or SNPs.
Primers
The allele-specific detection primers contained a 5′ NH2 group, a spacer sequence of 9 T residues 5′ of the actual gene specific sequence of 18–21 nucleotides in length and a 3′ nucleotide complementary either to the normal or mutant nucleotide. One PCR primer of each pair contained a 5′ RNA polymerase promoter sequence (TTC TAA TAC GAC TCA CTA TAG GGA G) and the other primer of each pair had a 5′ tail of random sequence (GCG GTC CCA AAA GGG TCA GT). The polarity of the latter primer was the same as that of the corresponding detection primer sequence. The length of the gene-specific sequence of the PCR primers was 18–23 nucleotides. All primer sequences are available at http://www.ktl.fi/molbio/wwwpub/. The primers were synthesized by Interactiva Biotechnologie GmbH (Ulm, Germany).
Preparation of Microarrays
The primer arrays were prepared on standard microscope glass slides. The slides were precleaned with 1% Alconox (Aldrich), followed by rinsing several times with distilled water and ethanol. The glass surface was activated with isothiocyanate and the NH2-modified oligonucleotides were immobilized as described by Guo et al. (1994) with the following modification: 3-aminopropyl triethoxysilane (A3648, Sigma) was used for silanization instead of its methoxy-derivative. Prior to spotting, the oligonucleotides were dissolved in a 400 mm sodium carbonate buffer (pH 9.0) to a final concentration of 20 μm, and immediately after spotting, the slides were exposed to vaporized NH3, followed by washing three times in distilled water. The arrays were stored at −70°C.
A custom-built spotter with Isel Automation Flachbettanlage 2 (Eiterfeld, Germany) mechanics controlled by an MCM-310 operating system and NUMO-6.0 software (Merval, Pietarsaari, Finland) was equipped with two TeleChem CPH-2 (Sunnyvale, California) printing pins. A customized, Windows-based software was used for designing the arrays. The spots were 125–150 μm in diameter and the center-to-center distance between them was 230 μm. Eighty identical arrays with 72, 66, or 62 primers per array were spotted on the glass slides in five columns and 16 rows with an array-to-array distance of 4.5 mm (see Fig. 2A). This pattern is compatible with a reaction chamber in a 384-well microtiter plate format (see Custom-made Reaction Chambers). The spotting was carried out at ambient humidity and temperature, resulting in batch-to-batch variation in spot uniformity, which occasionally interfered with quantitation (see below).
Multiplex PCR Amplification
The PCR primer pairs were grouped into multiplex PCR reactions with 7, 7, 8, and 8 primer pairs per reaction for the 31 mutations or with 2 and 3 primer pairs per reaction for the FV and HLA-H SNPs. The amplifications were carried out using 20 ng of DNA, 0.2 mm dNTPs, and 0.8 U of AmpliTaq Gold DNA polymerase (Perkin Elmer, Branchburg, New Jersey) in 15 μl of DNA polymerase buffer supplied with the enzyme. The primer concentrations varied from 0.1 μm to 1.5 μm and had been adjusted to give similar signal intensities in the reactions on the arrays. After initial activation of the polymerase at 95°C for 11 minutes, the thermocycling parameters were as follows: 95°C for 30 seconds and 65°–1°C per cycle for 4 minuts for 5 cycles; 95°C for 30 seconds, and 60°–0.5°C per cycle for 2 minutes, and 68°C for 2 minutes for 15 cycles; 95°C for 30 seconds, 53°C for 30 seconds, and 68°C for 2 minutes for 14 cycles; and 68°C for 6 minutes. The size of the amplicons ranged from 100- bp to 530-bp, including 46-bp of the nonspecific tail sequences. The multiplex PCR products from each product were pooled and then analyzed without concentration or purification steps.
Custom-made Reaction Chambers
Reusable miniaturized rubber reaction chambers were prepared in house using an inverted 384-well microtiter plate with V-shaped wells (Biometra) as a mold. Liquid silicon rubber (Elastosil RT 601 A/B, Wacker-Chemie GmbH, Munich, Germany) was poured into the mold, leaving about 1–2 mm of the tip of the wells uncovered. After allowing the rubber to harden over night, the grids containing 384 cone-shaped reaction chambers were cut to match the size of microscope slides. A rubber grid was placed over the 80 primer arrays to form 80 separate reaction chambers. The reaction chambers had a glass surface of about 2.8 mm in diameter, having the primer array as bottom and the molded cone-shaped silicon rubber as walls with open tops for pipet tips to fit into the chambers. Prior to adding the reaction mixtures, the rubber grid was firmly pressed against the glass surface in a custom-made aluminium rack with a Plexiglas cover containing drill holes for the pipet tips.
