

Abstract
Organisms with large genomes contain vast amounts of repetitive DNA sequences, much of which is composed of retrotransposons. Amplification of retrotransposons has been postulated to be a major mechanism increasing genome size and leading to “genomic obesity.” To gain insights into the relation between retrotransposons and genome expansion in a large genome, we have studied a 66-kb contiguous sequence at the Rar1 locus of barley in detail. Three genes were identified in the 66-kb contig, clustered within an interval of 18 kb. Inspection of sequences flanking the gene space unveiled four novel retroelements, designated Nikita, Sukkula, Sabrina, and BAGY-2 and several units of the known BARE-1 element. The retroelements identified are responsible for at least 15 integration events, predominantly arranged as multiple nested insertions. Strikingly, most of the retroelements exist as solo LTRs (Long Terminal Repeats), indicating that unequal crossing over and/or intrachromosomal recombination between LTRs is a common feature in barley. Our data suggest that intraelement recombination events deleted most of the original retrotransposon sequences, thereby providing a possible mechanism to counteract retroelement-driven genome expansion.
[The sequence data described in this paper have been submitted to the GenBank data library under accession no. AF254799.]
Genome size in eukaryotes differs considerably both across and within phyla whereas the number of genes in species of the same phyla is generally similar. This phenomenon has been referred to as C value paradox, i.e., variation in genome size (108 to 1011 bp) is not correlated with the complexity of the organism (Thomas 1971). For example, the size of the barley (Hordeum vulgare) genome is 11- and 35-fold larger than that of rice and Arabidopsis, respectively (Bennett and Leitch 1997). The paradox has been resolved by the realization that the share of the genome dedicated to genes is relatively constant in the eukaryotes, whereas the amount of repetitive DNA varies widely even within families of organisms and can represent >70% of some plant genomes (Flavell et al. 1977; Barakat et al. 1997). Much of the repetitive DNA is, in turn, retroelements, which represent 30% of the human and 50% of the maize genome (SanMiguel et al. 1996). To understand the relation of genome structure and repetitive DNAs to genome expansion/contraction, analysis of large contiguous genomic sequences is essential.
In maize, a plant with a large genome, “diagnostic sequencing” was taken to study the intergenic region of a 280-kb interval containing the Adh1-F locus (SanMiguel et al. 1996). Ten retroelement families compose >60% of this region. In contrast, retroelements including Ta11, Ty3-gypsy, Ty1-copia, and Athila families are mainly found at higher density toward the regions flanking centromeres in the small genome plant Arabidopsis (Copenhaver et al. 1999; Lin et al. 1999; Mayer et al. 1999). A comparison of the Adh regions of maize (Zea maize) and sorghum (Sorghum bicolor), having a 3.5-fold smaller genome than that of maize, revealed that an equivalent 78-kb sorghum sequence contains the same nine genes and five additional ones, as the 225-kb maize sequence (Tikhonov et al. 1999). The main difference is the presence in maize of 166 kb (74%) of retrotransposons and 33 miniature inverted-repeat transposable elements (MITEs) (6% of the sequence). These observations provide direct evidence that retroelement proliferation can account for an increase in genome size.
The replicative potential of retrotransposons and their possible role as factors in genome expansion raises the question of what mechanisms might counteract retrotransposon activity or even remove inserted copies to maintain or reduce genome size. Plants may resist retroelement amplification through such mechanisms as DNA methylation in repetitive sequences (Bennetzen et al. 1994) or gene silencing (Ketting et al. 1999). However, mechanisms for removing substantial parts of repetitive elements have not to date been shown in plants to play that role. In Drosophila, however, a high rate of DNA deletions inside retroelements was observed, and this process appears not to be restricted to these elements (Petrov et al. 1996). Recently, loss of retroelements was found to be greater than 40 times faster in Drosophila than Laupala crickets that have a genome 11 times larger than that of Drosophila (Petrov et al. 2000). This indicates that rate of DNA loss can contribute to large differences in genome size in closely related species. Unequal crossing over and/or intrachromosomal recombination between the LTRs (Long Terminal repeats) can remove the internal domain of LTR-containing retrotransposons (Bennetzen and Kellog 1997), creating a single, solo LTR at the excision site. However, a detailed analyses in the above mentioned maize Adh1-F region revealed very few solo LTRs compared to intact elements, suggesting that deletions inside retroelements might not contribute to genome size differences, at least in maize (SanMiguel et al. 1996).
In the barley genome, the BARE-1 retrotransposon is present in 16.6 ± .6 × 103 copies and composes ∼3% of the genome (Vicient et al. 1999). It is transcribed in somatic tissues (Suoniemi et al. 1996) and is translated, processed, and assembled into virus-like particles (Jääskeläinen et al. 1999). Dot blot data and combined restriction mapping and hybridization analyses of BAC (Bacterial Artificial Chromosome) clones indicated that, in barley and other Hordeum species, BARE-1 LTRs vastly outnumber (16 ± 2 ×) BARE-1 elements containing internal domains. (Jääskeläinen et al. 1999; Vicient et al. 1999). These data would be consistent with intraelement LTR recombination generating solo LTRs, at least of BARE-1. However, there are no sequence data yet available to determine the exact nature of these truncated copies nor the means by which they arose.
