

Abstract
In this work, we explore the use of representations in conjunction with DNA microarray technology to measure gene copy number changes in cancer. We demonstrate that arrays of DNA probes derived from low-complexity representations can be used to detect amplifications, deletions, and polymorphic differences when hybridized to representations of genomic DNA. The method is both reproducible and verifiable, and is applicable even to microscopic amounts of primary tumors. We also present a mathematical model for array performance that is useful for designing and understanding DNA microarray hybridization protocols. The future applications and challenges of this approach are discussed.
Karyotyping, determination of ploidy, and comparative genomic hybridization — although they are crude methods for genomic analysis — provide insight into the molecular basis of cancer as well as useful clinical guides to its diagnosis and treatment. Even more can be expected of molecular techniques that describe in great detail the many changes of the cancer cell. Profiling the RNA expression pattern in cancer cells by application of microarray technology is undoubtedly one such method (DeRisi et al. 1996; Khan et al. 1999; Sgroi et al. 1999; Wang et al. 1999). However, RNA profiling does have substantial and widely recognized limits: there is no standard RNA profile for comparison; RNA is not a stable molecule; the physiological state of the cancer at biopsy is very variable; and primary genetic lesions are not revealed. Thus, it is desirable to obtain a high-resolution image of the primary genetic changes that do occur in cancers. DNA is a far more stable component of the cancer cell, which does not vary as a function of the physiological state of the cell, and there is an absolute standard for comparison, namely the normal genome.
Two methods that can detect amplifications and deletions in the cancer cell at high resolution are in development, and their principles of operation now have been described (Pinkel et al. 1998; Pollack et al. 1999). We demonstrate here a third method that we believe has advantages over the first two.
The first method utilizes microarrayed bacterial artificial chromosome (BAC) DNAs (Pinkel et al. 1998). Total DNA from tumor and normal genomes, each labeled with different fluorochromes, are simultaneously hybridized to these arrays and the ratios of hybridization measured. Alterations in gene copy number in the tumor DNA are detected as a deviation of the ratio from the mean. The advantages of this method are that it is highly quantitative and sensitive, as a result of the integration of hybridization signal over very long probes; and that it utilizes reagents, namely elements of BAC libraries, that will at some time in the future be completely characterized and mapped. Its disadvantages are that the task of preparing a sufficient number of BAC DNAs to cover the genome is daunting; the method utilizes microarray printing technologies that are not commonplace; the theoretical resolution of the method is limited by the size of BACs; and, although the method can detect large deletions, it cannot detect loss of polymorphisms which are excellent and reliable markers for allelic loss.
The second method utilizes microarrayed cDNAs and ESTs (Pollack et al. 1999). Total genomic DNA from tumor and normal genomes is simultaneously hybridized to these arrays, and the ratios of hybridization signal are measured. Highly amplified regions of the genome, and perhaps some deletions, can be detected. This method uses commercially available collections of cDNAs and ESTs, along with a microarray printing technology that is readily available to the biomedical community. Many of the ESTs already have been mapped. The disadvantages of this method are that the signal-to-noise ratio is poor because of the complexity of the total human genome and the short length of arrayed probes. Hence, the signal is integrated over several probes with known map locations in order to allow a clear pattern of altered gene copy number to emerge. Although the theoretical limit of resolution is the gene itself, the practical resolving power of this method is likely to be poor because the signal is averaged over several adjacent probes; the cDNA collections are not complete; and not all of the ESTs currently are mapped. Our method, like the others, is ratiometric. However, rather than hybridizing total genomic DNA, we hybridize genomic representations of tumor and normal DNA. Representations are reproducible samplings of DNA populations in which the resulting DNA typically has a new format or reduced complexity or both (Lisitsyn et al. 1993; Lucito et al. 1998). The usefulness of genomic representations rests on five properties: representations with much lower nucleotide complexity than the entire genome can be made, and as such will have hybridization kinetics superior to that of the complete genome; they are reproducible; they can be prepared in large amounts from microscopic amounts of material; the parallel representations preserve gene ratios between genomes; and they can reflect genetic polymorphism because they can be made sensitive to restriction endonuclease cleavage. The experiments presented in this work confirm all these properties.
Most of our experience with representations derives from the use of this method in representational difference analysis (RDA), a method for cloning differences between two similar DNA populations (Lisitsyn et al. 1993, 1995). In most cases, we have made representations by cleaving DNA with a restriction endonuclease, ligation of the cleaved products to template oligonucleotides, and then polymerase chain reaction (PCR) amplification using complementary oligonucleotides. Complexity reduction results from the preferential amplification of small (<1 kb) DNA fragments during PCR. The degree of complexity reduction in a representation is determined mainly by the choice of restriction endonucleases used in its preparation. Using a restriction endonuclease that cleaves frequently, such as DpnII, one can derive high-complexity representations (HCRs) that contain about 70% of the genome (Lucito et al. 1998). When using restriction endonuclease, such as BglII, which cuts less frequently, we obtain low-complexity representations (LCRs) that contain about 2.5% of the genome.
