

Abstract
One of the first useful products from the human genome will be a set of predicted genes. Besides its intrinsic scientific interest, the accuracy and completeness of this data set is of considerable importance for human health and medicine. Though progress has been made on computational gene identification in terms of both methods and accuracy evaluation measures, most of the sequence sets in which the programs are tested are short genomic sequences, and there is concern that these accuracy measures may not extrapolate well to larger, more challenging data sets. Given the absence of experimentally verified large genomic data sets, we constructed a semiartificial test set comprising a number of short single-gene genomic sequences with randomly generated intergenic regions. This test set, which should still present an easier problem than real human genomic sequence, mimics the ∼200kb long BACs being sequenced. In our experiments with these longer genomic sequences, the accuracy of GENSCAN, one of the most accurate ab initio gene prediction programs, dropped significantly, although its sensitivity remained high. Conversely, the accuracy of similarity-based programs, such as GENEWISE, PROCRUSTES, and BLASTX, was not affected significantly by the presence of random intergenic sequence, but depended on the strength of the similarity to the protein homolog. As expected, the accuracy dropped if the models were built using more distant homologs, and we were able to quantitatively estimate this decline. However, the specificities of these techniques are still rather good even when the similarity is weak, which is a desirable characteristic for driving expensive follow-up experiments. Our experiments suggest that though gene prediction will improve with every new protein that is discovered and through improvements in the current set of tools, we still have a long way to go before we can decipher the precise exonic structure of every gene in the human genome using purely computational methodology.
The nucleotide genomic sequence is the primary product of the Human Genome Project, but a major short- and mid-term interest will be the amino acid sequences of the proteins encoded in the genome. Thus, methods that reliably predict the genes encoded in genomic sequence are essential, and computational gene identification continues to be an active field of research (for reviews, see Fickett 1996; Claverie 1997; Guigó 1997a; Burge and Karlin 1998; Haussler 1998). A new generation of gene prediction programs based on Hidden Markov Models (Burge and Karlin 1997) have shown significantly greater accuracy than previous programs based on other methodologies (Burset and Guigó 1996). Conversely, as the databases of known coding sequences increase in size, gene prediction methods based on sequence similarity to coding sequences, mainly proteins and ESTs, are becoming increasingly useful and are routinely used to identify putative genes in genomic sequences (The C. elegans Sequencing Consortium 1998). We have recently published an evaluation of sequence similarity-based gene prediction methods, in particular of EST-based gene prediction (Guigó et al. 2000). The accuracy of gene identification programs, however, has usually been estimated on controlled data sets made of short genomic sequences encoding a single and complete gene with a simple structure. Moreover, these data sets are often similar if not overlapping, to the sets of sequences on which the programs have been trained. Thus, these data sets are not representative of the sequences being produced at the genome centers, which are mostly large sequences of low coding density, encoding several genes or incomplete genes with complex gene structure. It is thus difficult to know how well the figures of accuracy estimated in the controlled benchmark data sets extrapolate to actual genomic sequences. Furthermore, programs that combine both sequence similarity and ab initio gene finding approaches, and those that predict genes by producing a splicing alignment between a genomic sequence and a candidate amino acid sequence have become recently available, such as PROCRUSTES (Gelfand et al. 1996) and GENEWISE (Birney and Durbin 1997), (http://www.sanger.ac.uk/Software/Wise2/). Programs that align genomic sequences with EST sequences, such as EST_GENOME (Mott 1997), could also be included in this category. These programs promise highly accurate predictions, but at the cost of greater computational time. However, this increase in accuracy has not been well-quantified on challenging data sets. The effects of the degree of similarity between the candidate homolog and the genomic sequence also deserve careful evaluation.
We believe a more realistic evaluation of the currently available gene prediction tools on challenging data sets would be useful. Ideally, one would like to benchmark the computational gene identification programs in real genomic sequences. The main problem is that most real sequences the structure of the genes has not been verified exhaustively by experimental means, and thus it is impossible to calibrate the accuracy of the predictions. Only recently, extensively annotated large genomic sequences from higher eukaryotic organisms have become available from the human genome (http://www.hgmp.mrc.ac.uk/Genesafe) and from the fly genome (http://www.fruitfly.org/GASP1/). In spite of the experimental analysis, the possibility of undetected genes in the sequence cannot be easily ruled out, which makes accuracy difficult to measure. Here, we attempt to overcome the lack of well-annotated large genomic sequences by constructing semiartificial ones. In these semiartificial sequences, known genomic sequences have been embedded in simulated intergenic DNA, and therefore, the location of all coding exons is known. Although the approach may seem unrealistic, we believe that the results obtained are instructive with regard to the accuracy of currently available gene identification tools.
We evaluate the accuracy of representatives of a wide variety of computational gene identification approaches: GENSCAN (Burge and Karlin 1997), an ab initio genefinder; BLASTX (Altschul et al. 1990; Gish and States 1993), a genefinding-oriented similarity search program; and PROCRUSTES (Gelfand et al. 1996) and GENEWISE (Birney and Durbin 1997), genefinders based on aligning a genomic DNA sequence fragment to a homologous protein sequence. We evaluate these programs on two benchmark data sets: A set of well-annotated single-gene DNA sequences, and a set of semiartificial genomic (SAG) sequences created by embedding the single-gene sequences from the first data set in simulated intergenic DNA.
RESULTS
We investigated the accuracy of the gene prediction tools (GENSCAN, PROCRUSTES, GENEWISE, BLASTX) described in Methods on two benchmark sets. In all cases, sequences were masked previously for repeated regions using REPEATMASKER (A. Smit and P. Green, unpubl.). The gene predictions obtained using the different tools were compared with the actual gene annotations using the accuracy measures described Methods.
