

Abstract
The gene that is mutated in ataxia-telangiectasia (A-T), ATM, is catalytically activated in response to DNA damage. Yet a full accounting for the CNS deficits in human A-T or its mouse models remains elusive. We have analyzed the CNS phenotypes of two mouse Atm alleles—Atmtm1Bal (Bal) and Atmtm1Awb (Awb). Neither mutant has detectable mRNA or protein in peripheral tissues. In brain, although Bal/Bal mice have no ATM protein, they have nearly normal amounts of Atm mRNA. Bal/Bal neurons exhibit extensive cell cycle reentry and degeneration in both cortex and cerebellum. Unexpectedly, in Awb/Awb mice a novel mRNA is found in which the engineered mutation is excised. This mRNA is apparently translated and produces a catalytically active ATM protein that responds to DNA damage by phosphorylating p53 and Chk2. Prompted by these results, we examined eight cases of human A-T and found evidence for residual ATM protein in seven of them. These findings offer important new insights into the human disease and the role of brain ATM activity in the severity of the neurological symptoms of A-T.
Introduction
Ataxia telangiectasia (A-T) is a hereditary multisystem disease resulting from mutations of the ATM gene (Sedgwick and Boder, 1960; Boder, 1975; Bundey, 1994; Savitsky et al., 1995). A-T is a progressive childhood neurodegenerative condition that results in cerebellar ataxia, choreoathetosis, and oculomotor dysfunction. By their second decade, affected children are usually wheelchair bound. The neurological deficits are associated with a massive loss of Purkinje and granule cells with striatal neurons and sensory ganglion cells involved later in the disease. Non-neurological phenotypes include immune system defects, hypersensitivity to ionizing radiation, and increased susceptibility to cancer (Boder and Sedgwick, 1958; Gatti et al., 1988; Bundey, 1994; Savitsky et al., 1995; Lavin and Shiloh, 1997).
The A-T gene (A-T Mutated—ATM) has been identified. The protein encoded by this gene is a 370 kDa member of the PI3-kinase family that functions as a key component of the cellular response to DNA damage. ATM is recruited to the DNA lesion by the MRN complex (Mre11/Rad50/Nbs1), where it autophosphorylates at serine 1981 in human (S1987 in mouse). This activates the kinase and allows ATM to phosphorylate downstream targets such as histone H2AX, p53, and the checkpoint kinase Chk2, among many other proteins. The result is a comprehensive program of cell cycle arrest and DNA repair that helps ensure the integrity of the genome.
The generation of many different mouse models of A-T has added greatly to our understanding of the disease (Savitsky et al., 1995; Barlow et al., 1996; Elson et al., 1996; Xu et al., 1996; Herzog et al., 1998; Sandoval et al., 1999; Borghesani et al., 2000). Most human ATM mutations result in protein truncations that destabilize the message, the protein, or both (Sandoval et al., 1999; Chun et al., 2003). The first reported mouse mutation, Atmtm1Awb, was engineered in the region of the 38th exon—the site of multiple independent human mutations (Barlow et al., 1996). Adult Atmtm1Awb (Awb) mice demonstrate radiosensitivity, immune deficiency, and cancer susceptibility. There is, however, no detectable loss of cerebellar neurons in these animals (Barlow et al., 1996). A second A-T mouse model, Atmtm1Bal (Bal), was engineered to delete the entire kinase domain (exon 58). Homozygous Bal/Bal mice are also radiosensitive, immune deficient, and prone to lymphomas. In this case, Purkinje cell vacuolization and degeneration has been reported (Xu et al., 1996; Kuljis et al., 1997), but other laboratories have failed to replicate this finding (Barlow et al., 1996, 2000).
In the current study, we report that the Bal/Bal mouse brain has significant levels of Atm mRNA but no detectable ATM protein. Bal/Bal neurons undergo cell cycle reentry and we confirm the presence of significant neurodegeneration. The Awb/Awb mouse brain also contains Atm message, but unlike Atmtm1Bal the Atmtm1Awb message undergoes a novel, brain-specific splicing event that leads to the production of nearly full-length Atm mRNA and a catalytically active protein that effectively responds to DNA damage. Consistent with these findings in mouse, human A-T samples also maintain residual ATM protein and mRNA.
Materials and Methods
Antibodies and chemical regents.
All antibodies used in this study were commercially available. ATM (2C1A1, 5C2, and Y170), P-Ser1987 ATM, P-Thr68-Chk2, PSD95, LC3, LC3-II, BrdU, and calbindin were from Abcam. phospho-S15-p53, caspase-3, and cleaved caspase-3 were purchased from both Abcam and Cell Signaling Technology; no difference in reactivity was found between the two lots. Cyclin A antibodies were purchased from Santa Cruz Biotechnology and Abcam; Cyclin D1 was from Santa Cruz Biotechnology. P-ser1981 ATM was from Rockland. Secondary antibodies used for immunocytochemistry were as follows: donkey anti-rabbit IgG and chicken anti-mouse IgG (Santa Cruz Biotechnology); chicken anti-mouse or rabbit Alexa 488; donkey anti-mouse or rabbit Alexa 594 (Invitrogen); all were used at a dilution of 1:500. DAPI (4′,6′-diamidino-2-phenylindole) was used as a nuclear counterstain.
Constructs and plasmids.
For GFP-WT-34-41 exons and GFP-Awb-34-41 exons, WT-34-41 exons and Awb-34-41 exons were cloned from wild-type and Awb/Awb mouse brain mRNA by SuperScript III one-step RT-PCR system (Invitrogen). RT-PCR products were subcloned in EGFP-C3 vector (Clontech).
Animals.
Our colony of Atmtm1Awb was maintained by intercrossing heterozygous animals obtained originally from Jackson laboratory; Atmtm1Bal mice were a gift from Dr. Yang Xu. Generation of mutants was achieved through the mating of heterozygous Atmtm1Awb males and Atmtm1Awb females as well as the mating of heterozygous Atmtm1Bal males and Atmtm1Bal females. Timed pregnancies were established from these matings; the date of appearance of a vaginal plug was considered embryonic day 0.5. Embryos were taken at embryonic day 16.5 (E16.5) for either cortical cultures or histology. All animals used in the experiments were 2–3 months old. All animal procedures were performed in accordance with Rutgers University Institutional Animal Care and Use Committee standards. The animal facilities at Rutgers University are fully AAALAC accredited.
Human subjects.