We array probes derived from a LCR of a standard human genome. We then hybridize these microarrays with LCRs of paired samples, one normal and one cancer. There are many advantages to this approach. Because LCRs have lower nucleotide complexity than total genomic DNA, we obtain a strong specific hybridization signal relative to nonspecific hybridization and noise, and are able to readily detect deletions, amplifications, and polymorphic differences in samples using short probes. Our resolution is limited only by the number of probes that can be microarrayed, and does not depend upon knowledge of the complete set of genes. Moreover, we can reliably detect allelic losses. Because the method is based on representations, samples can be prepared from microscopic amounts of tissue (Lucito et al. 1998). The probe collection can be maintained as cultures of individual bacterial clones, and produced for printing by PCR. Finally, the methods for arraying, labeling, and hybridizing are the same ones in common use for cDNA analysis.
Using two different pilot arrays of 1000–2000 small BglII fragments, we demonstrate that the method yields reproducible and verifiable results. We demonstrate the utility of our method for the analysis of microscopic amounts of material from a tumor biopsy, and examine the critical parameter of nucleotide complexity. We develop a useful mathematical model for predicting array performance, and discuss issues related to the application of our method on a larger scale.
RESULTS
Reproducibility of Array Hybridization Data
Any measuring tool must satisfy the criterion of reproducibility. Microarray hybridization has been extensively tested, and because we use it to measure gene ratios between two samples, it is particularly robust. However, we have introduced the added element of representation during the preparation of samples. We therefore have tested the reproducibility of our measurements when independent representations are made from the same DNA source and hybridized to microarrays.
For this series of experiments, we used DNA from a human breast cancer cell line, SKBR-3, and made multiple parallel BglII representations on separate days. These were separately labeled with Cy3 or Cy5, the two fluorochromes commonly used for this purpose, and hybridized in pairs to pilot arrays. The pilot arrays contained 1658 human BglII fragments, of size range 200–1000 bp, printed in duplicate, for a total of 3316 features. Figure 1A shows a plot of the normalized ratio of the channel intensities as a function of the intensity in one channel (Cy3) for each feature. For symmetry, we plotted the ratio of Cy5 to Cy3 channels above the median (if greater than one; otherwise, we plotted the inverse ratio below the median). The range of intensities is great, spanning at least 25-fold. We believe this spread is the result of a number of factors including inefficient and variable cross-linking of printed probes to the slide surface, variable retention of signal during the washing of the arrays, unequal amplification of certain sequences during representation, and the presence of repeat sequences in some of our probes.
Figure 1.

Results of microarray experiments graphed so that the intensity of one channel (usually the Cy3 channel) is the abscissa and the ratio of Cy5:Cy3 is the ordinate. (A) BglII representations were produced separately from the same source of genomic DNA, differentially labeled, and then hybridized to an array of 3316 features (1658 printed in duplicate). (B) One BglII representation was differentially labeled and then hybridized to the microarray described in A. (C) A breast primary tumor was separated into normal and tumor nuclei by sorting, and genomic DNA was prepared. BglII representations prepared from the genomic DNA were differentially labeled and then hybridized to the microarray described in A. Filled circled represent the limit of measurement for the scanner.
Nevertheless, there is a minimum scatter of ratios throughout a wide range of channel intensities; the ratios of channel intensity are approximately constant throughout the entire range. Only six ratios were outside of the range of 1.5, and none were outside the range of 2.0. Essentially the same results were obtained in three separate experiments.
For comparison, we hybridized the same representation to itself. A single BglII representation was labeled with Cy3 and then again with Cy5, mixed, and each hybridized to an array of the same probes. Figure 1B is plotted in the same manner as Figure 1A. Note that there is no greater variation from the mean in the comparison of parallel representations than occurred when we compared identical samples. These experiments validate the extreme reproducibility of representations, and suggest that making well-controlled parallel representations introduces no more noise than is inherent in the measurements made by this format of microarray as we practice it.
When we compare a differentially labeled sample with itself, the ratio of channel intensity for each feature ideally should be a constant. In fact, there is a deviation from this ideal, which is virtually independent of intensity (Fig. 1B). Possible sources for this variation include the differential behavior of the DNA samples after they are labeled with different fluorochromes, variation in hybridization and washing conditions over the surface of the array, gross regional changes in the physiochemical properties of the array surface, machine fluctuations, and software artifacts, among others. We refer to this “deviation from ideality” as ζ in the mathematical analysis presented below.
We also examined the reproducibility of our measurements of the differences between two different human breast cancer cell lines, SKBR-3 and MDA-MB-415. In these experiments, BglII representations of genomic DNA were made twice from each cell line. Pairs of representations were hybridized to 938 BglII probes, each printed in duplicate. We set minimum thresholds for channel intensity, averaged the Cy5/Cy3 ratios of duplicate features within each microarray, and graphed the values obtained from one experiment to those obtained from the other (Fig. 2). In this experimental series, we observed a >25-fold range of relative gene copy ratios, resulting from differences between the cell lines (see below). There is excellent concordance between independent microarray measurements. Essentially similar results have been obtained in four independent series of experiments using independent representations and independently printed microarrays. These experiments again attest to the reproducibility of representations and also to the reproducibility of printing, labeling, and hybridization.