Accuracy in Single Gene Sequences
Table 1 shows the accuracy of the different gene prediction tools on h178, the set of single gene sequences.
Table 1.
Accuracy of Gene Prediction Tools in the Set of Single Gene Sequences (h178)
| Program | No. |
Nucleotide
|
Exon
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Sn | Sp | CC | Sn | Sp | 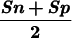 |
ME | WE | ||
| GenScan | 177 | 0.93 | 0.90 | 0.90 | 0.78 | 0.75 | 0.76 | 0.08 | 0.10 |
| Blastx default | 175 | 0.91 | 0.79 | 0.82 | 0.04 | 0.04 | 0.04 | 0.12 | 0.05 |
| Blastx topcomboN | 174 | 0.97 | 0.80 | 0.86 | 0.04 | 0.04 | 0.04 | 0.08 | 0.05 |
| Blastx 2 stages | 175 | 0.90 | 0.92 | 0.90 | 0.10 | 0.12 | 0.11 | 0.19 | 0.02 |
| GeneWise | 177 | 0.98 | 0.98 | 0.97 | 0.88 | 0.91 | 0.89 | 0.06 | 0.02 |
| Procrustes | 177 | 0.93 | 0.95 | 0.93 | 0.76 | 0.82 | 0.79 | 0.11 | 0.04 |
GENSCAN's accuracy is comparable to that reported earlier (Burge and Karlin 1997). On average, 90% of the coding nucleotides and 70% of the exons are predicted correctly by GENSCAN. Only 7% of the actual exons are missed completely, and only 9% of the predicted exons are wrong. We believe this is close to the maximum accuracy that can be achieved using currently available ab initio gene prediction programs.
The quality of the gene models inferred from BLASTX searches depends on the strategy used. Default usage of BLASTX produced poorer predictions than more sophisticated strategies. (Results for BLASTX default correspond to those published in Guigó et al. 2000.) Discrepancies between numbers in Table 1 and those reported in Guiqoet al. (2000) are due to the differences in the way the accuracy measures are summarized. In Guigó et al. 2000, we computed the accuracy measures on each test sequence, and averaged all of them. Here, we compute the accuracy measures globally from the total number of prediction successes and failures (at the base or exon level) on all sequences. The default BLASTX strategy produces reasonably high sensitivity (0.91) by projecting all HSPs over a given threshold along the query DNA sequence, but the sensitivity rises to an amazing 0.97, if the topcomboN feature is used. The topcomboN feature eliminates the need for low-complexity filters (seg + xnu), and for strict secondary HSP cutoff (S2 threshold). Surprisingly, its use does not appear to hurt specificity. The two-stage method (in which the top homolog with low-complexity filtering is chosen to build the BLASTX model with topcomboN in the second stage) increases specificity from 0.79 to 0.92. Using a single protein to build a model improves specificity because the noise from the less significant hits is reduced. But the two stage method does have lower sensitivity from a lack of information from the weaker secondary hits. However, this is still the best purely BLASTX-based strategy in terms of either specificity or overall accuracy, and the numbers are comparable to the accuracy of ab initio gene finders at the nucleotide level.
The proteins encoded by the sequences in h178 are mostly included in the nonredundant database of amino acid sequences (nr). However, BLASTX still does not produce perfect predictions. This certainly has an artefactual component: We have discovered a few annotation errors in h178. However, perfect gene predictions from BLASTX searches are intrinsically impossible because of the inability of BLASTX to predict the splice boundaries when they occur within codons (this especially affects its accuracy at the exon level, which is actually rather meaningless for BLASTX). In this regard, splicing alignment or sequence similarity-based gene prediction tools (SSBGP), such as GENEWISE and PROCRUSTES could, in principle, result in more accurate predictions. Thus, the protein sequence with the lowest P value after the BLASTX search was given to PROCRUSTES and GENEWISE to model their gene predictions. SSBGP tools improved the accuracy of the gene predictions inferred directly from BLASTX searches, and also slightly outperform GENSCAN in this set.GENEWISE predictions with an overall accuracy of 0.97, in particular, were close to perfect given the intrinsic inaccuracy of the database annotation considered to be the gold standard here. Of course, there is a price paid in computational time, and GENEWISE is expensive with its linear-memory dynamic programming technique.
GENSCAN accuracy, in theory, should be unaffected, whether the query sequence encodes genes for which a close homolog, remote homolog, or no homolog exists. GENEWISE and PROCRUSTES accuracy, on the other hand, should decrease as the homology becomes distant, and these programs have little utility if a homolog does not exist.
As we have already pointed out, nr database contains protein translations of most of the genes in our data set, which could be a significant drawback of the data set. It is difficult (if not impossible) to come up with criteria for eliminating just the translations. Mouse orthologs are often 100% identical at the protein level and variants of the same protein may be highly (98%–99%) identical. Thus, we chose to evaluate the effect of the similarity level (P value) of an available homolog on the accuracy of GENEWISE and PROCRUSTES by considering a variety of P value bins. Conceptually, identical or close to identical proteins would fall in the most significant P value bin, and other bins would be devoid of identical hits.
A set of Blast-probability (P value) thresholds was chosen to provide bins with varying levels of similarity (10−120, 10−80, 10−60, 10−40, 10−30, 10−20, 10−10, and 10−5). For each of these P values (10−80, for instance), we performed the following experiment. After running BLASTX against nr for the DNA sequences in h178, we discarded for each DNA sequence all HSPS corresponding to all protein sequences with a P value below cutoff (as if we were ignoring all known amino acid sequences over a given level of similarity to the protein encoded in the query DNA sequence). Then, the protein with the remaining top hit below the next higher P value threshold (10−60, in the case of the example) was used, if it existed, as a candidate homolog for the SSBGP tools. If there was no protein hit in the bin (10−80 to 10−60 in the example) then this gene was discarded for the evaluation of this bin.