Optimization of Allele-specific Extension Reactions
Multiplex PCR products carrying a 5′ T7-RNA polymerase promoter sequence were initially transcribed to RNA using the T7 Ampliscribe Kit (Epicentre Technologies, Madison, Wisconsin) at the conditions recommended by the manufacturer. Ten μl of the RNA target in 10 mm Tris-HCl (pH 7.4), 1 mm EDTA, 0.2 m NaCl, 0.1% Triton X-100 was allowed to anneal to allele-specific primers on the arrays at 37°C for 20 minutes, followed by a brief rinse in 0.1 m NaCl at RT. The AMV (Amersham-Pharmacia Biotech, Uppsala, Sweden), the MMLV (Epicentre Technologies), the SuperScript II (BRL), and the RetroTherm (Epicentre Technologies) RTs were then compared in allele-specific extension reactions on the arrays. Each RT enzyme was used at half the recommended unit concentrations, and the dNTPs (dATP, dGTP, CY5-dUTP, CY5-dCTP, Amersham-Pharmacia Biotech) were used at 6 μm concentrations in the reaction buffers provided with each enzyme. The reaction temperature was 42°C for the AMV, MMLV, and SuperScript II RTs and 52°C for RetroTherm. The performance of the MMLV RT in allele-specific primer extension was enhanced by increasing the reaction temperature to 52°C and including 0.4 m trehalose in the reaction buffer (Fig 7) (Mizuno et al. 1999; Carninci et al. 1998).
Figure 7.

Optimization of the conditions of the allele-specific primer extension reaction. (A) Comparison of four RT enzymes for their efficiency in incorporating labeled dNTPs and for their genotyping specificity in the array-based primer extension reaction. The MMLV RT is not as processibe as AMV and SuperScript because it yields lower signal intensities, but the MMLV RT showed the highest discrimination against misprimed extension. (MMLV = Moloney murine leukemia virus reverse transcriptase; AMV RT = avian myeloblastosis virus reverse transcriptase.) (B) Enhancement of the efficiency of nucleotide incorporation and genotyping specificity of the MMLV enzyme by increased reaction temperature and the presence of the disaccharide trehalose. Normalized signal intensities and signal intensity ratios are given on the y-axis.
Single-step Genotyping
For the simultaneous template preparation and allele-specific extension reactions, 0.5 μl of the total 60 μl of pooled multiplex PCR product, 6 mm of the four rNTPs, 6 μm dATP, dGTP, CY5-dUTP, and CY5-dCTP (Amersham-Pharmacia Biotech), 6 μm of the four ddNTPs, 6 U of MMLV RT (Epicentre Technologies), 0.5 μl of T7-RNA polymerase solution (Ampliscribe Kit) were combined in a total volume of 6 μl of T7 Ampliscribe reaction buffer supplemented with 0.4 m trehalose (T-5251, Sigma, St. Louis, Missouri) and 15% glycerol (w/v). The reactions were allowed to proceed for 1–2 hours at 52°C using the custom-made reaction chambers and rack. After the reaction, the silicon grids were removed, the slides were rinsed briefly with 6x SSC (0.9 m NaCl, 90 mm sodium citrate buffer, at pH 7.0), 0.1% Triton X-100, and distilled water, followed by a 5-minute wash in 50 mm NaOH and a brief rinse with distilled water.
Preparation of Templates for Complexity Testing
A 7-plex PCR product containing the DNA fragments spanning the INCL, FV, Batten, OAT402, OAT 181, AGU, and RS1/2 mutations served as the reference sample. The seven reference mutations were present in the normal homozygous form. This sample was mixed with multiplex PCR products containing the remaining 22 PCR products from the mutation screening assay. A third sample was prepared by adding a mixture of 77 individually amplified, purified, and concentrated human genomic PCR products (gifts from Drs. J. Saarela and J. Leppävuori). A fourth sample contained all the 99 PCR products, except the 7-plex reference PCR product. The mixtures contained similar amounts of each individual amplicon as estimated on EtBr-stained agarose gels.