Human autopsy tissue from four individuals diagnosed with A-T and four age-matched controls was obtained from UCLA Medical Center and Children's Hospital Los Angeles. Brain tissue was routinely fixed, paraffin embedded, and sectioned at 10 μm. All A-T cases were confirmed both clinically and by postmortem examination. The average age of the subjects was 21 years for the A-T patients and 25 years for non-A-T controls. The non-A-T controls were free of any known CNS neurological diseases at the time of death. Human frozen tissue was secured from four individuals diagnosed with A-T and four age-matched controls; the average age of the subjects was 18 years for the A-T patients and 20 years for non-A-T controls. Samples were obtained from NICHD Brain and Tissue Bank of Developmental Disorders at the University of Maryland, Baltimore, MD.
Immunohistochemistry.
For DAB/brightfield staining, all paraffin-embedded human sections were deparaffinized in xylene and then rehydrated through graded ethanol to water. The sections were pretreated in 0.3% hydrogen peroxide in methanol for 30 min to remove endogenous peroxidase activity, rinsed in Tris-buffered saline (TBS), and then treated with 0.1 m citrate buffer in a microwave at sufficient power to keep the solution at 100°C for 10 min. Sections were cooled in the same buffer at room temperature (RT) for 30 min and rinsed in TBS. Slides were incubated in 10% goat serum in PBS blocking solution for 1 h at RT, after which primary antibody was applied to the sections that were then incubated at 4°C overnight. The sections were washed three times in TBS before applying the secondary antibody (Vector Laboratories). Secondary antibody was applied for 1 h at RT. Afterward, sections were rinsed three times in TBS. Rinsed sections were then incubated in Vectastain ABC Elite reagent for 1 h and developed using diaminobenzidine, according to the protocol of the manufacturer (Vector Laboratories). The sections were counterstained with hematoxylin, and after dehydration, all sections were mounted in Permount under a glass coverslip. Control sections were subjected to the identical staining procedure, except for the omission of the primary antibody.
Immunofluorescence.
The mouse frozen brain sections were first rinsed in PBS, followed by pretreatment in antigen unmasking solution (low pH) for 30 min at 100°C. After the slides had cooled in buffer for 30 min at room temperature, slides were rinsed in PBS. Sections were incubated in 10% goat serum in PBS to block nonspecific binding for 1 h at room temperature. All primary antibodies were diluted in PBS containing 0.5% Triton X-100 and 5% goat serum and incubated with sections overnight at 4°C. After rinsing in PBS, they were incubated for 1 h with secondary antibody, which was conjugated with various fluorescence-labeled secondary antibodies. All sections were mounted in ProLong Gold antifade reagent with DAPI (Invitrogen) under a glass coverslip. All experiments were conducted in triplicates.
Mass spectrometry.
Protein extracts from mouse brain were immunoprecipitated with 2C1 ATM antibody. Immunoprecipitates were denatured and separated on a 6% SDS-PAGE gel. After Coomassie blue staining, the interesting gel slices were reduced with 10 mm DTT, alkylated by 20 mm iodoacetamide, and then digested by modified trypsin at 37°C overnight. The digested peptides were extracted and speed vac dried. Then the digestions were solubilized in 0.1% trifluoroacetic acid, and analyzed by HPLC-MSMS (Dionex Ultimate 3000 RSLCnano system with Thermo LTQ-Orbitrap-Velos mass spectrometer). The data were searched against mouse database by GPM Manager (Version 2.2.1).
RT-PCR.
Reverse-transcription reactions were performed using total RNA according to standard procedures. Briefly, total RNA from wild-type and mutant mouse brain, spleen, and thymus was prepared using PureLink micro-to-midi total RNA purification system (Invitrogen). RT-PCR was performed with Superscript III one-step RT-PCR system with platinum Taq High Fidelity (Invitrogen) to detect the splice positions of ATM. For mouse, the mRNA level of 36B4 was used as an internal control. The following sets of primers were used: 5′-gattacaagttctgaaaccc-3′ (sense) and 5′-gcttggtatgtatacacaagc-3′ (antisense) for ATM exons 14–15, 5′-ggaaggtgtttgggagaaatagg-3′ (sense) and 5′-ccgtattctagtaagggatgtaacattttc-3′ (antisense) for ATM exons 34–41, 5′-gaaaaggcatcaatattc-3′ (sense) and 5′c-ttcacaagctgtctcctttc-3′ (antisense) for exons 55–56, 5′-gatgtaagatggtgaaggacatgg-3′ (sense) and 5′-cacagcaccttctgaagaccaattc-3′ (antisense) for exons 55–61, 5′-ccaaaattctaaaatgtgct-3′ (sense) and 5′-cggcatatacgtgcaaattc-3′ (antisense) for PCNA, 5′-ttacttcaagtgcgtgcaga-3′ (sense) and 5′-atcttagaggccacgaacat-3′ (antisense) for cyclin D1, 5′-tgtccaatcctggt gatgtcc-3′ (sense) and 5′-tcagacaccagagtgcaagac-3′ (antisense) for p21, 5′-tcgacgggttgctcctcttaa-3′ (sense) and 5′-ctttccatgaaggaccagcag-3′ (antisense) for cyclin A, and 5′-cagctctggagaaactgctg-3′ (sense) and 5′-gtgtactcagtctccacaga-3 (antisense) for 36B4. For human samples, the mRNA level of tubulin was used as an internal control. The sequences of the primers used were as follows: 5′-ttacaaattcagaaactcttg-3′ (sense) and 5′-cttggtacagt tgctcaagca-3′ (antisense) for Atm exons 14–15, 5′-aggctgttggaagctgcttg-3′ (sense) and 5′-ctagtaatgg gttgtaacatc-3′ (antisense) for Atm exons 34–41, 5′-gtggaccacacaggagaatat-3′ (sense) and 5′-aatagaagaagtagctacact-3′ (antisense) for exons 55–58, and 5′-tggagccgggaataactg-3′ (sense) and 5′-gcctcgtcctcgccctcctc-3′ (antisense) for tubulin. The RT-PCR program was set to 55°C for 30 min and 94°C for 3 min, followed by 35 cycles of 94°C for 30 s, 55°C for 30 s, and 68°C for 1–2 min with an extension at 68°C for 10 min. The PCR products were analyzed on 1.5% agarose gels stained with ethidium bromide and visualized under ultraviolet light.
Western blotting.