Thus, the BLASTX gene models are based on all the protein homologs with probability higher than the threshold considered. The P value thresholds were chosen so as to generate roughly equal numbers of data points (sequences from h178) for each set. The minimum number of data points in any set is 73, large enough to avoid significant sampling bias.
The accuracy results as a function of P value of the homologs are shown in Figure 1. GENSCAN performance is expected to be constant, and was for the most part; the minor variations are because of changes in the data set. Only a fraction of the genes had homologs in each of the bins, thus the data set changed a little from bin to bin. The overall performance of SSBGP tools suffered substantially as the similarity decreased. Somewhat surprisingly, the performance of GENSCAN is superior to that of SSBGP tools even at rather high levels of similarity (P value between 10−80 and 10−60). When the similarity is strong, GENEWISE appears to outperform PROCRUSTES in the h178 sequence set. However, when the similarity is weak the difference in performance between the two tools at the nucleotide level is small, and for low levels of similarity PROCRUSTES seems to outperform GENEWISE, particularly at the exon level. This is not unexpected considering the design of these programs: GENEWISE is primarily a sequence alignment tool, and thus it performs very well when there is strong sequence similarity. PROCRUSTES is more of a gene prediction program; it possibly encodes a more sophisticated splice site and exon model, which allows for better exon prediction at low levels of similarity. As shown in Figure 3, a decrease in accuracy for sequence similarity-based methods is most likely a result of the decline in sensitivity, while specificity remains high, which is a very desirable feature.
Figure 1.

The accuracy of the gene prediction tools as a function of the similarity to the chosen homolog. For each P-value cutoff, the homolog with the lowest P value above the cutoff was chosen to build the gene prediction models. The table indicates the different ranges considered, the log-average of the P values in each range, and the number of sequences with acceptable homologs in the range. For example, there were 99 sequences in h178 for which after discarding all hits with P value < 10−120, the top remaining hit had a P value < 10−80. There were 73 sequences for which the top hit had a P value < 10−120, and 119 sequences for which the top hit had a P value > 10−5.
Figure 3.

If the candidate protein sequence is a remote homolog, direct gene modeling from BLAST-like database searches may have different predictions compared to more sophisticated SSBGP tools. (A) EMBL DNA sequence HSCKBG was compared with the protein sequences in the nr sequence database using BLASTX. Hits with P value < 10−20 were discarded, the top remaining corresponded to a fragmentary protein sequence gi:553231. Not surprisingly, only a small fraction of the actual gene was recovered using this homolog by either GENEWISE or PROCRUSTES. Other choices of homologs may have yielded different predictions but none of them by themselves appears to be perfect. Conversely, the gene model derived directly from the BLASTX search reproduces the exonic structure of the gene fairly well. Thus, even though upon discarding the close homologs, the remaining proteins individually showed only little overall similarity to the encoded protein product, as a collection they enable to walk its exonic structure. (B) If database protein sequences with hits below P-value = 10−20 are discarded, BLASTX is able to detect significant similarity between only one of the encoded exons in EMBL sequence HSPAC3G and the remaining protein sequences in the database. But with the top homolog among these, the SSBGP tools (GENEWISE in particular) are able to infer the correct exonic structure, picking up both the additional upstream exons. This is because the SSBGP tools are able to detect more distant sequence relationships than BLASTX with our choice of thresholds or because (as in this case) coding exons occur in low-complexity regions, which are usually masked when performing BLASTX searches to avoid large numbers of false positives. (C) In another case, direct gene modeling from BLASTX searches and SSBGP tools can complement each other to produce more accurate gene predictions. As in A and B, HSP hits below P-value = 10−20 were ignored after comparing EMBL sequence HSFOLA with the nonredundant protein sequence database.
Interestingly, when the similarity is weak (P value > 10−20), the advantage of sophisticated SSBGP tools as opposed to direct gene modeling from database searches such as those performed by BLASTX, seems to vanish. It is not unlikely that when the similarity is weak, the query DNA sequence and the top database search homolog share only a conserved domain. In such cases, SSBGP, relying on sequence similarity only to the top homolog, are only able to detect the part of the gene exonic structure encoding these domains. Direct gene modeling from BLASTX search results builds on all potential homologs (not only the top one); thus, weak homologs that share different conserved regions with the gene encoded in the DNA sequence may allow for better recovery of the overall exonic structure of the gene. In fairness to GENEWISE and PROCRUSTES, they can be used with multiple protein homologs and complete gene models synthesized, but that is computationally expensive and analytically problematic. Figure 1 illustrates an extreme example. A possible solution (at least when using GENEWISE) is to build a profile or an HMM based on the top few homologs and then align this profile with the target genomic sequence.
Conversely, when the similarity with the top homolog is weak, the BLASTX search picks up only the stronger regions of similarity between the homolog and the gene encoded in the query sequence, although lower levels of sequence similarity are shared in other regions between the protein and the query DNA sequences. These can be detected by the SSBGP tools (Fig. 1). Finally, in other cases, both situations occur simultaneously, and direct gene modeling from BLASTX search and SSBGP tools may complement each other to produce a more accurate overall prediction (Fig. 1).