Figure 2.

A comparison of two microarray experiments. Parallel representations were produced for the two cell lines MDA-MB-415 and SKBR-3. These representations were differentially labeled and hybridized to an array of 938 features printed in duplicate. The ratios of duplicates were averaged and then graphed, the abscissa being the ratios from Experiment 1 in ascending order (as an index) and the ordinate being the ratios from Experiment 2 indexed in the same order as the abscissa.
Verifiability of Microarray Data
Any measuring tool also must satisfy the criterion of independent verification. We therefore sought confirmation of microarray measurements by quantitative Southern blotting of representations and genomic DNAs. For these studies, we used the cell lines SKBR-3 and MDA-MB-415. We examined 36 nonrepetitive probes that were concordant between two microarray experiments: 11 probes that reported significant differences in gene copy number between the cell lines, 15 probes that detected little or no difference, and 10 probes taken from a yeast artificial chromosome (YAC) that contains a region in 8q23 (CEPH address 919g10) that we know to be amplified in SKBR-3. The blots were controlled for loading accuracy by stripping and rehybridization with control probes, and quantitated by scanning with a FUJIX BAS 2000 Bioimaging Analyzer (FUJI).
Figure 3 illustrates some of the blots that were scanned. In general, array probes that detect differences between the cell lines detect either of two types of events by Southern blotting: type I, increased copy number in one of the cell lines, where there is appreciable signal from both (Fig. 3A,D); or type II, the absence of signal from one cell line (Fig. 3C). Type I events are likely to be gene amplification. Type II events could result from a polymorphic difference between the two cell lines with a small BglII fragment in only one of the two cell lines, loss of heterozygosity at a polymorphic BglII site in one of the two cell lines, or homozygous deletion of a small BglII fragment. In fact, in five out of five cases of type II events, we concluded by PCR that the difference between the cell lines were a result of polymorphic differences and not homozygous deletion.
Figure 3.

Thirty-six probes that displayed copy number differences from the previous experiment shown in Fig. 2 were analyzed by Southern blotting representations and genomic DNA from the two cell lines MDA-MB-415 (designated by M) and SKBR-3 (designated by S). Some of the blots are shown. Southern blot of representations (A,C,D) or genomic DNA (B) are shown for probes designated with CHP names. CHP0187 was a probe that detected no difference in copy number by array hybridization.
For the comparison of microarray and blot hybridization (Fig. 4), we plotted the inverse ratios when Southern blot analysis indicated diminished signal in the cell line SKBR-3. Therefore, all type II events are plotted below one, and all type I events (amplifications) are plotted above one. We have fit a straight line to the data by linear regression. It is evident that microarray hybridization underestimates the change in copy number for gene deletions. This most likely results from nonspecific background hybridization in the absence of specific hybridization (see below).
Figure 4.

Shows the ratios of gene copy number obtained by microarray measurement on the X-axis with ratios obtained by quantitative blotting of representations on the Y-axis.
There was good agreement between microarray data and the blotting data for 35 out of 36 probes. Only one probe was significantly discordant with the blotting data, a probe that was consistently reported as amplified by microarray measurements but failed to report as amplified by Southern blotting of either representations or genomic DNA. We have no sure explanation for this anomalous probe, but it may detect a cross-hybridizing DNA under the stringency of array hybridization that is not detected under the stringency of blot hybridization.
Previous work has extensively demonstrated the faithfulness of representations in preserving gene copy ratios, (Lucito et al. 1998) and thus we were comfortable comparing microarray data with blots of representations. Nevertheless, we also compared blots of representations with the blots of genomic DNA. We confirmed the fidelity of representations for 13 of 13 probes that were successfully analyzed both ways. A comparison of five blots of representations and companion blots of genomic DNA are shown in Figure 3A and 3B.
Simulated Comparison of Low- and High-Complexity Array Hybridization
In principle (and in limited practice) high-complexity samples can be analyzed by arrays (Pollack et al. 1999), but signal-to-noise, nonspecific hybridization, and deviation from ideality are all problematic. In order to better understand the relationship between the nucleotide complexity of sample and array hybridization performance, we have made some simple mathematical analyses and simulations to enable us to compare the performance of a LCR BglII with that expected from total DNA.
In the ideal situation, we can express the intensity for a given probe in a given channel as
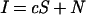 |
1 |
where I is the intensity, c is the copy number of the specific sequences complementary to the probe in the sample labeled with the given fluorochrome, S is the intensity contributed per diploid copy number of specific sequences, and N is the intensity contributed from nonspecific hybridization. One clearly recognized deviation from ideality is the background fluorescence of the array, which can be very variable. For the present study, we can effectively neglect this factor, because background can be determined from the scan of regions adjacent to the feature and subtracted from the measured intensity of the feature.