For Western blots, protein extracts from mouse tissue or primary cultures were separated with SDS-PAGE and electrophoretically transferred onto nitrocellulose membranes. Membranes were blocked with 5% nonfat milk in TBST and probed with primary antibodies in blocking buffer, followed by treatment with HRP-linked secondary antibodies and ECL Western blotting detection reagents (Pierce Fisher Scientific).
Primary neuronal cultures and siRNA.
Embryonic cortical neurons were isolated by standard procedures. For ATM-deficient cultures, all embryos from an Atmtm1Awb/+ × Atmtm1Awb/+ or AtmtmBal/+ × Atmtm1Bal/+ mating were harvested and treated separately. Isolated E16.5 embryonic cerebral cortices were treated with 0.25% trypsin-EDTA and dissociated into single cells by gentle trituration. Cells were suspended in Neurobasal medium supplemented with B27 and 2 mm glutamine, then plated on coverslips or dishes coated with poly-l-lysine (0.05 mg/ml) and laminin (5 μg/ml). All cultures were grown for a minimum of 5 d in vitro (DIV) before any treatment. siRNA transfection with Lipofectamine 2000 was performed at DIV4 or DIV5 in primary cultural neurons. siRNA duplexes against mouse ATM were purchased from Integrated DNA Technologies. The sequence used for ATM sense strand was as follows: 5′-ggagcaugcucuaaggacatt-3′. All experiments were performed a minimum of three times; each condition was examined in triplicate.
BrdU incorporation.
For BrdU labeling, 10 μm BrdU was added to the Neurobasal media at DIV6. After labeling for 24 h, the primary neurons were fixed, and DNA was hydrolyzed by exposing the cells to 2N HCl for 10 min. Specimens were then neutralizing in 0.1 m sodium borate, pH 8.6, for 10 min and then rinsed extensively in PBS (three times) for 45 min before treatment with blocking reagent. Nonspecific antibody binding was blocked by exposing the fixed cells to 5% normal goat serum in 0.1% Triton-X for 1 h before application of the BrdU primary antibody. DAPI staining was used as indicator for total cells number. Counts were made of BrdU-positive, DAPI-positive, and double-positive cells.
Statistical analyses.
All data are presented as the means ± SEMs. We evaluated statistical differences by Student's t test. For neuronal degeneration, bars represent the incidence as a percentage of whole cerebellum region in mouse, and we evaluated statistics by χ2 analysis. For all analyses, we considered p < 0.05 to be statistically significant.
Results
Neurodegeneration in Atmtm1Bal mouse cerebellum
We reexamined the neuropathology of both Awb/Awb and Bal/Bal cerebella. Cerebellar cryostat sections were stained with calbindin, activated caspase-3, and the autophagy marker LC3-II (Fig. 1A). As reported previously (Barlow et al., 1996), there were no signs of degenerative changes in any of the cells in the cerebellar cortex of Awb/Awb homozygotes. In Bal/Bal cerebellum, however, calbindin and H&E staining revealed the presence of large vacuoles in the Purkinje cell layer. This was inconsistent with previous reports (Barlow et al., 2000), and so we investigated this phenotype carefully. At higher magnification in the calbindin-stained preparations, Bal/Bal Purkinje cell spines appeared disordered and reduced in density. This finding was all the more significant since the Awb/Awb Purkinje cell dendrites appeared indistinguishable from wild type (Fig. 1A, right). These atrophic structural changes were accompanied by the appearance of several indicators of neurodegeneration. Immunocytochemical evidence for activated caspase-3 in both Purkinje and granule cell cytoplasm was observed in Bal/Bal, but not in Awb/Awb or wild-type mice. We also found substantially elevated levels of LC3-II (Fig. 1A), a marker for one of the key steps in the autophagy process. Western blots of protein extracts confirmed these findings. Activated caspase-3, as assessed by its cleavage to the smaller active fragment (Fig. 1B, upper gel) and the processing of LC3 to LC3-II (Fig. 1C) are both apparent in these blots. Further, the impression of a reduced Purkinje cell dendritic arbor was confirmed by a clear reduction in the levels of the postsynaptic marker protein, PSD95 (Fig. 1B, middle gel). Unexpectedly, evidence of a degenerative process extended to other regions of the Bal/Bal brain, notably the neocortex (Fig. 1D).
Figure 1.
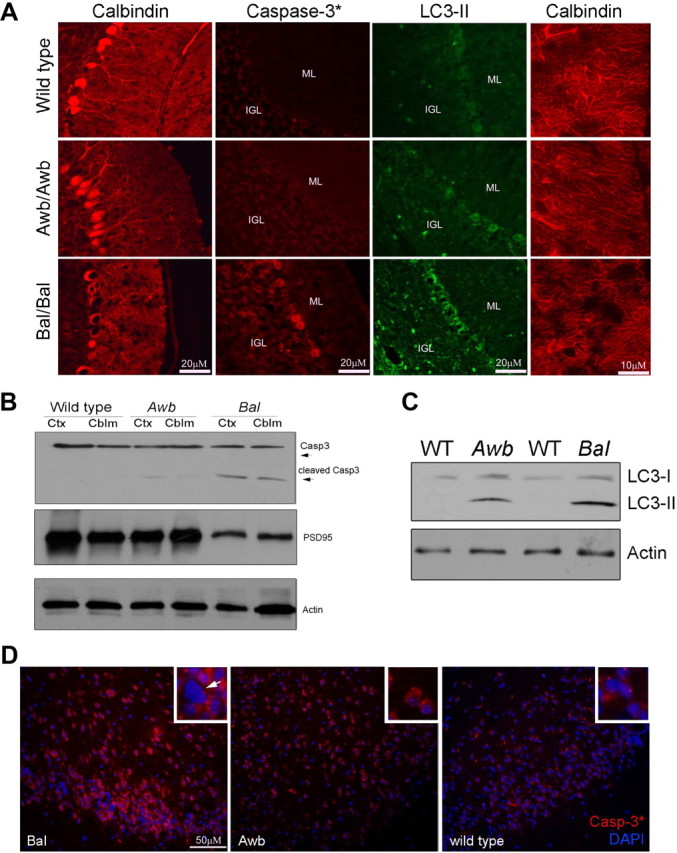
Degenerative changes found in Atmtm1Bal but not Atmtm1Awb Purkinje cells. A, Ten-micrometer cryostat sections of mouse cerebellum were immunostained with the antibodies indicated. Calbindin (first column) stains the entire Purkinje cell cytoplasm, activated caspase-3 (caspase-3*, second column) is a marker of ongoing apoptosis, and LC3-II (third column) is a marker for the autophagy process. Viewing the calbindin-stained Purkinje cell at higher magnification (fourth column) reveals the details of the Purkinje cell spiny dendrites. Three genotypes are shown, one in each row as indicated on the left (Bal, homozygous Atmtm1Bal; Awb, homozygous Atmtm1Awb). B, Western blot of lysates from wild type, Atmtm1Awb, and Atmtm1Bal probed with caspase-3 antibody [cleaved (Casp3*) and noncleaved (Casp3) isoforms are indicated with arrows] and PSD95. Both neocortex (Ctx) and cerebellum (Cblm) of Bal/Bal mice show evidence of apoptosis. Actin was used as a loading control. C, LC3-II levels are elevated in Bal/Bal mice. Western blots of wild-type, Atmtm1Awb, and Atmtm1Bal mouse whole-brain extracts were probed with LC3 antibody that recognizes both I and II isoforms. D, Immunostaining for activated caspase-3 in neocortex. Three genotypes were examined as indicated in the lower left of each main panel. Atmtm1Bal (Bal) neurons show enhanced immunostaining and frequent profiles suggestive of pyknotic nuclei (inset, white arrow). Comparable regions in the Atmtm1Awb mouse (Awb) have only modest increases in the levels of staining. The nuclei of even the most caspase-3-positive cells show no indication of nuclear condensation (inset). Wild-type neocortex has little to no immunoreactivity, although a few bright puncta of unknown identity are found. No suggestion of pyknosis (insets) was found in any region.