Examining the data in Table 1 and Figure 1, one may be tempted to conclude that the gene identification problem is almost solved. When a strong homolog exists, programs like GENEWISE and PROCRUSTES are likely to pick up the correct exon structure; when such a homolog does not exist, programs like GENSCAN will still be able to recover most of this structure. This, we believe, is rather optimistic, as the sequence set in which these programs have been tested is extremely easy. Although the results obtained are instructive of the comparative performance of the tools, they cannot necessarily be extrapolated to the performance of these tools in the large genomic sequences. In the next section, we present the results obtained on evaluating the tools on a set of simulated genomic sequences, which we believe provide a more realistic estimation of the actual accuracy of the gene prediction tools in large genomic sequences.
Accuracy in Semiartificial Genomic Sequences
A SAG data set containing known genes in random intergenic context (as described in Methods) was constructed to check if the accuracy measures from the previous section extrapolate to larger, more difficult data sets.
Because each SAG sequence contains multiple genes, the choice of the set of protein homologs to predict all the genes was no longer trivial. For ease of evaluation, we used the knowledge of the genes to pick these homologs, but there are other techniques that can be used to pick up a single candidate homolog for each gene-like region. In short, the top-scoring protein homolog from the BLASTX search for each of the genic sequences was used by GENEWISE and PROCRUSTES to predict the gene based on sequence similarity. For instance, artificial sequence AGS01 was obtained by embedding EMBL sequences HS10116, HSDNAAMHI, and HSNUCLEO in artificial intergenic DNA, with BLASTX top homologs being NCBI:gi 134635, 1136442, and 128841, respectively. The GENEWISE and PROCRUSTES predictions on the artificial sequence AGS01 were obtained by three independent executions of the programs, with each of the above top homolog proteins in turn. The programs were executed to predict genes on both strands and the model on the strand with the higher score was used to assess accuracy. This approach isolated the issue of the accuracy of these programs if the genomic sequence is large and the gene is encoded only in a small region of this sequence. There are other factors, such as the ability to choose the correct set of homologs that affect accuracy, but these factors were similar for all the programs, and other suboptimal (but perhaps more realistic) techniques would lead to lower accuracy. Thus, the accuracy numbers for the semiartificial sequences are not underestimated.
Table 2 shows the accuracy of the gene identification tools in Gen178, the set of simulated genomic sequences. As expected from theoretical considerations, SSBGP tools were mostly unaffected by the inclusion of genic sequences in the random intergenic-like DNA. PROCRUSTES appears to be less robust than GENEWISE when analyzing large genomic sequences. In particular, there is a significant decrease in specificity at the exon level (from 0.82 to 0.75), the likely result of predicting a relatively large number of small exons in otherwise noncoding DNA [wrong exons (WE) increasing from 0.04 to 0.16]. The comparatively low decrease in specificity at the nucleotide level, from 0.95 to 0.94, suggests that most of these false exons are rather short. Surprisingly, PROCRUSTES sensitivity at the exon level is slightly higher in the set of artificial sequences than in the set of single gene sequences.
Table 2.
Accuracy of Gene Prediction Tools in the Set of Semiartificial Genomic (SAG) Sequences (Gen178)
| Program | No. |
Nucleotide
|
Exon
|
Gene
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sn | Sp | CC | Sn | Sp | 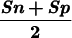 |
ME | WE | MG | WG | ||
| GenScan | 43 | 0.89 | 0.64 | 0.76 | 0.64 | 0.44 | 0.54 | 0.14 | 0.41 | 0.03 | 0.28 |
| 0.92 | 0.92 | 0.91 | 0.76 | 0.76 | 0.76 | 0.09 | 0.09 | ||||
| GeneWise | 43 | 0.98 | 0.98 | 0.97 | 0.88 | 0.91 | 0.89 | 0.06 | 0.02 | ||
| 0.98 | 0.98 | 0.97 | 0.88 | 0.91 | 0.89 | 0.06 | 0.02 | ||||
| Procrustes | 43 | 0.93 | 0.94 | 0.93 | 0.80 | 0.75 | 0.77 | 0.10 | 0.16 | ||
| 0.93 | 0.95 | 0.93 | 0.76 | 0.82 | 0.79 | 0.11 | 0.04 | ||||
(Italics) The accuracy values in the set of single gene sequences (from Table 1).
The accuracy of BLASTX was not affected by the intergenic context (data not shown) because no hits with a P value more significant than 10−10 were found in the simulated DNA.
Accuracy of ab initio gene finders suffered substantially in the set of artificial genomic sequences. Because of the tendency of gene finders to overpredict exons, one would expect that by placing the genic sequences in the simulated-intergenic context, some loss of specificity would be observed, with programs predicting perhaps a few extra exons in otherwise random DNA. On the other hand, one would expect the sensitivity to remain essentially constant as the exons predicted in the genic sequences should still be predicted when these are included in simulated-intergenic DNA. However, a significant decrease in specificity is observed (Table 2). For instance, GENSCAN specificity at the exon level drops to 0.64 from 0.92, and the proportion of WEs climbs to 41% from 9% in the single gene sequences. In addition, a significant decrease in sensitivity is also observed, with programs failing to predict exons that were correctly identified in the single gene sequences. For instance, the proportion of missing exons increases for GENSCAN from 9% to 14%. Almost 30% of the GENSCAN genes are predicted in the simulated-intergenic DNA. For ab initio gene finders, we believe these accuracy values (on SAG sequences) are more representative of their true accuracy on large genomic sequences than those obtained in the typical single gene benchmark experiments.
Figure 2 shows the predictions of the different programs in one of the artificially generated genomic sequences (∼157-kb long). As mentioned, SSBGPs predict the genic structure of the artificial genomic sequence rather well. Performance of ab initio gene finders, on the other hand, degrades substantially.
Figure 2.