Here, we report the analysis of a 65,979-bp contiguous sequence from barley chromosome 2HL. Three genes were found to be clustered whereas the majority of the flanking regions consists of highly complex nested retroelement configurations. Most of these configurations represent derivatives of the original elements that are likely to be products of intra- and interelement recombination events. This suggests that recombination between the LTRs of a single or multiple elements occurs frequently in many families of retrotransposons and may contribute to reducing genome size.
RESULTS
Gene Density
Three criteria were used to search for genes in the 66-kb contiguous sequence: homology with characterized genes or ESTs (Expressed Sequence Tags) in the public databases, occurrence of extended high coding probabilities, and application of gene finder programs. We identified three genes in the genomic interval that each match at least two of the three criteria. These were Rar1 (Shirasu et al. 1999) and two homologs of aquaporin genes at positions 56,694, 44,020, and 46,345, respectively (Fig. 1). The deduced amino acid sequences of the latter two proteins reveals a similarity of 71% and identity of 45% to each other. Both proteins are more similar to the δ-type of tonoplast intrinsic proteins from Arabidopsis (similarity 90% and 71%) than to the γ-type (79% and 65%) (Weig et al. 1997). Therefore, we designated these genes Hv-TIP1 (44,020–44,976) and Hv-TIP2 (45,365–46,345), respectively. Hv-TIP1 mRNA was detected in leaf and root tissues by reverse transcription–PCR (Polymerase Chain Reaction) analysis, whereas Hv-TIP2 was undetectable in these tissues (data not shown). Barley genes within the sequenced region were clustered because the Rar1 and aquaporin genes span only 18 kb of the 66-kb region.
Figure 1.

Arrangements of genes and retrotransposons at the barley Rar1 locus. Numbers above the black lines mark nt positions (kb) in the 66-kb contig. Gene exons are shown in blue. Retroelements are shown as color-coded thick lines, and arrows indicate sense direction of each element.
The region next to the two TIP genes (40,261–41,734) shows high sequence similarity to the tnp2 gene that is part of the En/Spm-type transposable element Tam1 from Antirrhinum (Fig. 1) (Nacken et al. 1991). A sequence similar to the reverse transcriptase of the non-LTR retrotransposon Ta11 was found between the TIPs and Rar1 (nt positions 50,003–53,152). These sequences contain multiple stop codons and reveal no other parts of En/Spm or Ta11-like elements, suggesting that they are nonfunctional transposable element genes.
BARE-1 Elements
The contiguous sequence presented here contains five BARE-1 units, four in inverted orientation regarding the sense direction of BARE-1 and one in direct orientation (Figs. 1 and 2B). Complete BARE-1 retrotransposons consist of 1.8-kb LTRs at either end, which contain the elements for transcription and mRNA processing, and an internal domain encoding a polyprotein that is processed into functional units. The organization of BARE-1 and other copia-like retrotransposons is: 5′ LTR — UTL — GAG — AP — IN — RT-RH — UTR (Untranslated Region) — LTR 3′, where UTL is the 5′ untranslated leader, GAG encodes the capsid protein of the virus-like particle, AP encodes an aspartic proteinase, IN encodes the integrase, RT-RH encodes both the reverse transcriptase and RNaseH, and UTR is the 3′ untranslated region (Fig. 2A). Retrotransposon insertion generates direct repeats at the integration site, due to repair of the staggered cuts made by the integrase. These direct repeats therefore are found at the outer edges of the inserted retrotransposon, on the left flank of the left LTR and the right flank of the right LTR.
Figure 2.

Retrotransposon BARE-1 insertions in the 66-kb contig. (A) Organization of a full-length 8.9-kb BARE-1 element as described in the text. The unit is drawn in the sense direction. (B) Organization of the BARE-1 domains on the contig. Arrows represent LTRs numbered as in the text. Black ovals (not to scale) indicate direct repeats found adjacent to the LTRs that were created upon BARE-1 insertion. (C) Proposed ancestral state of current genomic organization. Dotted lines indicate inter-/intrachromosomal recombination events resulting in loss of BARE-1 internal regions. The LTR-3 and -4 complex resulted from the insertion of one BARE-1 into another followed by recombination between two of the LTRs (shown by the arrow). LTR-5 and part of a UTL terminate the clone. (D) Model for recombination between two LTRs, resulting in a single recombinant LTR in the genome and a closed circle, bearing an LTR and the internal domain, which is then lost.
In the sequenced region, the first two BARE-1 units are solo LTRs, LTR-1 (nt 14,967–16,796) and LTR-2 (nt 24,049–25,865), and are flanked by the direct repeats TTCTT and GGTAG, respectively (Fig. 3A). Immediately internal to the direct repeats are the conserved, terminal inverted repeats characteristic of BARE-1 LTRs. The units are complete LTRs showing >90% identity to previously sequenced BARE-1 LTRs, a level of conservation typical for BARE-1 LTRs in the barley genome.
Figure 3.