When two differently labeled samples hybridize at the same time to a given probe and have essentially the same composition and concentration except for the sequences specifically complementary to the probe, and when the intensities of the two channels of fluorescent emission have been normalized properly, then the counterparts of S (and the counterparts of N) are equal in both channels. Furthermore, if we set the second channel as the normal, so that c is the copy number of the test sample in channel 1, then the ratio of the intensity of channel 1 to 2 then will be
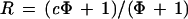 |
2 |
where Φ = S/N is the ratio of specific (per diploid copy number) to nonspecific hybridization for a given probe in a given experiment.
In fact, we do not observe ideality. When we measure essentially identical sequences, so that c = 1, we observe that the ratios of intensity between channels are not exactly constant, as in the data shown in Figure 1A and 1B. Rather, the ratios fluctuate about a constant and this fluctuation is roughly independent of channel intensity. We model this by
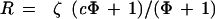 |
3 |
where ζ is a random variable, with mean one, independent of intensity.
In order to simulate array performance, we need to simulate ζ and Φ. ζ can be simulated from the experiments in which c = 1 and hence R = ζ. For the following, we take the experiment described in Figure 1A as our data set for ζ. We can estimate Φ from those experiments in which we have measured R for probes that we have determined to be deleted by blot hybridization (i.e., probes for which c = 0). Because the R values of these probes are biased by ascertainment (in that the probes with smallest R values were examined), this analysis suggests that Φ < 5 for BglII representations under the conditions used in the experiments reported in Figure 4.
In theory, Φ should be proportionate to the ratio of specific to nonspecific sequences. Because BglII yields a 2.5% representation, the average Φ of a probe hybridized to a BglII representation should be ∼40 times higher than when that probe is hybridized to total genomic DNA. Allowing for a more modest 20-fold effect, because of the somewhat uneven amplification in a representation (certain sequences are disfavored), we then may estimate an upper limit for Φ of ∼0.25 when arrays are hybridized with total genomic DNA.
We do not know the actual range of Φ, and it will have a different value for each probe. In addition to nucleotide complexity, factors such as probe length, G/C content, the presence of repeats, and the stringency of hybidization and washing conditions are also likely to influence Φ. For the purposes of modeling, we have assumed a fivefold range for Φ.
For each condition, we modeled total DNA or BglII representations, assuming 10,000 probes. We simulated samples having 20 targets with copy numbers of 4 (c = 4), and 20 targets homozygously deleted (c = 0). All target states can be interpreted clearly from a BglII representation, using a threshold for ratios of twofold relative to the mean (Fig. 5A). On the other hand, with total DNA as sample, if the proper threshold could be chosen, no more than one-third of all amplifications could be discerned without incurring many false positives (Fig. 5B). Moreover, knowing what threshold to set is very problematic. Detection of homozygous deletion would be virtually hopeless. Detection of amplification using total DNA can be improved significantly if nearby probes are “binned”, as described by Pollack et al. (1999). We will present simulations of this procedure in the Discussion.
Figure 5.

Simulations of microarray analysis of 10,000 probes where 20 probes are amplified fourfold and 20 probes are homozygously deleted. Either hybridization of BglII representations (A) or hybridization of total DNA (B–D) were modeled. The hybridization of total genomic DNA (B) also was modeled by moving averages of four (C) or replica measurements of four (D).
Experimental Comparison of Low- and High-Complexity Hybridization
We tested the role of complexity in array performance by a comparison of BglII and DpnII representations. Because all BglII sites (AGATCT) are also DpnII sites (GATC), our collection of microarrayed BglII fragments can be used as probes of DpnII representations, and because DpnII cleaves more frequently than BglII, a DpnII representation has higher complexity (∼70% of the genome) than a BglII representation (∼2.5%). These numbers were determined by cleaving in silico many megabases of known human genomic sequence, and determining the proportion of nucleotides in fragments ≤1.0 kbp, the sizes that are retained during representation. We compared BglII to DpnII representations of the two cell lines SKBR-3 and MDA-MB-415 by microarray hybridization. In these experiments, we used a different set of arrayed probes and a larger numbers of probes than used in the experiments reported in Figures 2, 3, and 4.
The results are strikingly clear when we make plots of ratios to single-channel intensity (see Fig. 6A–C). Note the difference in scale on the Y-axis in Figure 6C. In these figures, deviation from the main line represents a detected change in copy number, with points above the main line reflecting higher copy numbers in SKBR-3, and points below reflecting higher copy number in MDA-MB-415. There is a dramatic increase both in the number of probes that detect change and in the degree of change they detect, when the LCR is hybridized. Virtually none of the differences detected with BglII as decreased copy number in SKBR-3 can be detected with DpnII. Further analysis (data not shown) indicates that a clear minority of probes detect differences by both types of representation. These findings are consistent with our mathematical models and simulations as presented herein. It is possible that probes detecting changes in copy number as a result of polymorphic differences are detected by BglII representation and not by DpnII representations because of the polymorphism being in the outer nucleotide of the BglII site. These probes would no longer be polymorphic in a DpnII representation, and thus would have no detectable copy number fluctuation. Although this is a possibility for probes that detect polymorphism, it would not explain the lack of detectable ratio from the DpnII representations for probes that truly detect amplification as seen in Figure 3.