Dual-color Detection of Minority Alleles
The DNA concentrations of samples from known homozygotes for the eight mutations tested (see Fig. 4) and of a control sample known to be of normal homozygous genotype with respect to all the tested mutations were measured spectrophotometrically. DNA from each mutant sample (100% mutant allele) was mixed in 5%, 10%, and 20% proportions with the control sample (0% mutant allele) and the mixed samples were subjected to multiplex PCR as described above. An allele-specific extension reaction with the multiplex PCR product from the control sample as template was first performed over two whole microscope slide array surfaces facing each other using CY3-dUTP and CY3-dCTP (Amersham-Pharmacia) in 60 μl of the optimized reaction mixture detailed above. After rinsing with 6x SSC and distilled water, the same microscope slides were used to analyze the multiplex PCR products of the mixed samples in triplicate by standard allele-specific extension reactions with CY5 as label and using the rubber reaction chambers to separate the samples. The array was scanned for both CY5 and CY3 emissions. The CY5 signal intensity (test sample) was divided by the CY3 signal intensity (control sample) at all spots. These adjusted values were then used to calculate the signal intensity ratio between the test and control samples, which was further normalized to have a value 1 for the respective samples with 0% mutant allele. The test to control signal ratios in the mixed samples was then compared to this sample containing 0% of mutant alleles.
Array Scanning and Signal Quantitation
The microscope glass slides were scanned using the confocal ScanArray 4000 (GSI Lumonics, Watertown, Massachusetts), with excitation at 630 nm and emission at 670 nm for the one-color experiments (CY5) and at 540 nm, and 570 nm, respectively, for the dual-color experiment (CY3). Five or 10 μm resolution 16-bit TIFF images were analyzed using the Scanalyze 2.44 software (Eisen M., Stanford University). The average signals after subtraction of local background were used for calculating the signal ratios defining the genotypes. A critical feature in quantitation of the signals is that the spots are as uniform as possible. Some batches of arrays demonstrated nonuniform spots (e.g., donut-shaped spots) and the simple quantitation of signal averages after local background subtraction yielded less robust genotyping results. In the dual-color experiment the CY5/CY3 signal ratios were used to calculate the normalized intensity ratios.
Acknowledgments
We thank Drs. A. Palotie, A. Orpana, T. Alitalo, J. Hastbacka, J. Leisti, I. Sipila, P. Aula, T. Weber, F. Guttler, M. Lindlöf, M. Kestila, and J. Mykkanen for providing patient and carrier DNA samples; Drs J. Hastbacka, T. Alitalo, N. Aula, S. Ranta. and J. Mykkanen for providing unpublished sequence information; Dr. J. Ignatius for valuable discussions regarding the mutation screening array; P. Niini for technical expertise with the arrayer; and M. Levander for excellent laboratory assistance. The following grants made this study possible: the Technology Development Centre of Finland, the Instrumentarium Foundation, EC Biomed2 Contract no. BMH4–972013, The Hjelt Fond of the Pediatric Research Foundation, the Emil Aaltonen Foundation and the Maud Kuistila Foundation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL tomi@orion.ri.mgh.mcgill.ca; FAX (514) 934-8353.