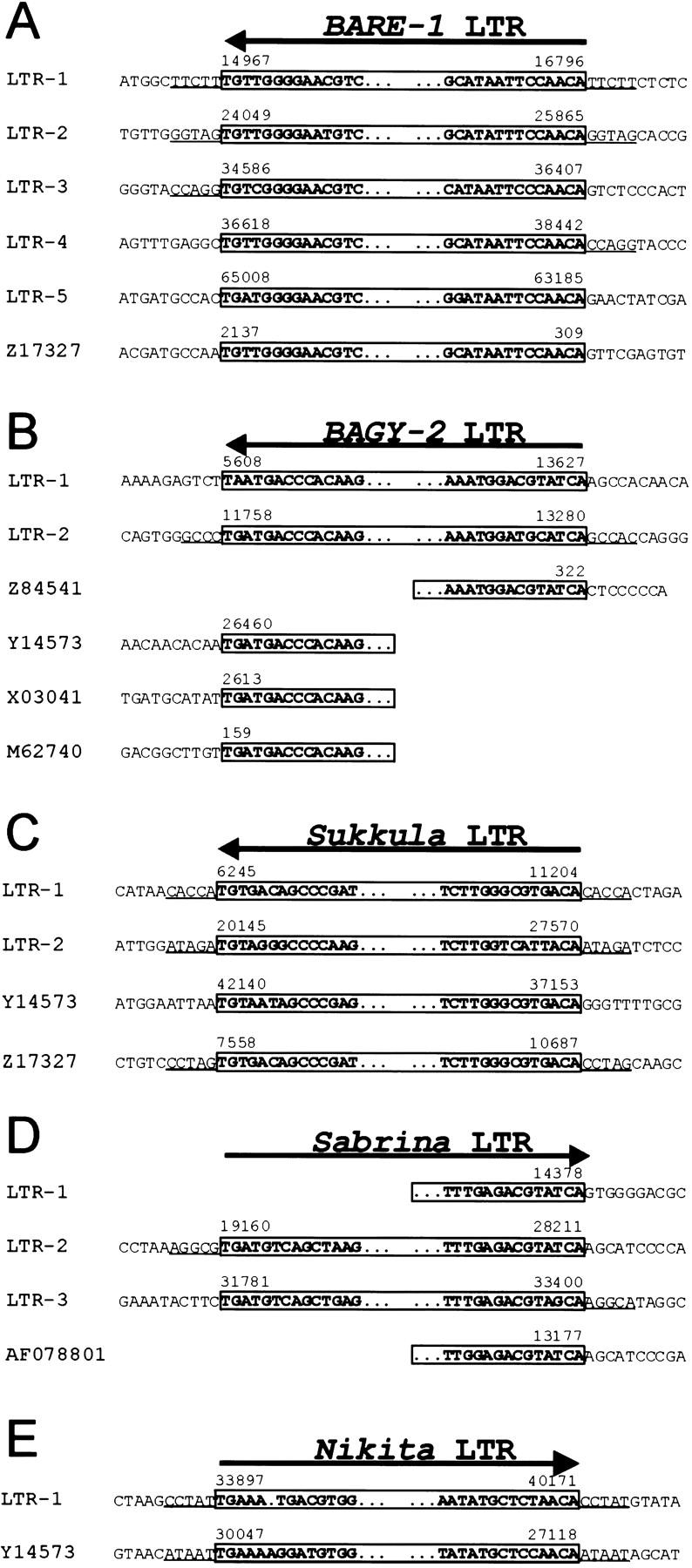
Sequence compilation of terminal LTR sequences. Terminal LTR sequences of BARE-1 (A), BAGY-2 (B), Sukkula (C), Sabrina (D), and Nikita (E) are boxed. Five–-base pair direct repeats flanking the LTRs are underlined. Numbering of LTRs is identical to Figure 1and indicated on the left of each sequence. GenBank accession numbers are given on the left for those LTRs that are not present in the 66-kb contig. Numbers above the DNA sequences refer to nt positions in the 66-kb contig or in other deposited GenBank sequences. Arrows indicate sense direction of each element.
The third and fourth BARE-1 LTRs, LTR-3 and LTR-4, directly flank BARE-1 sequence that consists of the 3′ UTR and part of the RH region (position 34,586–36,407) (Fig. 2B). All of the components of the unit LTR-3 — UTR — RH — LTR-4 are arranged in the same order and orientation as in a full-length BARE-1 element. However, because the junction between the interrupted RH region and LTR-4 occurs precisely at the end of the LTR sequence, it is unlikely that the structure results from an internal deletion of a full-length BARE-1 element. Instead, this observation points to an origin involving an insertion of one BARE-1 element into another, followed by a deletion resulting from recombination between two LTRs, as depicted in Figure 2C. Consistent with this origin is the occurrence of 5-bp direct repeats flanking the entire structure (CCAGG, Fig. 3A), generated by the proposed first insertion event.
The fifth BARE-1 unit (position 63,185–65,979) consists of an LTR (LTR-5) followed by a UTL, highly conserved with respect to BARE-1a (GenBank accession no. Z17327), which is interrupted by the edge of the cloned genomic region.