Figure 6.

Shows the comparision of hybridizations of BglII representations to that of DpnII representations. Microarrays of 1658 features were hybridized, scanned, and threshed for intensity and the data were graphed in the same format as the data in Figs. 1A–C, with ratios (or inverse ratios) plotted as a function of single-channel intensity. (A) BglII representations of the two cell lines MDA-MB-415 and SKBR-3 were differentially labeled and hybridized to arrays and graphed as described. (B) DpnII representations of the above cell lines were differentially labeled and hybridized to arrays analyzed and graphed as described. (C) The data from Fig. 6B were graphed at a smaller range to show scatter.
We then compared the specific performance of probes derived from a YAC (919g10) that localizes to 8q23 (Table 1). This YAC derives from one of two regions residing near to but distinct from c-myc that we find commonly amplified in breast cancers (M. Nakamura, unpubl.). As can be seen from the data derived from the LCR (BglII), there are probes from this region that are highly amplified in SKBR-3 and probes that are not. One could use such data to delimit the epicenter of this amplification. One can infer from the HCR (DpnII) that this region has undergone amplification, because the great majority of probes register ratios above the median. However, from the HCR data we do not have a clear indication of the degree of amplification that has occurred, and would be at a complete loss to delimit the epicenter.
Table 1.
Comparison of Ratios Obtained from Hybridizations of BglII and DpnII Representations
| Name | Bgl Ratio | Dpn Ratio |
|---|---|---|
| CHP0140 | 5.43 | 0.98 |
| CHP0125 | 5.33 | 1.37 |
| CHP0218 | 3.86 | 1.25 |
| CHP0138 | 3.75 | 1.05 |
| CHP0121 | 3.37 | 1.23 |
| CHP0131 | 3.27 | 0.68 |
| CHP0134 | 3.25 | 1.06 |
| CHP0142 | 3.20 | 1.15 |
| CHP0120 | 2.97 | 1.38 |
| CHP0123 | 2.93 | 1.04 |
| CHP0215 | 2.53 | 1.04 |
| CHP0137 | 2.45 | 1.24 |
| CHP0132 | 1.76 | 1.03 |
| CHP0119 | 1.53 | 0.99 |
| CHP0136 | 0.90 | 0.96 |
Comparison of ratios shown for features located within one yeast artificial chromosome (YAC). This YAC maps to 8q23, a region amplified in the cell line SKBR-3.
Analysis of Microscopic Amounts of Tumor Biopsies
We tested whether we could analyze small amounts of human tumor biopsies by microarray measurements. We chose a breast tumor, CHTN9, for which we also had data from RDA, Southern blotting of representations, and quantitative PCR (using TaqMan probes and ABI 7700 sequence detector). Because biopsies are a mixture of tumor and normal stroma, we flow-sorted the nuclei from the biopsy into aneuploid and diploid fractions, and prepared BglII LCRs from 10,000 nuclei of each fraction.
We compared gene copy number between aneuploid (presumed tumor) and diploid (presumed normal) representations. In Figure 1C, we plotted the ratio of the channel intensities as a function of channel intensity in the normal channel for each feature (open circles). As in Figure 1A, for symmetry, we plotted the tumor:normal ratio above the median if >1.0, otherwise the normal:tumor ratio below the median. Thus, amplifications are found above the main line and losses below the line. Because the scanner does not record above an intensity of 65,000 units, amplification will be underestimated at features that give strong signal in the normal channel. Lower luminosity excitation would collect more accurate data from these features. For the excitation luminosity setting of the experiment depicted in Figure 1C, the points designated by closed diamonds delimit the high intensity measurements of the scanner.
If we set a twofold difference in the ratio of median channel intensities for a feature to indicate probes that have undergone either amplification or deletion, there is excellent correlation between our microarray results and what we know about this tumor. All 15 amplified probes that were found in these tumors by RDA, and confirmed by other means, were confirmed as amplified by our microarray analysis. Additional probes that derive from known amplified loci, but that have not yet been individually confirmed by other means, also are found amplified by microarray analysis. Moreover, probes that derive from loci that we know are not amplified in these tumors do not show amplification by microarray hybridization. Finally, five out of six probes found to be deleted by RDA also were found to be deleted by microarray hybridization. Clearly, for CHTN9, our array data detects more amplifications than deletions. This is because the arrayed probes were weighted with probes from several loci that we know to be amplified in this tumor.