Enhanced cell cycle reentry in Atmtm1Bal mouse cerebellum
We have reported previously on the appearance of cell cycle markers in Purkinje cells of human A-T as well as in the Awb mouse model (Yang and Herrup, 2005). Expanding these findings to Bal/Bal mice, we found that the number of Purkinje cells that were positive for the presence of cell cycle proteins was substantially increased, compared to both wild-type and Awb/Awb mice (Fig. 2A, quantified in Fig. 2B). Semiquantitative RT-PCR analysis confirmed these results; the levels of p21, cyclin A, PCNA, and cyclin D1 mRNA were all greater in Bal/Bal cerebellum than in either Awb/Awb or wild type (Fig. 2C, quantified in Fig. 2D). We next examined the behavior of the three mouse Atm alleles in cultured neurons in vitro. In primary neuronal cultures of E16.5 cerebral cortex, no significant neuronal cell cycle activity (<5%) was found in either wild-type or Awb/Awb cultures as measured by BrdU incorporation after a 24 h exposure (Fig. 2E). By contrast, >20% of the neurons in Bal/Bal cultures incorporated BrdU during the same time period. The cell-autonomous difference in the ability to suppress the cell cycle in cortical neurons was unexpected. To determine the Atm dependence of this difference, Atm siRNA (Fig. 2E) was applied to cultures of each of the three genotypes. While the suppression of ATM synthesis led to little change in Bal/Bal cultures, BrdU incorporation increased in both wild-type and Awb/Awb neurons in the presence of Atm siRNA. The strong suggestion that emerges from these results is that Awb/Awb cortical neurons in vitro retain the ability to suppress the cell cycle and this ability is ATM dependent.
Figure 2.

Cell cycle reentry found in Atm mouse cerebellum. A, Images of mouse brain sections immunostained with anti-cyclin A and anti-cyclin D1 antibodies (red). Nuclei were counterstained with DAPI (blue). Genotypes and antibodies as indicated on the panel. A, Cyclin A; D1, cyclin D1. B, Immunostained Purkinje cells were counted in material similar to that shown in A. Results were expressed as a percentage of the total of calbindin-immunopositive cells. C, Cerebella from 3-month-old Atm+/+, Atmtm1Awb, and Atmtm1Bal mice were subjected to total RNA extraction, reverse transcription, and PCR amplification as described. Representative blots of the amplified PCR fragments of various cell-cycle genes from wild-type, Atmtm1Awb, and Atmtm1Bal mice cerebella at 20, 25, and 30 cycles of are shown. D, Quantifications of RT-PCR products from 3–5 animals of each genotype are displayed in the histogram. Band quantifications were normalized to 36B4 levels. Data are displayed as average ± SEM fold increase over Atm+/+, with Atm+/+ samples set to 1. p values (*p < 0.05) were calculated from multiple animals by Student's t test. E, E16.5 cortical neurons of the genotypes indicated were cultured for 4–5 d before exposure to either control siRNA (left) or siRNA against ATM. Twenty-four hours later, BrdU was added to the medium to assess cell cycle activity. Values represent the percentage of the total cell population (DAPI counts) that was BrdU immunolabeled. Each bar represents the average of three independent experiments; error bars denote SEM.
Brain-specific mutant ATM protein found in Awb/Awb but not Bal/Bal mice
The variable phenotype of the two mutants led us to ask whether the difference might be attributable in part to the presence of residual ATM protein in the Awb/Awb brain. In wild-type mice, multiple ATM antibodies each recognized a band corresponding to full-length ATM in every tissue (Fig. 3A, lanes 1, 4, 7). In Bal/Bal mice, no immunoreactive bands were detectable (Fig. 3A, lanes 3, 6, 9). In Awb/Awb brain, the situation was more complex. We used three different ATM antibodies—2C1A1, Y170, and 5C2—to probe Western blots of tissue lysates. In spleen or thymus, none of the antibodies labeled a band in Awb/Awb protein extracts. In the brain, while the Y170 antibody labeled no band (Fig. 3A, lane 8, lower gel), the 2C1A1 (Fig. 3A, lane 8, upper gel) and 5C2 (Fig. 3A, lane 8, middle gel) antibodies both labeled a weak ATM band. We confirmed these results in brain sections and primary cultures (Fig. 3B). Y170 immunofluorescence was absent in Awb/Awb neurons, but 2C1A1 gave robust neuronal staining both in vivo and in vitro. These findings suggest that a brain-specific protein product of the Atmtm1Awb gene (ATMAWB) exists in the Awb/Awb mouse, but it is missing the Y-170 epitope (or the epitope is masked in the mutant protein).
Figure 3.