Gypsy-like Retrotransposons
The region between nt positions 405 and 5501 encodes a protein with similarity to polyproteins from Ty3/Gypsy-like plant retrotransposons, which have the domain order GAG — AP — RT — RH — IN (Fig. 1) (Suoniemi et al. 1998). Because the deduced protein sequence of the polyprotein is 47% similar to the internal domain of the previously described barley BAGY-1 (Panstruga et al. 1998), we designated this element BAGY-2. To make the polyprotein domains into a single contiguous open reading frame, we had to introduce three frame shifts (positions 3071, 3193, and 3648) and a nonsense mutation (position 4160), suggesting that this retroelement does not encode functional enzymes. Sequences in the 3′ direction from the BAGY-2 polyprotein are interrupted by the edge of the 66-kb sequence contig. However, the likely 5′ LTR of BAGY-2 was located between nt 5608 and 13,627 (Fig. 3B). Identical end sequences were found in a number of other deposited barley and wheat sequences, supporting the idea that this is indeed one LTR of BAGY-2 (Fig. 3B). The 5 LTR of BAGY-2 is interrupted by a Sukkula element (see below, between nt 6245 and 11,204) and by the LTR of a second copy of BAGY-2 (between nt 11,758 and 13,280). This second copy seems to represent a solo LTR because its near-identical flanking sequences, GCC(A)C and GCCAC, are likely to represent the 5-bp direct repeats of the original insertion event (Figs. 1 and 3B).
Sukkula Elements
The interval between nt positions 6245 and 11,204 shows sequence similarity to intergenic sequences at the barley Mlo locus and to an insertion sequence present in the 3′ LTR of one BARE-1 element (GenBank accession nos. Y14573 and Z17327, respectively; Fig. 1) (Manninen and Schulman 1993). A compilation of these three ∼5-kb long sequences revealed stretches with varying degrees of sequence similarity (data not shown). A segment of 1788 bp appears to be internally deleted from the copy that resides in the BARE-1 LTR. The insertion in the BARE-1 LTR is flanked by a 5-bp direct repeat, CCTAG, typical of retroelement integration sites (Fig. 3C). Likewise, the 5-kb sequence at the Rar1 locus is also flanked by 5-bp direct repeats, in this case CACCA (Fig. 3C), suggesting that the related 5-kb sequences represent retrotransposon derivatives. Here, we name the insertion sequences Sukkula (pronounced sook-koo-la), which means “shuttle” in Finnish. The terminal sequences of Sukkula are strikingly similar to LTR terminal regions of gypsy-like retrotransposons (RIRE) described recently in rice (Kemekawa et al. 1999). Although none of the Sukkula copies in the contig contains sequences encoding corresponding gypsy-like polyproteins adjacent to the LTR sequences, we have identified Sukkula internal domains elsewhere in the barley genome (data not shown). Thus, it is likely that the Sukkula sequences in the contig represent solo LTRs of a novel barley retrotransposon that were generated by recombination between two LTRs of the original element.
More evidence for the insertional activity of Sukkula was found between nt 20,145 and 27,570, where another remnant of this retroelement interrupts a Sabrina element (see below, Fig. 1). A 5-bp direct repeat, ATAGA, was identified at positions 20,145 and 27,570 suggesting that this Sukkula copy represents a further solo LTR (Fig. 3C). This Sukkula copy was, in turn, interrupted by a BARE-1 solo LTR and by a large region between nt 20,145 and 24,049 with no sequence similarities to any other elements, suggesting a set of nested solo-LTRs and possibly other insertions in this region.
Sabrina Elements
Sequences in a number of regions (nt 14,378–17,590, 19,160–28,211, and 31,781–33,400) are highly similar to sequences flanking the insertion site of a Cerebra retroelement in barley (AF078801) (Figs. 1 and 3D). Several lines of evidence suggest that these are fragments of yet another retroelement that we designate Sabrina. Imperfect 5-bp direct repeats (AGGCG/AGGCA) directly flank Sabrina sequences present between positions 19,160 and 33,401, thereby possibly defining a Sabrina integration site. Indeed, sequences next to the 5-bp direct repeats are related but in inverse orientation, appropriately positioned to be the diverged terminal LTR sequences of this element (Fig. 3D). Two further putative Sabrina terminal LTR sequences (positions 28,211 and 31,781) were found within the ∼14-kb interval bordered by the imperfect 5-bp direct repeats (Fig. 3D). The orientation of these terminal sequences relative to those next to the 5-bp direct repeats indicates the presence of two Sabrina LTRs, designated LTR-2 and LTR-3 (Fig. 1). The Sabrina LTR-2 is disrupted by Sukkula LTR-2 and by BARE-1 LTR-2. A third potential Sabrina LTR, LTR-1, was identified at positions 14,378–17,590 although the 5′ end of the LTR appears to be truncated (Figs. 1 and 3D). This Sabrina LTR was found to be interrupted by the BARE-1 LTR-1 (Fig. 1). Finally, almost identical Sabrina terminal LTR sequences were found in the deposited genomic sequence adjacent to the insertion of a Cerebra retroelement (AF078801; Fig. 3D).
Nikita Elements
Another likely solo LTR element was located between nt positions 33,897 and 40,171. These sequences are highly related to an ∼3-kb stretch at the Mlo locus (nt positions 30,047 and 27,118 in Y14573) (Fig. 3E). We designated the element Nikita. In both cases, 5-bp direct repeats were found at the insertion sites (CCTAT and ATAAT, respectively; Fig. 3E). The Nikita element at the Rar1 locus is interrupted by two BARE-1 LTRs (LTR-3 and LTR-4) at nt positions 34,586 and 38,442 (Fig. 1), whereas it is contiguous at the Mlo locus.