(AGS17, top) Gene predictions in one of the artificial genomic sequences. The row EMBL indicates the coordinates of the actual genes. Exons corresponding to the same gene (or predicted to be in the same gene) are linked by a box. (AGS17, middle) Predictions of GENSCAN finders in the region 23,000 to 41,000 from the semiartificial genomic sequence. (HSIL9RA, bottom) The predictions improve if GENSCAN is provided only the 18,000-bp long genic sequence that has been inserted in this region. This figure, as well as Fig. 1, has been prepared using gff2ps. (Abril and Guigó 2000)
Although all genes predicted by GENSCAN overlap real genes, it still predicts a large number of false positive exons. In addition, even when predicting the exons correctly, their assembly into genes is often incorrect. For instance, in the sequence in Figure 2, GENSCAN has difficulty in predicting the correct gene boundaries, and it expands the gene beyond its actual limits. In the lower portion of the Figure 2, we compare the predictions in the region between positions 23,000 and 41,000 from the SAG sequence to the predictions on just the actual genic sequence (without the random context). GENSCAN performance suffers substantially from this inclusion in pseudointergenic context. One explanation is that GENSCAN uses the wrong isochore model for this sequence: the actual isochore structure being destroyed by the usage of artificial intergenic context. In such a case, decrease in performance would be an artifact of our SAG sequences rather than a feature of GENSCAN. Experiments with gene finders other than GENSCAN (data not shown) indicate that such a decrease in performance is not specific to GENSCAN, but rather a general feature of ab initio gene finders.
As with the set of single gene sequences, the comparison of GENSCAN with SSBGP tools is not strictly fair. The SSBGPs are affected by the existence of closer homologs, while GENSCAN is not affected. To study the effects of the range of similarity on the accuracy of gene prediction in the SAG data set, we extracted two different sets of SAG sequences. In the first set, each gene in each SAG sequence has a strong homolog (BLASTX P value < 10−50), and in the other set, each gene in each sequence had a moderate homolog (BLASTX P value between 10−50 and 10−6). Some of the genes in the second set also had better homologs which were ignored for this analysis. The results are shown in Table 3. If the similarity is strong, the sequence similarity-based methods perform very well, outperforming ab initio tools (as in Table 2). However, if the average similarity between the genes encoded and the known proteins is only moderate (though perhaps, still better than expected for real genomic sequences), the performance of these tools is similar to the performance of GENSCAN. At the exon level, the overall accuracy stays at ∼50%. A very similar accuracy has also been observed independently on test sets on actual genomic sequences (http://predict.sanger.ac.uk/th/brca2/; see Discussion). We believe this is still an overestimation of the actual accuracy of these tools in real genomic sequences.
Table 3.
Accuracy of Gene Prediction Tools in the Set of Semiartificial Genomic Sequences, When Either Strongly or Moderately Similar Sequences are Used to Model the Genes
| Program |
Strong similarity P Value < 10−50 17 SAG sequences
|
Moderate similarity 10−50 < P value < 10−6 26 SAG sequences
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Nucleotide
|
Exon
|
Nucleotide
|
Exon
|
|||||||||
| Sn | Sp | CC | Sn | Sp | 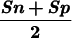 |
Sn | Sp | CC | Sn | Sp | 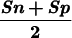 |
|
| GenScan | 0.91 | 0.66 | 0.77 | 0.67 | 0.46 | 0.56 | 0.91 | 0.61 | 0.74 | 0.67 | 0.43 | 0.55 |
| GeneWise | 0.99 | 0.99 | 0.99 | 0.90 | 0.93 | 0.91 | 0.68 | 0.98 | 0.81 | 0.46 | 0.63 | 0.54 |
| Procrustes | 0.92 | 0.96 | 0.94 | 0.80 | 0.75 | 0.77 | 0.66 | 0.79 | 0.72 | 0.48 | 0.32 | 0.40 |
The geometric mean of the P values of the strong similarity sequences was 10−135 and for the weaker similarity group it was 10−39.
DISCUSSION
Computational genefinders produce acceptable predictions of the exonic structure of the genes when analyzing single gene sequences with very little flanking intergenic sequence, but are unable to correctly infer the exonic structure of multigene genomic sequences. In particular, ab initio genefinders predict and utilize intergenic boundaries poorly. Conversely, as our results indicate, sequence similarity searches on databases of known coding sequences are extremely helpful in deciphering the exonic structure for the genes that have known homologs. For very strong similarity, SSBGP tools appear to be the most useful. Surprisingly even for genes predicted based on homologs with a moderate degree of similarity (10−50 < P value < 10−6), GENSCAN performs comparably to SSBGP programs. It appears that at such levels of similarity, potential splice signals and statistical biases in the sequence composition carry information comparable to sequence similarity for the purposes of identifying coding regions. It is possible that the use of SAG sequences does not provide a realistic scenario to test the accuracy of computational gene finders. Ideally, one would like to use large genomic sequences with gene structure experimentally verified. However, experimentally verifying each and every gene along with alternative splice structures in a large genomic sequence remains a difficult challenge. Techniques such as exon-trapping (Church et al. 1994) have high sensitivity but poor specificity, while RT–PCR or identifying a cDNA clone for every transcript can be fairly specific (Hochgeschwender 1992), but have less than perfect sensitivity and are dependent on finding a tissue in a developmental stage under an environmental condition in which that gene (or alternative gene product) is expressed. In particular, proving that a piece of sequence (that appears coding to gene-prediction programs) is not coding is extremely difficult. Thus, even though there are a number of attempts to consolidate genomic gene prediction data sets [Banbury Cross (http://igs-server.cnrs-mrs.fr/igs/banbury), GeneSafe (http://www.hgmp.mrc.ac.uk/Genesafe), GASP (http://www.fruitfly.org/GASP1/)], the number of experimentally well-annotated large genomic sequences remains small, and even in those cases, the reliability of the annotation is difficult to assess (Reese et al. 2000). To compensate for the lack of these verified data sets, we have built semiartificial data sets with known genes placed in the context of random intergenic sequence. This ensures that all the genes in these sequences are known. In fact, most of these genes have fairly small genomic spread (i.e., none of the introns is very large), and a number of the ab initio gene prediction programs have been trained on them. This should make this data set easy for most programs. However, our model for intergenic sequence is possibly imperfect for at least two reasons: The genes are not necessarily placed in the correct isochore context; and the apparent codon composition in the simulated intergenic DNA may be different from that of actual intergenic sequence. These imperfections may conceivably make gene prediction more difficult on this data set for ab initio programs, but we think these are more than offset at least in part by the small genes and the fact that the programs have partly trained on these genes. Overall, the sensitivity and specificity numbers are most instructive in the relative context. The sensitivity of most tools remains high even when confronted with large intergenic sequences, but the specificity of the ab initio tools drops because of large intergenic regions.