REFERENCES
- Aittomaki K, Lucena JLD, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, Kaskikari R, Sankila EM, Lehväslaiho H, Engel AR, et al. Mutation in the follicle stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82:959–968. doi: 10.1016/0092-8674(95)90275-9. [DOI] [PubMed] [Google Scholar]
- Bell GI, Karam JH, Rutter WJ. Polymorphic DNA region adjacent to the 5′ end of the human insulin gene. Proc Natl Acad Sci. 1981;78:5759–5763. doi: 10.1073/pnas.78.9.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertina RM, Koeleman BPC, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, van der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- Beutler E, West C, Gelbart T. HLA-H and associated proteins in patients with hemochromatosis. Mol Med. 1997;3:397–402. [PMC free article] [PubMed] [Google Scholar]
- Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet. 1999;21:S33–S37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- Bunge S, Kleijer WJ, Steglich C, Beck M, Zuther C, Morris CP, Schwinger, Hopwood JJ, Scott HS, Gal A. Mucopolysaccharidosis type I: Identification of 8 novel mutations and determination of the frequency of the two common α-L-iduronidase mutations (W402X and Q70X) among European patients. Hum Mol Genet. 1994;3:861–866. doi: 10.1093/hmg/3.6.861. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Carninci P, Nishiyama Y, Westover A, Itoh M, Nagaoka S, Sasaki N, Okazaki Y, Muramatsu M, Hayashizaki Y. Thermostabilization and thermoactivation of thermolabile enzymes by trehalose and its application for the synthesis of full length cDNA. Proc Natl Acad Sci. 1998;95:520–524. doi: 10.1073/pnas.95.2.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, Mindrinos M, Richards DR, Sapolsky RJ, Anderson M, Drenkard E, Dewdney J, Reuber TL, Stammers M, Federspiel N, et al. Genome-wide mapping with biallelic markers in Arabidopsis thaliana. Nat Genet. 1999;23:203–207. doi: 10.1038/13833. [DOI] [PubMed] [Google Scholar]
- Collins A, Lonjou C, Newton NE. Genetic epidemiology of single nucleotide polymorphisms. Proc Natl Acad Sci. 1999;96:15173–15177. doi: 10.1073/pnas.96.26.15173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner BJ, Reyes AA, Morin C, Itakura K, Teplitz RL, Wallace RB. Detection of sickle cell beta S-globin allele by hybridization with synthetic oligonucleotides. Proc Natl Acad Sci. 1983;80:278–282. doi: 10.1073/pnas.80.1.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DW. In: The Metabolic Basis of Inherited Disease. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. 1989. pp. 2409–2437. [Google Scholar]
- Cronin MT, Fucini RV, Kim SM, Masino RS, Wespi RM, Miyada CG. Cystic fibrosis mutation detection by hybridization to light-generated DNA probe arrays. Hum Mutat. 1996;7:244–55. doi: 10.1002/(SICI)1098-1004(1996)7:3<244::AID-HUMU9>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Cross NCP, de Franchis R, Sebastio G, Dazzo C, Tolan DR, Gregori C, Odievre M, Vidailhet M, Romano V, Mascali G, et al. Molecular analysis of aldolase B genes in hereditary fructose intolerance. Lancet. 1990;335:306–309. doi: 10.1016/0140-6736(90)90603-3. [DOI] [PubMed] [Google Scholar]
- Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, Osborn AH, Dahl HH, Middleton A, Houseman MJ, et al. Prelingual deafness: High prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet. 1997;6:2173–2177. doi: 10.1093/hmg/6.12.2173. [DOI] [PubMed] [Google Scholar]
- Drobyshev A, Mologina N, Shik V, Pobedimskaya D, Yershov G, Mirzabekov A. Sequence analysis by hybridization with oligonucleotide microchip: identification of β-thalassemia mutations. Gene. 1997;188:45–52. doi: 10.1016/s0378-1119(96)00775-5. [DOI] [PubMed] [Google Scholar]