Stowaway Elements
In addition to retrotransposon-like sequences, a 160-bp inverted repeat element was found flanking the 3′ region of TIP2 (nt 47,279–47,443). On the basis of sequence similarity, this element belongs to the Stowaway family (Bureau and Wessler 1994). Like Tourist elements, Stowaway family members are small elements (ranging from ∼50 to 300 bp) that lack coding potential but share conserved terminal inverted repeats and the potential to form DNA secondary structures (Bureau and Wessler 1994). They are thought to be deletion derivatives of autonomous type II (DNA) transposable elements. A further Stowaway element (112 bp) was found directly beyond the 3′ end of the Rar1 gene (nt 61,060–61,170). The location of the two Stowaway elements in the sequence contig is consistent with findings in other plant species indicating a tight association with genes (Bureau and Wessler 1994). Indeed, analysis of two other large genomic barley sequence contigs, the Mlo (Y14573) and the Mla locus (Wei et al. 1999), identified Stowaway elements only in tight association with genes (data not shown).
DISCUSSION
In-depth analysis of a 66-kb contiguous stretch of barley chromosome 2HL has produced insights into gene density, gene organization, and a complex structure of intergenic sequences. These data allow us to infer models for the evolution of complex grass genomes.
Our analysis has identified at least three genes within the 66-kb contig, yielding a density of approximately one gene per 20 kb. This is almost identical to the density described in the only other published large contiguous barley genomic interval, a 60-kb stretch at Mlo on chromosome 4HL (Panstruga et al. 1998). This 60-kb stretch harbors three genes that are clustered within an interval of 18 kb. Despite the limitations of computer-aided gene identification (Fickett 1996) and the uncertainties of whether the two genomic intervals are representative for the barley genome, patterns are recognizable from the sequence contigs. First, the observed gene density at both loci is 6–10-fold higher than expected from an equidistant gene distribution in the 5300-Mb barley genome (Bennett and Leitch 1997; Bevan et al. 1998; Panstruga et al. 1998). Second, if one considers only the intervals of the sequence contigs harboring genes at Rar1 and Mlo, the density is approximately one gene per 6 kb. This is only marginally different from the 4.8 kb calculated as the mean of a 1.9-Mb contiguous sequence of the small-size genome of Arabidopsis (Bevan et al. 1998) and similar to the density seen at the orthologous Lrk/Tak loci in wheat, barley, and rice (Feuillet and Keller 1999). Third, with the exception of a Ta11-like reverse trascriptase sequence and a Nikita element that were located between genes at the Rar1 and Mlo loci, respectively, it appears that the gene space is largely void of transposable element sequences. Our data therefore imply a clustering of genes and support the idea of ‘gene islands’ in the barley genome, given the fact that the Arabidopsis genome is ∼35-fold less complex and estimates of gene number in higher plants vary only between 25,000 and 43,000 (Miklos and Rubin 1996).
Analysis of the 66-kb contiguous stretch on barley chromosome 2HL reveals an extraordinarily complex structure of sequences flanking the gene island. At least four of the five identified BARE-1 elements have undergone recombination events, leaving only 1.8-kb remnants of the large 8.9-kb retroelement at the Rar1 locus. Solo LTRs may result from unequal crossing over or from intrachromosomal recombination between nearby LTRs. In the latter, recombination between the LTRs would result in circularization and deletion of the internal region and simultaneous production of a hybrid LTR containing the left end of the left LTR and the right end of the right LTR (Fig. 2D). The sequence analysis supports the large LTR excesses recently reported for the barley and other genus Hordeum genomes (Vicient et al. 1999). The structures of the BARE-1 retroelements found on the LTR-3 — LTR-4 stretch of chromosome 2H reflect both the phenomena of nested retrotransposon insertion (SanMiguel et al. 1996; Suoniemi et al. 1997).
However, analysis of the contig indicates that not only BARE-1 but also the novel retroelements Sukkula, Sabrina, Nikita, and BAGY-2 all have undergone recombination events at the Rar1 locus, leaving in each case only remains of the original elements as solo LTRs and the products of nested insertions. In contrast to BARE-1 elements, the newly identified elements are generally interrupted by nonrelated retrotransposons. For example, the arrangement of the unit Sabrina LTR-2 — LTR-3 can be interpreted as an internal deletion of a full-length Sabrina element, as evidenced by the flanking imperfect 5-bp direct repeats (AGGCG/A) (Fig. 3D). Alternatively, the unit may reflect a nested Sabrina insertion that was followed by an inter-/intrachromosomal recombination between the LTR of the original element and the LTR of the newly inserted Sabrina element (Fig. 4). Thus, the unit Sabrina LTR-2 — LTR-3 may represent a structure equivalent to the one described above for the BARE-1 unit LTR-3 — LTR-4. If this is the case, then we predict that the unaccounted sequence space between nt positions 28,212 and 31,780 is likely to represent non-LTR Sabrina DNA.
Figure 4.