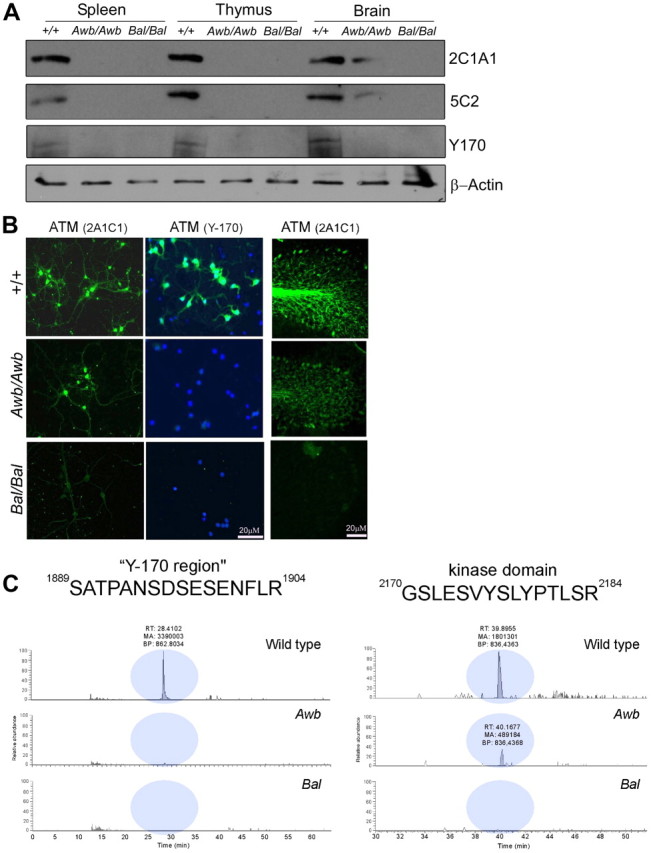
Atmtm1Awb but not Atmtm1Bal produces a brain-specific mutant ATM protein. A, Western blots of wild-type, Awb/Awb, and Bal/Bal spleen extracts, thymus extracts, and brain extracts were probed with three different ATM antibodies: 2C1A1, 5C2, and Y-170. Actin levels were used as a loading control. B, Immunofluorescence of cultured E16.5 cortical neurons immunostained with either 2C1A1 (left column) or Y-170 (middle column) ATM antibody. The images in the right column are cryostat sections of cerebellar cortex immunostained with 2C1A1 ATM antibody. The genotypes of the cultures or sections are indicated on the left. No staining of Bal/Bal cells is found with any ATM antibody, in vivo or in vitro. Awb/Awb cells stain with 2C1A1 but not Y-170. C, Extracted ion chromatograph for 2+ ion of the brain samples subjected to mass spectroscopy. The regions of the chromatograph corresponding to two ATM peptides are highlighted by the light blue circles in the three genotypes indicated on each trace. Mass window is 10 ppm. Two peptide fragment sequences are illustrated, one in each column. The amino acid sequence of each is shown at the top. Note that the Awb/Awb mouse has a reduced but clear peak in the region of the kinase domain, but lacks the peptide found in the region of the Y-170 epitope. Wild-type samples had these two peaks (any many more); Bal/Bal mice had none.
Our conclusion that the Awb/Awb mouse brain is capable of producing an immunoreactive peptide rests heavily on the quality of the antibodies we used. Although we and others had previously validated the specificity of the 2C1A1 antibody (Kozlov et al., 2006; Li et al., 2009), we wished to reconfirm that the ATMAWB protein is truly derived from the same gene as wild-type ATM. Immunoprecipitates of ATM were from brain lysates of the three Atm genotypes using the 2C1A1 antibody. Peptides from these immunoprecipitates were then sequenced by mass spectroscopy in the Rutgers Proteomics Center. In wild-type samples, 40 peptide fragments were found whose amino acid sequence was identical to that published for ATM protein; representative peptides are shown in Figure 3C (top). As expected, no peptides from any portion of the ATM protein could be found in Bal/Bal mouse brain (Fig. 3C, bottom). However, in Awb/Awb mouse brain (Fig. 3C, middle), 12 ATM peptides were found. Although their number was smaller than wild type, their sequences were 100% identical to those published for ATM; this confirms that Awb/Awb brain produces a true protein product from its Atm gene. Two peptides were particularly noteworthy. The first, from the region of the ATM peptide in the vicinity of the Y-170 epitope was present in wild type, but missing in both mutants (Fig. 3C, left traces). The second, from the kinase domain was present in the wild type, missing in the Bal/Bal mice, but present in Awb/Awb.
Brain-specific Atm mRNA is found in both Atmtm1Awb and Atmtm1Bal
We next explored the status of Atm mRNA in brain and other tissues. In wild-type mice strong RT-PCR bands were found in all tissues. In the mutants, no message could be detected in spleen or thymus (Fig. 4A). In brain, however, weak RT-PCR products could be amplified from both mutants. In the Bal/Bal brain, the exon 14–15 and 34–41 bands were normal in size, but the exon 55–61 band (the region where the Atmtm1Bal mutation was engineered) was shorter (Fig. 4A, red box). DNA sequencing of the mutant message in this region revealed a normal ATM message up to the triplet encoding L2693, after which the sequence diverged. In wild-type message, the PI3 kinase domain begins 39 residues later, at L2751. Thus, if protein were made from Atmtm1Bal mRNA it would be predicted to contain no kinase domain. As immunoreactive ATM protein is not detected (Fig. 3A), either the Atmtm1Bal message is not translated or the translated protein is not stable.
Figure 4.

Brain-specific Atm mRNA found in both Atmtm1Awb and Atmtm1Bal mice. A, RT-PCR reveals alternative message forms in Atmtm1Awb and Atmtm1Bal mouse brain. RNA was extracted from wild-type and Atmtm1Awb and Atmtm1Bal tissues as indicated, reverse transcribed, and amplified by primers located in the exons indicated at the left. 36B4 mRNA was used as a control. The red arrow indicates that the brain Atmtm1Awb message is reduced in size, relative to wild type in the region between exons 34 and 41; the purple arrow indicates that the Atmtm1Bal message is also reduced in size, relative to wild type in the region between exons 55 and 61. B, Based on DNA sequencing, the predicted protein sequence of the RT-PCR products is shown. The portion of the wild-type message that is deleted in the Atmtm1Awb message is shown in green; the 5 aa added to the Atmtm1Awb message are shown in red. The grayed amino acid sequence to the right shows the common continuation of both mutant and wild-type message through the codon for the autophosphorylated serine 1987 (magenta) and beyond. C, Images of GFP fusions with short ATM peptides encoded by the PCR amplified product of exons 34 through 41 of mouse ATM cDNA. Wild-type and Atmtm1Awb GFP-fusion peptides distribute primarily to the cytoplasm of transfected N2a cells (second and third rows), compared with a GFP-only construct (top row). In each experiment, mCherry was cotransfected to identify all transfected cells. D, Western blots of cell lysates from N2a cells transfected with GFP, and the GFP fusion peptides described in C. Blots were probed with GFP antibody.