Interestingly, the accuracy reported here for GENSCAN is very similar to the accuracy found in the BRCA2 region (Chruch et al. 1994; Couch et al. 1996); probably the best annotated human genomic region from an experimental standpoint. BRCA2 region is a large genomic tract with multiple genes, thus, a difficult data set for most gene prediction programs. At the exon level, Tim Hubbard and Richard Bruskiewich (Sanger Center, UK) report for GENSCAN in this region a sensitivity of 0.63 (termed coverage there) and a specificity of 0.38 (termed accuracy there) (http://predict.sanger.ac.uk/th/brca2/). As anticipated, these values are slightly worse than the ones we have found here in the SAG data set (0.64 and 0.44, respectively). This seems to indicate that the approach of building artificial genomic sequences is not too unrealistic, and that it could be useful both for training and testing gene prediction programs. Results in these sequences, however, should be taken as an upper bound estimate of the accuracy of the programs in real genomic sequences.
There is a growing class of gene identification programs that combine both sequence similarity and traditional coding potential measures, such as Genie (Kulp et al. 1996 1997), HMMgene (Krogh 1997), and GSA (Huang et al. 1997). Unfortunately, because of a lack of public availability at the time of the initiation of this study, their evaluation will have to await a future analysis.
EST similarity can also provide useful information regarding gene structure for ∼85% of the common genes (Guigó et al. 2000). A set of single gene sequences in h178 was used to optimize a method for deriving exonic structures from EST matches. When using the EST sequences in the public databases, the method yielded an accuracy of Sn = 0.72, Sp = 0.87, and CC = 0.69 at the nucleotide level, when predicted gene structures were compared to the annotated mRNA (not the coding) exonic structure. Other secondary questions regarding EST-based gene prediction may also be important, such as the extent to which EST matches help in delineating the gene boundaries.
Though there is considerable variation in the accuracy of various gene prediction programs depending on data sets and the availability and choice of homolog, we believe that a judicious use of these programs in combination can result in highly accurate gene structures for genes with known homologs. There is, however, still considerable progress to be made on predicting alternative spliced structures and genes with no known homologs.
METHODS
Computational Gene Identification Tools
Gene identification tools may be categorized into ab initio tools (those not utilizing sequence similarity and relying on intrinsic gene measures such as coding potential and splice signals), and those based (at least partly) on sequence similarity.
Ab initio Gene Identification Tools
The ab initio gene identification tools use information from both the gene signals in the genomic DNA (such as splice sites, start and stop codons, and promoter elements), and the statistical biases in DNA composition that is characteristic of coding regions. There are a number of such programs (for reveiws, see Fickett 1996; Claverie 1997; Guigó 1997a; Burge and Karlin 1998; Haussler 1998). GENSCAN (Burge and Karlin 1997) is one of the most accurate and widely used programs in this category, and we use it as a representative.
SSBGP Tools
A number of recent programs predict genes by aligning genomic sequences with candidate homologous protein sequences. These programs may include a splice site model, coding potential, and sequence similarity to known proteins to infer gene predictions. We evaluated two of these programs, PROCRUSTES (Gelfand et al. 1996), and GENEWISE (Birney and Durbin 1997) (http://www.sanger.ac.uk/Software/Wise2/).
These programs require as input a candidate homologous protein sequence; therefore, in typical use, a sequence similarity database search with the query genomic sequence is performed a priori and the top hit is used as the candidate (or top hits are used as candidates, in the case of a query sequence encoding multiple genes). The database similarity searches were performed against the nonredundant protein sequence database from NCBI, nr, using BLASTX (Altschul et al. 1990; Gish and Sates 1993). BLASTX performs a translation of the query sequence into the six frames, and searches for similarities between each of these translations and the protein sequences in the database.
BLASTX was designed as a similarity-based gene prediction tool, and it is possible to model a gene directly from the database search results. BLASTX, however, does not confine its similarity to exon;, thus the similarity region is not constrained to begin or end on splice sites. Moreover, BLASTX does not explicitly predict genes in genomic sequences, and some postprocessing of its output is required to infer gene predictions from the search results. Indeed, while computational gene finders predict genes, that is pairs of positions (corresponding to exon starts and ends) along the query genomic sequence, database searches only produce lists of sequence database hits along the query sequence. Each hit above a given similarity threshold may be assumed to be a coding exon. For different database entries, however the set of hits may be different. The problem is then to infer a gene model from the set of database hits. A simple solution is to project the hits into a single axis along the genomic sequence, and to assume the union of these projections to be the coding exons.