- Dubiley S, Kirillov E, Mirzabekov A. Polymorphism analysis and gene detection by minisequencing on an array of gel-immobilized primers. Nucleic Acids Res. 1999;27:e19. doi: 10.1093/nar/27.18.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P. Analysis of gene expression in single live neurons. Proc Natl Acad Sci. 1992;92:3010–3014. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estivill X, Bancells C, Ramos C the Biomed CF Mutation Analysis Consortium. Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations. Hum Mutat. 1997;10:135–154. doi: 10.1002/(SICI)1098-1004(1997)10:2<135::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- Farr CJ, Saiki RK, Erlich HA, McCormick F, Marshall CJ. Analysis of RAS gene mutations in acute myeloid leukemia by polymerase chain reaction and oligonucleotide probes. Proc Natl Acad Sci. 1988;85:1629–1633. doi: 10.1073/pnas.85.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Jr, Ellis MC, Fullan A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- The Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet. 1997;17:339–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- Gerry NP, Witowski NE, Day J, Hammer RP, Barany G, Barany F. Universal DNA microarray method for multiplex detection of low abundance point mutations. J Mol Biol. 1999;292:251–262. doi: 10.1006/jmbi.1999.3063. [DOI] [PubMed] [Google Scholar]
- Gilles PN, Wu DJ, Foster CB, Dillon PJ, Chanock SJ. Single nucleotide polymorphic discrimination by an electronic dot blot assay on semiconductor microchips. Nat Biotech. 1999;17:365–370. doi: 10.1038/7921. [DOI] [PubMed] [Google Scholar]
- Guldberg P, Friis Henriksen K, Sipila I, Guttler F, de la Chapelle A. Phenylketonuria in a low incidence population: molecular characterization of mutations in Finland. J Med Genet. 1995;32:976–978. doi: 10.1136/jmg.32.12.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson KL, Huang XC, Morris MS, Lipshutz RJ, Lockhart DJ, Chee MS. Mutation detection by ligation to complete n-mer DNA arrays. Genome Res. 1998;8:1142–1153. doi: 10.1101/gr.8.11.1142. [DOI] [PubMed] [Google Scholar]
- Gunthard HF, Wong JK, Ignacio CC, Havlir DV, Richman DD. Comparative performance of high-density oligonucleotide sequencing and dideoxynucleotide sequencing of HIV type 1 pol from clinical samples. AIDS Res Hum Retrov. 1998;14:869–76. doi: 10.1089/aid.1998.14.869. [DOI] [PubMed] [Google Scholar]
- Guo Z, Guilfoyle RA, Thiel AJ, Wang R, Smith LM. Direct fluorescence analysis of genetic polymorphisms by hybridization with oligonucleotide arrays on glass supports. Nucleic Acids Res. 1994;22:5456–5465. doi: 10.1093/nar/22.24.5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacia JG, Sun B, Hunt N, Edgemon K, Mosbrook D, Robbins C, Fodor SPA, Tagle DA, Collins FS. Strategies for mutational analysis of the large multiexon ATM gene using high-density oligonucleotide arrays. Genome Res. 1998a;8:1245–58. doi: 10.1101/gr.8.12.1245. [DOI] [PubMed] [Google Scholar]
- Hacia JG, Woski SA, Fidanza J, Edgemon K, Hunt N, McGall G, Fodor SPA, Collins FS. Enhanced high density oligonucleotide array-based sequence analysis using modified nucleoside triphosphates. Nucleic Acids Res. 1998b;26:4975–4982. doi: 10.1093/nar/26.21.4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacia JG. Resequencing and mutational analysis using oligonucleotide microarrays. Nat Genet. 1999a;21:42–47. doi: 10.1038/4469. [DOI] [PubMed] [Google Scholar]
- Hacia JG, Fan J-B, Ryder O, Jin L, Edgemon K, Ghandour G, Mayer RA, Sun B, Hsie L, Robbins CM, et al. Determination of ancesteral alleles for human single nucleotide polymorphisms using high-density oligonucleotide arrays. Nat Genet. 1999b;22:164–167. doi: 10.1038/9674. [DOI] [PubMed] [Google Scholar]
- Head SR, Rogers YH, Parikh K, Lan G, Anderson S, Goelet P, Boyce-Jacino MT. Nested genetic bit analysis (N-GBA) for mutation detection in the p53 tumor suppressor gene. Nucleic Acids Res. 1997;25:5065–5071. doi: 10.1093/nar/25.24.5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglund P, Haila S, Socha J, Tomaszewski L, Saarialho-Kere U, Karjalainen-Lindsberg ML, Airola K, Holmberg C, de la Chapelle A, Kere J. Mutations of the down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nat Genet. 1996;14:316–319. doi: 10.1038/ng1196-316. [DOI] [PubMed] [Google Scholar]
- Huopaniemi L, Rantala A, Forsius H, Somer M, de la Chapelle A, Alitalo T. Three widespread founder mutations contribute to high incidence of X-linked juvenile retinoschisis in Finland. Eur J Hum Genet. 1999;7:368–376. doi: 10.1038/sj.ejhg.5200300. [DOI] [PubMed] [Google Scholar]