RT-PCR products were also successfully amplified from mRNA of the Awb/Awb brain. The bands from exons 14–15 and 55–61 were normal, but the region of the message between exons 34–41 (the location of the Atmtm1Awb mutation) was ∼150 bp smaller than predicted (Fig. 4A, purple box). Sequencing of the PCR product from this region revealed an unusual splice event using nucleotide 5639 as a splice donor and 5832 as a splice acceptor. The resulting mRNA is predicted to encode a protein identical to wild type up to L1833. From K1834 to F1897, a 15 bp insert encoding KKRSY replaces the wild-type sequence. Beginning with S1898, however, the correct reading frame is restored (Fig. 4B). If translated into protein, this message would be predicted to retain kinase activity as well as the autophosphorylation site (S1987) (highlighted in purple in Fig. 4B). To examine whether this brain-specific splicing of Atm in Awb mouse can be translated into protein, we inserted RT-PCR products from wild-type and Awb mouse brain mRNA into EGFP-C3 vector to create GFP:ATM peptide fusion constructs. When the sequences from exon 34–41 of the wild-type PCR fragment were inserted and the construct transfected into N2a cells, GFP fluorescence was found in a predominantly cytoplasmic location compared with a control GFP-only construct (Fig. 4C). Surprisingly, the GFP fusion with Awb exons 34–41 showed lower expression and a distinct pattern of aggregation in the cytoplasm (Fig. 4C,D).
The brain-specific mutant ATM protein in Atmtm1Awb retains kinase activity
The prediction of residual kinase activity for the ATMAWB protein led us to reassess the response of the neurons of the Awb/Awb brain to the induction of DNA damage. Mutant and wild-type mice were irradiated (5 Gy) and killed 6 h later to allow a full DNA damage response to be established. The brains of irradiated wild-type mice contained evidence of the expected phosphorylation of ATM-S1987, p53-S15, and Chk2-T68 all known ATM targets (Fig. 5A, lane 2). When Bal/Bal mice were similarly irradiated, none of these phosphorylations occurred (Fig. 5A, lane 6). By contrast, in irradiated Awb/Awb brain, all three phosphoproteins were detected, although their levels were less than wild type (Fig. 5A, lane 4). Immunolabeling of brain sections from irradiated animals confirmed the retention of ATMAWB kinase activity (Fig. 5B), as did the presence of the phosphorylated proteins in neurons in culture after irradiation (Fig. 5C). Similar evidence for the retention of ATMAWB kinase activity was observed in neocortex (data not shown). As earlier reports failed to detect ATM autophosphorylation kinase activity in Atmtm1Awb brain (Dar et al., 2006), we tested a second 1981S phospho-specific antibody (Rockland). We found that this second reagent was less sensitive and less likely to reveal the phosphorylation of ATMAWB protein (data not shown). To prove that the events we observed in our samples were indeed ATM dependent, we exposed wild-type and Awb/Awb neuronal cultures to the DNA damaging agent, etoposide, in the presence or absence of siRNA to mouse ATM. The siRNA effectively lowered the ATM levels and rendered both wild-type and Awb/Awb cultures nonresponsive to etoposide (Fig. 5D, lanes 5 and 7).
Figure 5.

ATMAWB protein retains kinase activity. A, Mice of wild-type, Bal/Bal, and Awb/Awb genotypes were exposed to 5 Gy whole-body ionizing radiation (IR) (+) or left untreated (−). Six hours later, mice were killed and cerebellar extracts subjected to Western analysis with phospho-specific antibodies against ATM phospho-S1987, p53 phospho-S15, or Chk2 phospho-T68. In wild type, all substrates show evidence of radiation-induced labeling. In Bal/Bal mice, by contrast, none was labeled after IR-induced DNA damage. Despite their genotype, Awb/Awb mice showed a robust activation of all three substrates after ionizing radiation. B, Fluorescent images of wild-type and Atmtm1Awb brain sections from mice treated as in A and immunostained with anti-ATM phospho-S1987, Chk2 phospho-T68, or p53 phospho-S15 (red). In vivo, ATMAWB tissue is as responsive as wild type to DNA damage. C, E16.5 cortical neurons were cultured from wild-type and Awb/Awb brains as indicated. All cultures were exposed to 5 Gy IR, fixed 6 h later, and stained with the phospho-specific antibodies indicated at the left. D, DNA damage-induced phosphorylations in both wild-type and Awb/Awb cells are ATM dependent. E16.5 cortical neurons were cultured from wild-type and Awb/Awb brains as indicated. Cells were then exposed to either control siRNA or ATM-directed siRNA at DIV5. At DIV7, 20 μm etoposide (Etop) was added for 1 h (+) or cultures were left untreated (−). Cells were harvested, and lysates were prepared and subjected to Western analysis with the antibodies indicated on the right. The effectiveness of the siRNA treatment can be seen by the near absence of ATM protein in treated cultures of both genotypes (lanes 5–8, row 4). The effectiveness of the etoposide treatment can be seen in the phosphorylations of the typical ATM substrates in treated (lane 1) but not in untreated (lane 2) cells. Note that the Awb/Awb and wild-type cells respond identically (lanes 3 and 4).