In total, three strategies based on BLAST were tested:
default — A procedure consisting of projecting the HSPs onto the genomic sequences was used (see Guigó et al. 2000). BLASTX was run with E = 1e-10 − filter xnu + seg S2 = 60, and all HSPs with identity <40% were discarded. The choices of S2 and percentage identity were influenced by the need to restrict false matches.
topcomboN — BLASTX was used with default parameters except for −filter xnu + seg topcomboN = 1. HSPs with P value > 10−20 were discarded, and the projections along the query sequence of the remaining HSPs assumed to be the predicted coding exons. WashU–BLAST has a parameter topcomboN that limits all HSPs generated to be in one consistent group. For example, for BLASTX searches, each region of the nucleotide sequence is only aligned to a single region on the protein sequence and the ordering of these HSPs has to be consistent along both the nucleotide and protein sequences. This restricts spurious matches arising from repetitive domains with query sequences, and from low scoring hits in introns and flanking regions.
two-stage — BLASTX was used in a two stage process that first identifies one or more candidate protein sequences in the presence of a low-complexity filter. In the second stage, BLASTX is used to align the candidates individually with the genomic sequence, this time without the filter and with topcomboN = 1. This two pass technique is closer to the strategy used with GENEWISE and PROCRUSTES, where a first BLASTX search pinpoints the protein homolog to be used, and a subsequent GENEWISE uses this protein homolog.
Both GENEWISE and PROCRUSTES were run with mostly standard parameters: GENEWISE v2.1.16b -both -gff -pretty -para -cdna -genes -quiet and PROCRUSTES was run in the local mode with MIN_EXN 20, MIN_IVS 50, GAP 2, INI_GAP 10, MATRIX pam120.mtx. GENSCAN was run with default parameters.
Benchmark Sets
Two sets of sequences have been used to evaluate the programs discussed above. First, a typical benchmark set made of sequences from the EMBL database release 50 (1997) that included 178 human genomic sequences coding for single complete genes for which both the mRNA and the coding exons are known. The procedure used to extract the sequences is described in Burset and Guigó (1996) and Guigó (1997b). We will refer to this set here as h178. All the genes in this data set are on the forward strand. Other characteristics of h178 are provided in Table 4.
Table 4.
Characteristics of the Benchmark Sequence Sets
| Set | No. | G + C |
Sequence length
|
Genes (average)
|
CDS (average)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| average | min | max | no. | length | density | no. exons | length | density | ||||
| h178 | 178 | 50% | 7169 | 622 | 86640 | 1 | 3657 | 53% | 7169 | 5.1 | 968 | 21% |
| Gen178 | 42 | 40% | 177160 | 70037 | 282097 | 4.1 | 15136 | 8.6% | 43000 | 21 | 4007 | 2.3% |
The columns Genes (average) and CDS (average) provide values averaged over all the sequences (178 in h178 and 42 in Gen178). Gene density provides the percentage of nucleotides that occur in genic regions (exons, introns, and UTRs), and the number of kilobases per gene. CDS no. exons is the average number of coding exons per sequence, and CDS density is the percentage of nucleotides that occur in coding regions.
For the reasons discussed in this paper, this does not appear to be a challenging benchmark set for estimating the accuracy of gene identification programs in the larger genomic sequences. Unfortunately, very few large genomic sequences have been studied extensively to produce complete experimental determinations of the exact structure of each gene. To overcome this limitation, we generated a semiartificial set of genomic sequences in which accurate gene annotation can be guaranteed.
In essence, a set of annotated genic sequences are placed randomly in a background of random intergenic DNA. The length of the semiartificial sequence is generated randomly according to a normal distribution. Genomic fragments containing genes and random-sized segments of intergenic sequence are then concatenated until their combined lengths exceed the target. The strands are also chosen at random for each genic subsequence.
Table 4 shows the characteristics of the generated sequences when the method is applied to the sequences in h178 and the intergenic background is generated using a Markov Model of order 5 as described in Guigó and Fickett (1995) assuming an average intergenic G + C content of 38%. The 178 genic sequences were collapsed into 42 SAG sequences. Some of the resulting parameters, such as average G + C content of 40%, a gene every 43 Kb, and a coding density of 2.3% are in agreement with that for the overall human genome. This data set has flaws and is not a perfect representative of the human genome. Some of the ignored characteristics include the isochore organization of the human genome, known and unknown repeats in the intergenic regions, presence of pseudogenes and other evolutionary remnants, genes with huge introns, and tandem gene clusters. Most of the missing properties (pseudogenes, repeats, huge introns) make gene prediction much more difficult. Thus, we expect the accuracy results on Gen178 to still be an overestimate of the true accuracy.
Evaluating Accuracy
The measures of accuracy used here are discussed extensively in Burset and Guigó (1996). We will restate them briefly. Accuracy is measured at three different levels: nucleotide, exon, and gene. At the nucleotide and exon levels, we essentially compute the proportion of actual coding nucleotides/exons that have been predicted correctly–(which we call Sensitivity) and the proportion of predicted coding nucleotides/exons that are actually coding nucleotides/exons (which we call Specificity). To compute these measures at the exon level, we will assume that an exon has been predicted correctly only when both its boundaries have been predicted correctly. To summarize both Sensitivity and Specificity, we compute the Correlation Coefficient at the nucleotide level, and the average of Sensitivity and Specificity at the exon level. At the exon and gene level, we also compute the Missing Exons/Genes (the proportion of actual exons/genes that overlap no predicted exon/gene) and the Wrong Exons/Genes (the proportion of predicted exons/genes that overlap no actual exon/gene).
The measures are computed globally from the total number of prediction successes and failures (at the base and exon level) on all sequences. Accuracy in Table 1 is computed ignoring predictions in the reverse (wrong) strand. The first column in Tables 1 and 2 indicates the number of sequences for which the progams produced predictions.