DISCUSSION
We have demonstrated that hybridization of arrays of short (<1 kbp) DNA probes based on LCRs provides a method for detecting amplifications, deletions, and polymorphic differences that are both reproducible and independently verifiable. We have demonstrated the analysis of microscopic amounts of tumor biopsy material using this method. In this report, we have made LCRs using amplification of DNA following BglII cleavage, but our results are applicable to any system in which representations of samples that reduce complexity are matched with an appropriate array of probes. In fact, other researchers have made arrays from inter-Alu probes, and hybridized these arrays to whole genomes sampled by inter-Alu PCR (Geschwind et al. 1998).
We believe that our method has advantages in sample preparation, flexibility, and resolution. Because representations are used to prepare samples, only very minute amounts of starting material are needed. The flexibility derives from having a virtually inexhaustible set of probes to use, so that probes with desirable characteristics can be selected. The resolution results from generally high-specific to nonspecific hybridization signals for probes and is therefore limited only by the density of probes that can be printed. The maximum possible resolution of any array is determined by the number of probes that can be arrayed. For example, a 30,000-member array has a potential resolution of 100 kb if probes were uniformly spaced. The theoretical resolution with a BglII representation is ∼20 kb (150,000 small fragments per genome), and thus the maximum density of a chip is more likely to determine resolution than is the use of this particular type of representation.
Obviously, array performance can be improved by any method that improves signal-to-noise. We have described a method for increasing signal, namely making complexity reductions of genomic samples. Technical improvements in printing, sample labeling, array surface properties, hybridization conditions, and so on, that reduce noise (Φ and ζ) serve the same end. Statistical methods also can be used to reduce noise. For example, high-complexity samples can be analyzed by arrays of cDNA probes (Pinkel et al. 1998; Pollack et al. 1999), but signal-to-noise is problematic, and additional measures are required to establish reliability. In particular, Pollack et al. (1999) use moving averages, which entails averaging signal over chromosomally linked probes. This, in effect, “squeezes” the background noise.
We have simulated moving averages by 4 and 16 adjacent probes, and used both Φ and ζ modeled from our experimental results (Fig. 5C). In these simulations, each data point is the average of four independent probes, for which we assume independent values for both Φ and ζ. Moving averages of four gives a significant improvement in the detection of amplified sequences, but detection of deletion is still very problematic. Assuming the proper threshold could be determined, most amplifications can be safely discerned. Few, if any, homozygous deletions could be called safely without also calling many false positives. Moving averages of 16 (data not shown), however, enables deletions to be recognized readily, and are comparable to analysis of BglII representations.
Although moving averages require knowledge of the linkage of probes, similar enhancement could be achieved, in principle, merely by replica hybridizations, provided of course that ζ is truly random and not a property of a particular probe. Replica measurements require no knowledge of probe linkage. Replica measurements of four are simulated in Figure 5D, and assumes independent values for ζ for each replica (but constant Φ). The result is very similar to moving averages of four. However, there is a price that must be paid for either moving averages or replica measurements, either as a loss of genomic resolution in the detection of lesions; an increase in the number of probes used in the design of the chip; or, an increase in the number of replica hybridizations that must be performed.
One advantage of hybridizing arrays to representations made by amplifications after restriction endonuclease cleavage is the ease of detecting allelic loss: representations are sensitive to nucleotide polymorphisms at the restriction endonuclease sites used in their preparation. For example, if normal DNA is heterozygous for a BglII site that creates a small BglII fragment, the loss of this site in the tumor is readily seen as a gene deletion. Because representations also can be made to be sensitive to polymorphisms at internal restriction endonuclease sites, it should be possible to intensively survey the cancer genome for allelic losses, or even mutational load. The same principles could be applied for whole-genome genotyping of individuals by array hybridization. In fact, we showed that some of the gene copy number differences we detected between representations of two cell lines arise because of BglII polymorphisms.
The sensitivity of our method to polymorphism is an asset, but can also be a liability. We estimate from data about the frequency of polymorphism in the human genome that one out of 60–120 BglII fragments will detect a polymorphism at a BglII site. Thus, in comparing tumor and normal DNA from different individuals, most differences would be from polymorphism. Therefore, tumor and normal DNA ordinarily should be from the same individual. Even then, a difference between normal and tumor might reflect loss of heterozygosity (LOH) rather than homozygous deletion. Since homozygous loss will tend to locate a presumptive tumor suppressor with greater precision than LOH, it is desirable to distinguish such events. Resolution of these events can be accomplished if we can establish dense probe “neighborhoods”, that is, a linkage of nearby probes. Loss of heterozygosity will be detected as a loss of signal from only a small subset of our probes, namely those that are capable of detecting BglII polymorphisms, and such probes will be sparsely distributed. Therefore, LOH generally will not cause the conjoint loss of signal from closely linked probes. On the other hand, if our probes are sufficiently dense, homozygous deletion will be marked by the conjoint loss of signal from closely linked probes.