Possible integration and recombination events generating Sabrina unit LTR-2 — LTR-3. Two Sabrina elements are shown in dark and bright gray with the arrows denoting the LTRs. Nonelement DNA is in black with the ovals representing 5-bp direct repeats flanking Sabrina LTR-2 and LTR-3. The broken arrow line shows a deduced intraelement recombination event; dotted lines indicate corresponding positions; thin black lines denote a nested Sabrina insertion.
Unlike the BARE-1 unit LTR-3 — LTR-4, a Sukkula insertion has interrupted the Sabrina LTR-2, which, in turn, is interrupted by a BARE-1 insertion (BARE-1 LTR-2) (Fig. 1). Both Sukkula LTR-2 and BARE-1 LTR-2 insertions have flanking 5-bp direct repeats, strongly suggesting that both LTR structures are the result of intraelement recombination events that deleted most of the original retrotransposon sequences. However, we were unable to annotate sequences between nt positions 20,200 and 24,048, but it is possible that they represent the insertion of a further, as yet unknown, element into Sukkula LTR-2.
Taken together, our data identified at least 15 insertion events from retrotransposons and transposon-like elements within the 66-kb contig (Fig. 5). As many as four elements were found to be nested into each other, comparable to nested insertions seen in the maize genome (SanMiguel et al. 1996). It appears that the frequency of nested insertions of distinct retroelements is similar to that of nested insertions of sequence-homologous partners into each other (five versus three, respectively). The insertions of unrelated elements into each other allows, by the nesting order depicted in the simplified diagram in Figure 5, a consideration of the relative insertional activity and abundance of certain elements at various times. None of the five BARE-1 insertion sequences in the contig is interrupted by heterologous retroelement sequences. The BARE-1 retroelement family is abundant throughout Hordeum (Vicient et al. 1999), is present and insertionally polymorphic in other Triticeae genera (Gribbon et al. 1999) and is closely related to RIRE-1 in the phylogenetically distant rice (Noma et al. 1997). Hence, BARE-1 appears to be both an ancient and an active retrotransposon. Its recent activity is supported by the observation that all available flanking 5-bp direct repeats are perfectly conserved (Fig. 3B). In contrast, we infer that Sabrina elements represent ancient but recently inactive elements because they were found to be disrupted by both Sukkula and BARE-1 elements (Fig. 5).
Figure 5.

Deduced nesting order of retroelements. Symbols and color codes correspond to Figure 1.
Although nested retrotransposon insertion appears also characteristic of the maize genome (SanMiguel et al. 1996), the findings here are remarkable for the prevalence of solo LTRs. This implies that in barley the conversion to solo LTRs occurs more rapidly than integration. Regional recombinational differences are known to occur in complex plant genomes. However, at least for BARE-1, amounting to 2.9% of the barley genome, there is direct evidence that the rapid conversion to solo LTRs is not restricted to the Rar1 locus but operates throughout the whole genome (Vicient et al. 1999). Work with Nicotiana with repeats of varying length (Puchta and Hohn 1991) indicates that recombination between elements as long as BARE-1 LTRs (1.8 kb) may be quite efficient. A detailed analysis of a 280-kb interval in maize flanking the Adh1 and u22 genes indicates very few solo LTRs relative to intact elements (SanMiguel et al. 1996). Of the 16 named families of maize retrotranspsosons in the current GenBank database, only five have LTRs longer than 750 bases, the others averaging 450 ± 65 bp (s.e.m.). This contrasts markedly with the LTRs on the barley contig (BARE-1 1.8 kb; BAGY-2 1.5 kb; Sukkula 4.9 kb; Sabrina 1.6 kb; Nikita 2.9 kb). The two solo LTRs reported for maize (SanMiguel et al. 1996) both are of Ji elements, having LTRs of 1156 bp (GenBank accession no. U68405). Hence, one explanation for the low frequency of solo LTRs in the maize genome may be a lower recombinational efficiency between the comparatively short LTRs of maize retroelements.
Recombination between nearby LTRs offers a means to reverse the increase in genome size caused by successive integrations of large retrotransposons. Were recombination to occur between homologous LTRs of different retroelement copies, the intervening genomic DNA would also be removed irrespective of whether the recombination is inter- or intrachromosomal. Indeed, the units BARE-1 LTR-3 — LTR-4 and Sabrina LTR-2 —LTR-3 provide strong evidence that entire elements have been eliminated at Rar1. Although this might be deleterious if a gene was between the copies, nested retrotransposon insertion or insertion into repetitive DNA would not present this hindrance. The organization of the contig suggests that extensive intra-/interchromosomal recombination has acted to delete integrated retroelements. These data may help explain the observation that, in the phylogeny of diploid grasses, decreases in genome size have possibly occurred with the same frequency as size increases (Bennetzen and Kellog 1997).