The presence of ATM immunoreactivity in human A-T cerebellum
The finding of residual ATM protein in Awb/Awb mice prompted us to examine the situation in the human. We immunostained control cerebellum from unaffected individuals with several different ATM antibodies, and in each case, we found labeling in both the nucleus and cytoplasm of the cerebellar neurons (Fig. 6A,B). Four cases of A-T were immunostained with the same panel of antibodies. Unexpectedly, when the 2C1A1 antibody was used, Purkinje cells in all four A-T cases were also immunostained (two representative cases are shown in Fig. 6C,D). Similar results were obtained when a fluorescence-conjugated secondary antibody was used (data not shown). The 2C1A1 antibody staining specificity was confirmed by preabsorption of the antibody with the immunizing peptide (Fig. 6E,F). The presence of the 2C1A1 epitope in human A-T Purkinje cells combined with the mouse data from the Awb mutant prompted us to reexamine the same cases of A-T using the Y-170 antibody. In an exact parallel to the situation in the Awb/Awb mouse cerebellum, all four A-T cases appeared to be missing the Y-170 epitope (Fig. 6G,H). Similar results were confirmed with immunofluorescent staining (data not shown). The persistence of ATM protein led us to ask about the levels of ATM mRNA in A-T brain. Frozen cerebellar tissues were obtained from four new cases of A-T and four new age-matched controls, and total RNA was extracted. RT-PCR of tubulin cDNA demonstrated that we had successfully extracted brain mRNA. Additional RT-PCRs were performed with primers corresponding to two different regions of the ATM message. The region of the message encoding the kinase domain (exons 55–58) was reliably detected in all four samples. The region between exons 14 and 15, however, was missing in one subject. This analysis confirms that, as in mouse, a stable pool of ATM message is present in the A-T cerebellum. The absence of an amplified 14–15 band in one subject suggests that the differential Atm mRNA isoforms also occur in humans (Fig. 6I, red cycle). We then analyzed the ATM protein levels in these four subjects. Immunoprecipitation was performed with the 2C1A1 antibody followed by Western blots with 2C1A1 or Y170. As in mouse, and consistent with the immunocytochemistry shown in Figure 6A–H, a clear band of slightly lower molecular weight was present in the 2C1A1 blot but not in Y170 blot (Fig. 6J). To ensure that the 2C1A1 immunoprecipitates contained genuine ATM protein, we performed amino acid sequencing by mass spectrometry of three samples. In two subjects, no ATM peptides were detected (Fig. 6K–N, dashed lines). Mass spectroscopy of the third subject, however (Fig. 6J, lane 3), revealed the unambiguous presence of ATM protein (Fig. 6K–N, blue circles, row 3).
Figure 6.
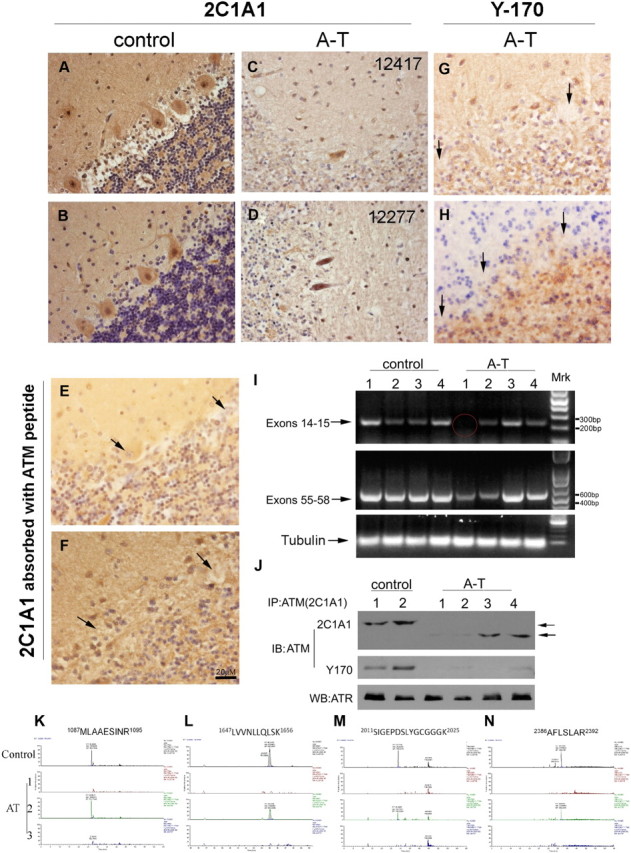
The diversity of ATM immunoreactivity found in human A-T. A, B, Paraffin sections of human cerebellar cortex from individuals who died with no known brain disease were immunostained with ATM antibody (2C1A1). Note the robust nuclear and cytoplasmic staining. C, D, Paraffin sections of cerebellar cortex from two patients who died with A-T were immunostained as in A and B. Note the presence of ATM immunoreactivity in the A-T cerebellar Purkinje cells from all cases. E, F, Paraffin sections of control and A-T human cerebellum were immunostained with ATM antibody (2C1A1) after absorption with the human ATM peptide used for immunization (Abcam, ab40436). No ATM staining was found in this situation in either control or A-T sections (arrows). G, H, Paraffin sections of cerebellar cortex from two patients who died with A-T were immunostained with the Y-170 ATM antibody. No ATM staining is visible (arrows). I, RT-PCR of ATM message in A-T patient cerebella. RNA was extracted from control and A-T patients, reverse transcribed, and amplified by primers located in the exons indicated at the left. Tubulin mRNA was used as a control. Message encoding ATM exons 55–58 was present in all eight samples. The portion exon of the message encoding exons 14–15, however, is missing in one A-T case (A-T, subject 1). J, Lysates from frozen cerebellar samples of two control (lanes 1–2) and four individuals with A-T (lanes 3–6) were immunoprecipitated with 2C1A1 antibody. Blots were probed with either 2C1A1 (top) or Y170 (middle) antibody; an ATR blot served as a control demonstrating that comparable amounts of protein were extracted from the samples. K–N, Extracted ion chromatograph for 2+ ion corresponding to four human ATM peptides in one control and three A-T patients noted on the left side of each trace. The mass window is 10 ppm. Four peptide fragment sequences are illustrated. The amino acid sequence of each is shown at the left. Note that one A-T case (AT2), as well as the control, had detectable amounts ATM protein in cerebellum (blue filled circles). In the other two cases (AT1, AT3), no comparable peptides were detected (dashed circles).
Discussion
Our findings offer important new insights into the etiology of A-T. First, the results illustrate that in both human and mouse, the handling of Atm message and protein in brain is substantially different from that found in most other tissues. In mouse spleen and thymus, both the Awb and Bal mutations lead to undetectable levels of Atm message and protein. In brain, by contrast, the levels of Atm mRNA in both mutants are comparable to wild type. More importantly, the brain appears able to generate several unique RNA splicing patterns in the presence of a mutation. In Awb message, the use of nucleotide 5639 as a splice donor and 5832 as a splice acceptor is a pattern we have been unable to detect in wild-type message populations. The methods used by the neurons to achieve this unusual splice pattern are unknown.