The full value of genomic array hybridization emerges from linking data about the arrayed probes to the physical, genetic, and ultimately, the transcription map of the genome. In this critical respect, representational arrays are at an initial disadvantage with respect to the other genomic array methods: cDNAs, of course, correspond to a transcriptional unit and many have been mapped; BACs will be precisely mapped in the near future. Random representational probes do not have associated physical, genetic, or transcriptional mapping information. However, representational probes can be mapped efficiently and placed into association in a variety of ways by hybridizing arrays of these probes to collections of YACs, BACs, or radiation hybrids (preliminary results). Array hybridization to even unordered and unmapped pools of BACs, given sufficient numbers of probes and BACs, results in the assemblage of contigs of BACs and neighborhoods of probes with associated inferred physical distances. A manuscript describing these mapping methods and inferred linkage metrics is in preparation.
A more direct approach, that leverages the sequence information of the human genome, can be taken to build a collection of mapped representational probes. This depends on a property of representations that we have noted, namely that almost all predicted small BglII fragments of the genome are elements of BglII representations. Therefore, probes can be selected from the genome databases that have known map information, such as inclusion in BACs with known chromosomal positions. A collection of such probes can be generated from predicted small BglII fragments, either as oligonucleotides or longer fragments generated by PCR from representations using designed oligonucleotide primer pairs.
We have described and illustrated the use of representational microarrays for the detection of gene copy number fluctuations in cancer. This tool also has other potential uses, including measuring mutational load in cancers, monitoring DNA methylation patterns, genome-wide genetic typing, and detection of de novo mutations in humans. Hopefully, we will be able to illustrate these applications in subsequent manuscripts.
METHODS
Materials
We obtained 96-well sterile and nonsterile plates from Corning-Costar; 96-well PCR plates were obtained from Marsh; Escherichia coli strain XL1 Blue was obtained from Stratagene; BglII, DpnII, and Ligase were supplied by New England Biolabs; silanated glass slides were obtained from CEL Associates. Taq polymerase was purchased from Perkin Elmer, and oligo-nucleotides were obtained from Operon Technologies. Pins (Chipmaker 2) used for the arrayer, and the hybridization chamber were purchased from Telechem International. Klenow fragment, Cy3 and Cy5, and dNTPs were obtained from Amersham Pharmacia Biotech.
Arraying
We used the Cartesian PixSys 5500 (Cartesian Technologies) to array our probe collections onto slides. We used a 2x2 pin configuration, and printed each probe in a center-to-center spacing of 280 μm in duplicate, yielding eight quadrants or blocks. The dimensions of each printed array was 2 cm2. Arrays were printed on commercially prepared silanated slides.
Probe Collection
BglII probes were obtained by several procedures. Initially, we obtained BglII probes that were the products of RDA experiments. Subsequently, we cloned small (<1.0 kbp) BglII fragments from BACs, P1s, and YACs obtained from various library resources (Research Genetics). Finally, we added to our collection by random cloning of small BglII fragments from the human genome. Probe fragments were maintained as pUC19 inserts in the E. coli strain XL1 Blue.
Preparation of Probes for Arraying
Arrays were made from two sets of probes, an early set with ∼800 members, and a later set of ∼2000 members. Glycerol stocks of the E. coli hosts were arrayed in 96-well plates. Probe preparation was started by PCR amplification of the insert directly from the lysed E. coli host, using primers set 1: pUC(for) aaggcgattaagttgggtaac and pUC(rev) caatttcacacaggaaa cagc. Twenty cycles of PCR (95°C for 1 sec, 55°C for 30 sec, and 72°C for 1 min) were followed by an extension of 10 min at 72°C. This created a stock for further amplifications. One microliter of this reaction then was used for a second PCR amplification to produce the probe fragments for arraying. PCR amplification was carried out with primer set 2: M13 ttgtaaaacgacggccagtg and M13 Rev ggaaacagctatgaccatga. These are internal to primer set 1, decreasing the possibility of E. coli contamination. The same PCR conditions were followed. PCR reactions were precipitated by addition of one-tenth volume of 3M NaAcetate (pH 5.3) and one volume of isopropanol. After 30 min at –20°C, the plates were centrifuged at 1500 rpm in a tabletop centrifuge. The supernatant was removed and the pellet was washed with 70% ethanol, centrifuged at 1500 rpm in a tabletop centrifuge for 5 min, and again the supernatant removed. The plates were dried in a vacuum oven, and then resuspended in 15 μL of 3x SSC for arraying.
Sample Preparation
Representations were prepared as described in Lucito et al. (1998). Briefly, DNA of choice was digested to completion with either BglII or DpnII, and cohesive adapters were ligated to the digested ends. PCR primers complimentary to the adapter ligated then were used for amplification by PCR. This product then was used for hybridization.