METHODS
DNA Manipulations and Computer Analysis
Physical delimitation of the Rar1 locus has been performed previously (Lahaye et al. 1998). A fivefold redundancy of independent YAC (Yeast Artificial Chromosome) clones and a greater than fourfold redundancy of BAC clones cover the relevant genomic region (Shirasu et al. 1999). DNA fingerprinting of multiple overlapping and independent YAC and BAC clones revealed absence of any rearrangements in each tested clone in the sequenced area. The contiguous DNA sequence of 65,979 bp from barley chromosome 2HL has been deposited at GenBank under accession number AF254799. Briefly, BAC 1B2 and BAC12 covering this interval were completely sequenced by means of random shotgun cloning in the sequencing vector, pBluescript II KS+. A 49-bp gap between BAC 1B2 and BAC12 was closed by using PCR amplification with end sequences of these two BACs, on template DNA of BAC 3H6, and subsequent direct sequencing of the amplicon. Sequence homology analyses were performed using BLAST2 software available from the National Center for Biotechnology Information (http://www.ncbi.nim.nih.gov). Putative exons were predicted by the programs GENSCAN 1.0 (http://gnomic.stanford.edu/∼chris/GENSCANW.html), NetPlantGene V2.0 (http://www.cbs.dtu.dk/NetPlantGene.html), and Grail (http://compbio.ornl.gov/Grail-1.3).
Acknowledgments
We thank Nicholas Collins and Louise Jones for constructive criticism of this manuscript and Margaret Shailer for technical assistance. This work was supported by grants from the GATSBY Charitable Foundation and the Biotechnology and Biological Sciences Research Council to P. S.-L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL schlef@bbsrc.ac.uk; FAX 44 1603 450011.
REFERENCES
- Barakat A, Carels N, Bernardi G. The distribution of genes in the genomes of Gramineae. Proc Natl Acad Sci USA. 1997;94:6857–6861. doi: 10.1073/pnas.94.13.6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MD, Leitch IJ. Nuclear DNA amounts in angiosperms—583 new estimates. Ann Bot. 1997;80:169–196. [Google Scholar]
- Bennetzen JL, Kellog EA. Do plants have a one-way tickets to genomic obesity? Plant Cell. 1997;9:1509–1514. doi: 10.1105/tpc.9.9.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Schrick K, Springer PS, Brown WE, SanMiguel P. Active maize genes are unmodified and flanked by diverse classes of modified, highly repetitive DNA. Genome. 1994;37:565–576. doi: 10.1139/g94-081. [DOI] [PubMed] [Google Scholar]
- Bevan M, Bancroft I, Bent E, Love K, Goodman H, Dean C, Bergkamp R, Dirkse W, Van Staveren M, Stiekema W, et al. Analysis of 1.9 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature. 1998;391:485–488. doi: 10.1038/35140. [DOI] [PubMed] [Google Scholar]
- Bureau TE, Wessler SR. Stowaway—a new family of inverted repeat elements associated with the genes of both monocotyledonous and dicotyledonous plants. Plant Cell. 1994;6:907–916. doi: 10.1105/tpc.6.6.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver GP, Nickel K, Kuromori T, Benito MI, Kaul S, Lin XY, Bevan M, Murphy G, Harris B, Parnell LD, et al. Genetic definition and sequence analysis of Arabidopsis centromeres. Science. 1999;286:2468–2474. doi: 10.1126/science.286.5449.2468. [DOI] [PubMed] [Google Scholar]
- Feuillet C, Keller B. High gene density is conserved at syntenic loci of small and large grass genomes. Proc Natl Acad Sci USA. 1999;96:8265–8270. doi: 10.1073/pnas.96.14.8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickett JW. Finding genes by computer: The state of the art. Trends Genet. 1996;12:316–320. doi: 10.1016/0168-9525(96)10038-x. [DOI] [PubMed] [Google Scholar]
- Flavell RB, Rimpau J, Smith DB. Repeated sequence DNA relationships in four cereal genomes. Chromosoma. 1977;63:205–222. [Google Scholar]
- Gribbon BM, Pearce SR, Kalendar R, Schulman AH, Paulin L, Jack P, Kumar A, Flavell AJ. Phylogeny and transpositional activity of Ty1-copia group retrotransposons in cereal genomes. Mol Gen Genet. 1999;261:883–891. doi: 10.1007/pl00008635. [DOI] [PubMed] [Google Scholar]
- Jääskeläinen M, Mykkänen A-H, Arna T, Vicient C, Suoniemi A, Kalendar R, Savilahti H, Schulman AH. Retrotransposon BARE-1: Expression of encoded proteins and formation of virus-like particles in barley cells. Plant J. 1999;20:413–422. doi: 10.1046/j.1365-313x.1999.00616.x. [DOI] [PubMed] [Google Scholar]
- Kemekawa N, Ohtsubo H, Horiuchi T, Ohtsubo E. Identification and characterization of novel retrotransposons of the gypsy type in rice. Mol Gen Genet. 1999;260:593–602. doi: 10.1007/s004380050933. [DOI] [PubMed] [Google Scholar]
- Ketting RF, Haverkamp THA, van Luenen HGAM, Plasterk RHA. mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner Syndrome helicase and RNaseD. Cell. 1999;99:133–141. doi: 10.1016/s0092-8674(00)81645-1. [DOI] [PubMed] [Google Scholar]
- Lahaye T, Shirasu K, Schulze-Lefert P. Chromosome landing at the barley Rar1 locus. Mol Gen Genet. 1998;260:92–101. doi: 10.1007/s004380050874. [DOI] [PubMed] [Google Scholar]