- Ijist L, Ruiter JPN, Hoovers JMN, Jakobs ME, Wanders RJA. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Clin Invest. 1996;98:1028–1033. doi: 10.1172/JCI118863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E, Baumann M, Grön K, Syvänen A-C, Enomaa N, Halila R, Aula P, Peltonen L. Aspartylglucosaminuria: cDNA encoding human aspartylglucosaminidase and the missense mutation causing the disease. EMBO J. 1991;10:51–58. doi: 10.1002/j.1460-2075.1991.tb07920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Batten Disease Consortium. Isolation of a novel gene underlying Batten disease, CLN3. Cell. 1995;82:949–957. doi: 10.1016/0092-8674(95)90274-0. [DOI] [PubMed] [Google Scholar]
- Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JCF, Trent JM, Staudt LM, Hudson J, Jr, Boguski MS, Lashkari D, Shalon D, Botstein D, Brown PO. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- Jeffrey GP, Chakrabarti S, Hegele RA, Adams PC. Polymorphism in intron 4 of HFE may cause overestimation of C282Y homozygote prevalence in haemochromatosis. Nat Genet. 1999;22:325–326. doi: 10.1038/11892. [DOI] [PubMed] [Google Scholar]
- Kere J, Estivill X, Chillon M, Morral N, Nunes V, Norio R, Savilahti E, de la Chapelle A. Cystic fibrosis in a low-incidence population: two major mutations in Finland. Hum Genet. 1994;93:162–166. doi: 10.1007/BF00210603. [DOI] [PubMed] [Google Scholar]
- Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Molec Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- Kure S, Takayanagi M, Narisawa K, Tada K, Leisti J. Identification of a common mutation in Finnish patients with nonketotic hyperglycinemia. J Clin Invest. 1992;90:160–164. doi: 10.1172/JCI115831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara Y, Narisawa K, Miyabayashi S, Tada K, Coates M, Bachmann C, Elsas LJ, II, Pollitt RJ, Rhead WJ, Roe CR. Identification of a common mutation in patients with medium-chain acyl-CoA dehydrogenase deficiency. Biochem Biophys Res Commun. 1990;171:498–505. doi: 10.1016/0006-291x(90)91421-n. [DOI] [PubMed] [Google Scholar]
- Mitchell GA, Brody LC, Sipila I, Looney JE, Wong C, Engelhardt JF, Patel AS, Steel G, Obie C, Kaiser-Kupfer M, et al. At least two mutant alleles of ornithine δ-aminotransferase cause gyrate atrophy of the choroid and retina in Finns. Proc Natl Acad Sci USA. 1989;86:197–201. doi: 10.1073/pnas.86.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Carninci P, Okazaki Y, Tateno M, Kawai J, Amanuma H, Muramatsu M, Hayashizaki Y. Increased specificity of reverse transcription priming by trehalose and oligo-blockers allows high-efficiency window separation of mRNA display. Nucleic Acids Res. 1999;27:1345–1349. doi: 10.1093/nar/27.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HK, Fournier O, Asseline U, Dupret D, Ngueyn TT. Smoothing of the thermal stability of DNA duplexes by using modified nucleosides and chaotropic agents. Nucleic Acids Res. 1999;27:1492–1498. doi: 10.1093/nar/27.6.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov TT, Rendle RB, Goelet P, Rogers YH, Kotewicz ML, Anderson S, Trainor GL, Knapp MR. Genetic Bit Analysis: a solid phase method for typing single nucleotide polymorphisms. Nucleic Acids Res. 1994;22:4167–4175. doi: 10.1093/nar/22.20.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastinen T, Kurg A, Metspalu A, Peltonen L, Syvänen A-C. Minisequencing: A specific tool for DNA analysis and diagnostics on oligonucleotide arrays. Genome Res. 1997;7:606–614. doi: 10.1101/gr.7.6.606. [DOI] [PubMed] [Google Scholar]
- Pastinen T, Perola M, Niini P, Terwilliger J, Salomaa V, Vartiainen E, Peltonen L, Syvänen A-C. Array-based multiplex analysis of candidate genes reveals two independent and additive genetic risk factors for myocardial infarction in the Finnish population. Hum Mol Genet. 1998a;7:1453–1462. doi: 10.1093/hmg/7.9.1453. [DOI] [PubMed] [Google Scholar]
- Pastinen T, Liitsola K, Niini P, Salminen M, Syvänen A-C. Contribution of the CCR5 and MBL genes in susceptibility to HIV-1 infection in Finnish population. AIDS Res Hum Retrov. 1998b;14:695–698. doi: 10.1089/aid.1998.14.695. [DOI] [PubMed] [Google Scholar]