Data Availability
Both the set of single gene sequences and the set of semiartificially generated genomic sequences will be available from http://www1.imim.es/databases/gpecal2000/.
Acknowledgments
We thank Randall F. Smith, Ewan Birney, Chris Burge, and Warren Gish, and the anonymous referees (one in particular for pointing out the topcomboN feature in WU-BLAST) for useful comments. This work was partially supported by a grant from Plan Nacional de I + D, BIO98-0443-C02-01, and from the Ministerio de Educación y Ciencia (Spain) to R.G. M.B. is supported by a Formación de Personal Investigador fellowship, FP95-38817943, from the Ministerio de Educación y Ciencia (Spain), J.F.A. is supported by a predoctoral fellowship, 99/9345, from the Instituto de Salud Carlos III (Spain).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rguigo@imim.es; FAX 3493-221-3237.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.122800.
REFERENCES
- Abril, J.F. and Guigó, R. 2000. gff2ps: A tool for visualizing genomic annotations. Bioinformatics in press. [DOI] [PubMed]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman D. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Birney E, Durbin R. Dynamite: A flexible code generating language for dynamic programming methods used in sequence comparison. Ismb. 1997;5:56–64. [PubMed] [Google Scholar]
- Burge CB, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- ————— Finding the genes in genomic DNA. Curr Opin Struc Biol. 1998;8:346–354. doi: 10.1016/s0959-440x(98)80069-9. [DOI] [PubMed] [Google Scholar]
- Burset M, Guigó R. Evaluation of gene structure prediction programs. Genomics. 1996;34:353–357. doi: 10.1006/geno.1996.0298. [DOI] [PubMed] [Google Scholar]
- Church DM, Stotler CJ, Rutter JL, Murrell JR, Trofatter JA, Buckler AJ. Isolation of genes from complex sources of mammalian genomic DNA using exon amplification. Nat Genet. 1994;6:98–105. doi: 10.1038/ng0194-98. [DOI] [PubMed] [Google Scholar]
- Claverie J-M. Computational methods for the identification of genes in vertebrate genomic sequences. Hum Mol Genet. 1997;6:1735–1744. doi: 10.1093/hmg/6.10.1735. [DOI] [PubMed] [Google Scholar]
- Couch FJ, Rommens JM, Neuhausen SL, Couch EJ, Rommens JM, Neuhausen SL, Belanger C, Dumont M, Abel K, Bell R, Berry S, Bogden R, Cannon-Albright L. Generation of an integrated transcription map of the BRCA2 region on chromosome 13q12-q13. Genomics. 1996;36:86–99. doi: 10.1006/geno.1996.0428. [DOI] [PubMed] [Google Scholar]
- Fickett JW. Finding genes by computer: the state of the art. Trends Genet. 1996;12:316–320. doi: 10.1016/0168-9525(96)10038-x. [DOI] [PubMed] [Google Scholar]
- Gelfand MS, Mironov AA, Pevzner PA. Gene recognition via spliced alignment. PNAS. 1996;93:9061–9066. doi: 10.1073/pnas.93.17.9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gish W, States D. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- Guigó R. Computational gene identification. J Mol Med. 1997a;75:389–393. doi: 10.1007/s001099770003. [DOI] [PubMed] [Google Scholar]
- ————— Computational gene identification: An open problem. Comput Chem. 1997b;21:215–222. doi: 10.1016/s0097-8485(97)00008-9. [DOI] [PubMed] [Google Scholar]
- Guigó R, Fickett JW. Distinctive sequence features in protein coding, genic non-coding, and inter-genic human DNA. J Mol Biol. 1995;253:51–60. doi: 10.1006/jmbi.1995.0535. [DOI] [PubMed] [Google Scholar]
- Guigó R, Burset M, Agarwal P, Abril JF, Smith RF, Fickett JW. Sequence similarity based gene prediction. In: Suhai S, editor. Genomics and proteomics: Functional and computational aspects. New York, NY: Kluwer Academic / Plenum Publishing; 2000. pp. 95–105. [Google Scholar]
- Haussler D. Trends Biochem. Sci., supplementary guide to bioinformatics. 1998. Computational genefinding; pp. 12–15. [Google Scholar]
- Hochgeschwender U. Toward a transcriptional map of the human genome. Trends Genet. 1992;8:41–44. doi: 10.1016/0168-9525(92)90336-3. [DOI] [PubMed] [Google Scholar]
- Huang X, Adams MD, Zhou H, Kerlavage AR. A tool for analyzing and annotating genomic sequences. Genomics. 1997;46:37–45. doi: 10.1006/geno.1997.4984. [DOI] [PubMed] [Google Scholar]
- Krogh A. Two methods for improving performance of an HMM and their application for gene finding. ISMB. 1997;5:179–186. [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden markov model for the recognition of human genes in DNA. In: States DJ, Agarwal P, Gaasterland T, Hunter L, Smith R, editors. Intelligent systems for molecular biology. Menlo Park, CA: AAAI Press; 1996. pp. 134–142. [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. Integrating database homology in a probabilistic gene structure mode. In: Altman RB, Dunke AK, Hunter L, Klein TE, editors. Biocomputing: Proceedings of the 1997 Pacific Symposium. New York, NY: World Scientific; 1997. pp. 232–244. [PubMed] [Google Scholar]
- Mott R. EST_GENOME: A program to align spliced DNA sequences to unspliced genomic DNA. Comput Appl Biosci. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Both the set of single gene sequences and the set of semiartificially generated genomic sequences will be available from http://www1.imim.es/databases/gpecal2000/.