- Lin XY, Kaul SS, Rounsley S, Shea TP, Benito MI, Town CD, Fujii CY, Mason T, Bowman CL, Barnstead M, et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999;402:761–768. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- Manninen I, Schulman AH. BARE-1, a copia-like retroelemnt in barley (Hordeum vulgare L.) Plant Mol Biol. 1993;22:829–846. doi: 10.1007/BF00027369. [DOI] [PubMed] [Google Scholar]
- Mayer K, Schuller C, Wambutt R, Murphy G, Volckaert G, Pohl T, Dusterhoft A, Stiekema W, Entian KD, Terryn N, et al. Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana. Nature. 1999;402:769–777. doi: 10.1038/47134. [DOI] [PubMed] [Google Scholar]
- Miklos GLG, Rubin GM. The role of the genome project in determining gene function: Insights from model organisms. Cell. 1996;86:521–529. doi: 10.1016/s0092-8674(00)80126-9. [DOI] [PubMed] [Google Scholar]
- Nacken WKF, Piotrowiak R, Saedler H, Sommer H. The transposable element Tam1 from Antirrhinum majus shows structural homology to the maize transposon En/Spm and has no sequence sepcificity of insertion. Mol Gen Genet. 1991;228:201–208. doi: 10.1007/BF00282466. [DOI] [PubMed] [Google Scholar]
- Noma K, Nakajima R, Ohtsubo H, Ohtsubo E. RIRE1, a retrotransposon from wild rice Oryza australiensis. Gen Genet Syst. 1997;72:131–140. doi: 10.1266/ggs.72.131. [DOI] [PubMed] [Google Scholar]
- Panstruga R, Büschges R, Piffanelli P, Schulze-Lefert P. A contiguous 60 kb genomic stretch from barley reveals molecular evidence for gene islands in a monocot genome. Nucleic Acids Res. 1998;26:156–1062. doi: 10.1093/nar/26.4.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov DA, Lozovskaya ER, Hartl DL. High intrinsic rate of DNA loss in Drosophila. Nature. 1996;384:346–349. doi: 10.1038/384346a0. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Sangster TA, Johnston JS, Hartl DL, Shaw KL. Evidence for DNA loss as a determinant of genome size. Science. 2000;287:1060–1062. doi: 10.1126/science.287.5455.1060. [DOI] [PubMed] [Google Scholar]
- Puchta H, Hohn B. A transient assay in plant cells reveals a positive correlation between extrachromosomal recombination rates and the length of homologous overlap. Nucleic Acids Res. 1991;19:2693–2700. doi: 10.1093/nar/19.10.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— From centiMorgans to base pairs: Homologous recombination in plants. Trends Plant Sci. 1996;1:340–348. [Google Scholar]
- SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, et al. Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996;274:765–768. doi: 10.1126/science.274.5288.765. [DOI] [PubMed] [Google Scholar]
- Shirasu K, Lahaye L, Tan M-W, Zhou F, Azevedo C, Schulze-Lefert P. A novel class of eukaryotic zinc-binding protein is required for disease resistance signaling in barley and development in C. elegans. Cell. 1999;99:355–366. doi: 10.1016/s0092-8674(00)81522-6. [DOI] [PubMed] [Google Scholar]
- Suoniemi A, Narvanto A, Schulman A. The BARE-1 retrotransposon is transcribed in barley from an LTR promoter active in transient assays. Plant Mol Biol. 1996;31:295–306. doi: 10.1007/BF00021791. [DOI] [PubMed] [Google Scholar]
- Suoniemi A, Schmidt D, Schulman AH. BARE-1 insertion site preferences and evolutionary conservation of RNA and cDNA processing sites. Genetica. 1997;100:219–230. [PubMed] [Google Scholar]
- Suoniemi A, Tanskanen J, Schulman AH. Gypsy-like retrotransposons are widespread in the plant kingdom. Plant J. 1998;13:699–705. doi: 10.1046/j.1365-313x.1998.00071.x. [DOI] [PubMed] [Google Scholar]
- Thomas CA. The genetic organization of chromosomes. Annu Rev Genet. 1971;5:237–256. doi: 10.1146/annurev.ge.05.120171.001321. [DOI] [PubMed] [Google Scholar]
- Tikhonov AP, SanMiguel PJ, Nakajima Y, Gorenstein NM, Bennetzen JL, Avramova Z. Colinearity and its exceptions in orthologous adh regions of maize and sorghum. Proc Natl Acad Sci USA. 1999;96:7409–7414. doi: 10.1073/pnas.96.13.7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicient CM, Suoniemi A, AnamthamatJonsson K, Tanskanen J, Beharav A, Nevo E, Schulman AH. Retrotransposon BARE-1 and its role in genome evolution in the genus Hordeum. Plant Cell. 1999;11:1769–1784. doi: 10.1105/tpc.11.9.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei FS, Gobelman-Werner K, Morroll SM, Kurth J, Mao L, Wing R, Leister D, Schulze-Lefert P, Wise RP. The Mla (powdery mildew) resistance cluster is associated with three NBS-LRR gene families and suppressed recombination within a 240-kb DNA interval on chromosome 5S (1HS) of barley. Genetics. 1999;153:1929–1948. doi: 10.1093/genetics/153.4.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weig A, Deswarte C, Chrispeels MJ. The major intrinsic protein family of Arabidopsis has 23 members that form three distinct groups with functional aquaporins in each group. Plant Physiol. 1997;114:1347–1357. doi: 10.1104/pp.114.4.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]