- Pease AC, Solas D, Sullivan EJ, Cronin MT, Holmes CP, Fodor SP. Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc Natl Acad Sci. 1994;91:5022–5026. doi: 10.1073/pnas.91.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen L, Jalanko A, Varilo T. Molecular genetics of the Finnish disease heritage. Hum Mol Genet. 1999;8:1913–1923. doi: 10.1093/hmg/8.10.1913. [DOI] [PubMed] [Google Scholar]
- Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3′-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88:3698–3703. [PubMed] [Google Scholar]
- Ranta S, Zhang Y, Ross B, Lonka L, Takkunen E, Messer A, Sharp J, Wheeler R, Kusumi K, Mole S, et al. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat Genet. 1999;23:233–236. doi: 10.1038/13868. [DOI] [PubMed] [Google Scholar]
- Sapolsky RJ, Hsie L, Berno A, Ghandour G, Mittmann M, Fan JB. High-throughput polymorphism screening and genotyping with high-density oligonucleotide arrays. Gen Anal. 1999;14:187–192. doi: 10.1016/s1050-3862(98)00026-6. [DOI] [PubMed] [Google Scholar]
- Savukoski M, Klockars T, Holmberg V, Santavuori P, Lander ES, Peltonen L. CLN5, a novel gene encoding a putative transmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis. Nat Genet. 1998;19:286–288. doi: 10.1038/975. [DOI] [PubMed] [Google Scholar]
- Schafer AJ, Hawkins JR. DNA variation and the future of human genetics. Nat Biotech. 1998;16:33–39. doi: 10.1038/nbt0198-33. [DOI] [PubMed] [Google Scholar]
- Shalon D, Smith SJ, Brown PO. A DNA microarray system for analyzing complex DNA samples using two-color fluorescent probe hybridization. Genome Res. 1996;6:639–45. doi: 10.1101/gr.6.7.639. [DOI] [PubMed] [Google Scholar]
- Shumaker JM, Metspalu A, Caskey CT. Mutation detection by solid phase primer extension. Hum Mutat. 1996;7:346–54. doi: 10.1002/(SICI)1098-1004(1996)7:4<346::AID-HUMU9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Southern EM, Maskos U, Elder JK. Analyzing and comparing nucleic acid sequences by hybridization to arrays of oligonucleotides: evaluation using experimental models. Genomics. 1992;13:1008–1017. doi: 10.1016/0888-7543(92)90014-j. [DOI] [PubMed] [Google Scholar]
- Southern EM. DNA chips: analysing sequence by hybridization to oligonucleotides on a large scale. Trends in Genet. 1996;14:1123–1128. doi: 10.1016/0168-9525(96)81422-3. [DOI] [PubMed] [Google Scholar]
- St-Louis M, Leclerc B, Laine J, Salo MK, Holmberg C, Tanguay RM. Identification of a stop mutation in five Finnish patients suffering from hereditary tyrosinemia type I. Hum Mol Genet. 1994;3:69–72. doi: 10.1093/hmg/3.1.69. [DOI] [PubMed] [Google Scholar]
- Syvänen A-C, Ikonen E, Manninen T, Bengtstrom M, Soderlund H, Aula P, Peltonen L. Convenient and quantitative determination of the frequency of a mutant allele using solid-phase minisequencing: application to aspartylglucosaminuria in Finland. Genomics. 1992;12:590–5. doi: 10.1016/0888-7543(92)90452-x. [DOI] [PubMed] [Google Scholar]
- Tang K, Fu D-J, Julien D, Braun A, Cantor CR, Koster H. Chip-based genotyping by mass spectrometry. Proc Natl Acad Sci. 1999;96:10016–10020. doi: 10.1073/pnas.96.18.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrents D, Mykkänen J, Pineda M, Fediubadalo L, Estevez R, de Cid R, Sanjurjo P, Zorzano A, Nunes V, Huoponen K, et al. Identification of SLC7A7, encoding y + LAT-1, as the lysinuric protein intolerance gene. Nat Genet. 1999;21:293–296. doi: 10.1038/6809. [DOI] [PubMed] [Google Scholar]
- Verheijen FW, Verbeek E, Aula N, Beerens CEMT, Havelaar AC, Joosse M, Peltonen L, Aula P, Galjaard H, van der Spek PJ, et al. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet. 1999;23:462–465. doi: 10.1038/70585. [DOI] [PubMed] [Google Scholar]
- Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, Hoffmann SL, Peltonen L. Mutations in the palmitoyl protein thioesterase gene causing infantile neuronal ceroid lipofuscinosis. Nature. 1995;376:584–587. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- Wang DG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, Ghandour G, Perkins N, Winchester E, Spencer J, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science. 1998;280:1077–1082. doi: 10.1126/science.280.5366.1077. [DOI] [PubMed] [Google Scholar]