Labeling of Sample
Ten micrograms of representation was denatured by heating to 95°C in the presence of 5 μg of random nonamer in a total of 100 μL. After 5 min, the sample was removed from heat and 20 μL of 5x buffer was added (50mM Tris-HCL [pH 7.5], 25 mM MgCl2, 40mM DTT, supplemented with 33 μM dNTPs), 10 nmol of either Cy3 or Cy5 was added, and the four units of Klenow fragment were added. After incubation of the reaction at 37°C for 2 h, the reactions were combined and the incorporated probe was separated from the free nucleotide by centrifugation through a Microcon YM-30 column. The labeled sample then was brought up to 15 μL, at a concentration of 3 x SSC and 0.2% SDS, denatured, and then hybridized to the array.
Processing of the Array
The array was placed in a humidified chamber for 3–5 min until spots became hydrated. The slide was crosslinked by ultraviolet irradiation of 60 mJ in a Stratagene Stratlinker. The slide then was hydrated again in the humidified chamber and snap-dried by heating on the surface of a hot plate for several seconds. The array then was washed in 0.1% SDS for ∼10 sec, in deionized water for ∼10 sec, and then denatured in boiling deionized water for ∼1–2 min. After denaturation the array was quickly immersed in ice-cold benzene free ethanol for several seconds, taken out, and allowed to dry. Cover slips for the arrays were put through the same wash procedure from the SDS to the ice-cold ethanol. The 15 μL of sample then was placed on the array and a cover slip slowly placed on the array.
Scanning, Informatics and Data Handling
Arrays were scanned by either GSI Lumonics ScanArray3000 or Axon GenePix4000. Feature ratios were calculated after background subtraction using either ScanAlyze (Stanford University) or Axon GenePix2.0. Background values for each feature were calculated as the median fluorescence signal for nonfeature pixels for each channel. The resulting tab-delimitated text files then were imported into S-plus 2000, a mathematics and statistical software package (MathSoft, www.mathsoft.com), with which we normalized the data and threshed by minimum intensity value of 300–500, depending on the average background pixel intensity. We implemented databases in Microsoft Access and used Per1 for data extraction and reformatting.
Acknowledgments
This work was supported by grants to M.W. from the National Institutes of Health (NIH) (OIG-CA39829 and 5P50-CA-68425); the U.S. Army (DAMD17–94-J-4247); NIH (R01 CA81674-R2); NIH SPORE 2P50CA68425; 1 In 9: The Long Island Breast Cancer Action Coalition; St. Giles Foundation; The Lillian Goldman Charitable Trust and Mrs. Lillian Goldman through The Breast Cancer Research Foundation; and Tularik Inc. M.W. is an American Cancer Society research professor.
We thank C. Yen and M. Nakamura for valuable information and reagents, and L. Rodgers for help with informatics. Also, special thanks to D. Bottstein, R. Kucherlapati, G. Childs, A. Massimi, T. Harris, J. Latter, J. Gergel, and P.O. Brown for useful encouragement and the sharing of their expertise and unpublished data.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lucito@cshl.org; FAX (516) 367-8381.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr138300
REFERENCES
- DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet. 1996;14:457–460. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Gregg J, Boone A, Karrim H, Pawalikowska-Haddal A, Ellison J, Ciccodicola A, D'Urso M, Woods R, Rappold GA, et al. Klinefelter's syndrome as a model of anomalous cerebral laterality: Testing gene dosage in the X chromosome pseudoautosomal region using a DNA microarray. Dev Genet. 1998;23:215–229. doi: 10.1002/(SICI)1520-6408(1998)23:3<215::AID-DVG7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Khan J, Saal LH, Bittner ML, Chen Y, Trent JM, Meltzer PS. Expression profiling in cancer using cDNA microarrays. Electrophoresis. 1999;20:223–229. doi: 10.1002/(SICI)1522-2683(19990201)20:2<223::AID-ELPS223>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Lisitsyn N, Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;258:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- Lisitsyn NA, Lisitsina NM, Dalbagni G, Barker P, Sanchez CA, Gnarra J, Linehan WM, Reid BJ, Wigler MH. Comparative genomic analysis of tumors: Detection of DNA losses and amplification. Proc Nat Acad Sci. 1995;92:151–155. doi: 10.1073/pnas.92.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucito R, Nakimura M, West JA, Han Y, Chin K, Jensen K, McCombie R, Gray JW, Wigler M. Genetic analysis using genomic representations. Proc Nat Acad Sci. 1998;95:4487–4492. doi: 10.1073/pnas.95.8.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- Sgroi DC, Teng S, Robinson G, Le Vangie R, Hudson JR, Jr, Elkahloun AG. In vivo gene expression profile analysis of human breast cancer progression. Cancer Res. 1999;15:5656–5661. [PubMed] [Google Scholar]
- Wang K, Gan L, Jeffrey E, Gayle M, Gown AM, Skelly M, Nelson PS, Ng WV, Schummer M, Hood L, et al. Monitoring gene expression profile changes in ovarian carcinomas using cDNA microarray. Gene. 1999;18:101–108. doi: 10.1016/s0378-1119(99)00035-9. [DOI] [PubMed] [Google Scholar]