One consequence of the persistence of an atypical message population in brain is the occasional production of a brain-specific protein. While Atmtm1Bal mRNA does not lead to detectable ATMBAL protein, the Atmtm1Awb allele leads to an ATMAWB protein that is nearly normal in size and retains detectable kinase activity. Based on our cDNA sequencing, the ATMawb kinase domain is predicted to be retained. We find evidence of this in the responsiveness of Atmtm1Awb brain to etoposide and radiation, as revealed by the phosphorylation of ATM-S1987, Chk2-T68, and p53-S15. Detection of mouse ATM autoactivation has been explored previously (Dar et al., 2006), but this study used a phospho-S1981 antibody, which is less sensitive on Atmtm1Awb tissue. The exact reason for this is unknown, but it suggests that the configuration of the ATMAWB protein occludes the S1981 epitope, in a way that the wild-type protein does not. The 1987 epitope, by contrast, is accessible in both genotypes.
Our human studies illustrate that the situation in Awb/Awb mice is not an anomaly. All four of the A-T subjects we examined by immunocytochemistry had detectable ATM in their Purkinje cells when visualized with the 2C1A1 antibody. These same cases were negative for the presence of the Y-170 epitope. The subject illustrated in Figure 6D had a homozygous splicing mutation at IVS32–12A>G 21(Gatti et al., 1989). Based on the robust presence of the 2C1A1 epitope in her Purkinje cells, we would predict that her brain was able to produce a peptide from a stable alternatively spliced ATM message. Given the absence of the Y-170 epitope, however, we would also predict that the mutant peptide was nonfunctional in brain. The case illustrated in Figure 6C was a compound heterozygote for mutations at two sites: 2T>C (abolishing the ATG initiation site) and 3245ATC>AGAT. In this case, we detected only modest immunoreactivity for the 2C1A1 epitope, compared to controls or the other A-T cases. We would predict that the ATG mutation produces a true null, even in brain, while the second mutation produces a stable peptide in brain. As the progression of disease in both cases was more or less typical, we would further predict that the mutant ATM protein in the second subject was not functional in brain. Unfortunately, no frozen samples were saved from these autopsies precluding further analysis. In the context of this tissue-specific “rescue” of ATM protein in A-T brain cells, it is worth noting the previous report that in peripheral mononuclear cells from human A-T subjects, a mutant protein can regularly be found (Chun et al., 2003).
The consistent absence of the Y-170 epitope in the A-T brain raises the question of whether this particular region of the ATM protein has a unique contribution to normal CNS function. The phenotypes of the two mouse models we have examined are potentially informative in this regard. Both mutants lack any detectable ATM protein in peripheral tissues, consistent with the faithful replication of the A-T phenotypes in these regions. In brain, despite the presence of Atmtm1Bal mRNA, Bal/Bal animals lack a stable protein and thus their brain represents the state expected of a true null allele. The severe degeneration found in Bal/Bal cerebella (Fig. 1) is consistent with this suggestion. By contrast, Awb/Awb animals produce a peptide (ATMAWB) that is functional in its response to DNA damage (Fig. 5) and is capable of suppressing the neuronal cell cycle in vitro (Fig. 2D). While some cell cycle activity is found in vivo (Fig. 2A–C), the near total absence of cerebellar degeneration suggests that the Purkinje cell survival function of ATMAWB is not significantly impaired. Nonetheless, we have recently shown that the ATMAWB protein fails in its function as a vesicle protein binding protein in the cytoplasm of neurons. The result is that, in the brains of Awb/Awb mice, hippocampal LTP and spontaneous vesicle dye release are both defective (Li et al., 2009). It is the Y-170 region of ATM that is lost in Awb/Awb mice and in three of four human A-T cases that we have examined. This would argue that certain CNS-specific functions, that may well be central to the neurological symptoms of A-T, require the Y-170 region of the ATM protein to be carried out. Further, the clear response of Awb/Awb neurons to etoposide and ionizing radiation suggests that this activity of ATM is most likely distinct from its function as a DNA damage response protein. The diversity of neurological symptoms found clinically (Li and Swift, 2000; Alterman et al., 2007), and the existence of cases A-T with mild CNS features but profound peripheral defects suggests that these two distinct functions are at play in the human brain as well as in the mouse.
Footnotes
We thank Rutgers University, the A-T Children Project, and the NIH (R01-NS20591) for funding this work. We thank Haiyan Zhen and Caifeng Zhao for expert help in analysis of mass spectrometry data (Biological Mass Spectrometry Facility Center for Advanced Biotechnology and Medicine, University of Medicine and Dentistry of New Jersey–Robert Wood Johnson Medical School).
References
- Alterman N, Fattal-Valevski A, Moyal L, Crawford TO, Lederman HM, Ziv Y, Shiloh Y. Ataxia-telangiectasia: mild neurological presentation despite null ATM mutation and severe cellular phenotype. Am J Med Genet A. 2007;143A:1827–1834. doi: 10.1002/ajmg.a.31853. [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Barlow C, Ribaut-Barassin C, Zwingman TA, Pope AJ, Brown KD, Owens JW, Larson D, Harrington EA, Haeberle AM, Mariani J, Eckhaus M, Herrup K, Bailly Y, Wynshaw-Boris A. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci U S A. 2000;97:871–876. doi: 10.1073/pnas.97.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boder E. Ataxia-telangiectasia: some historic, clinical and pathologic observations. Birth Defects Orig Artic Ser. 1975;11:255–270. [PubMed] [Google Scholar]
- Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. 1958;21:526–554. [PubMed] [Google Scholar]
- Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, Roberts TM, Swat W, Segal RA, Gu Y. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci U S A. 2000;97:3336–3341. doi: 10.1073/pnas.050584897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundey S. Clinical and genetic features of ataxia-telangiectasia. Int J Radiat Biol. 1994;66:S23–29. [PubMed] [Google Scholar]
- Chun HH, Sun X, Nahas SA, Teraoka S, Lai CH, Concannon P, Gatti RA. Improved diagnostic testing for ataxia-telangiectasia by immunoblotting of nuclear lysates for ATM protein expression. Mol Genet Metab. 2003;80:437–443. doi: 10.1016/j.ymgme.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Dar I, Biton S, Shiloh Y, Barzilai A. Analysis of the ataxia telangiectasia mutated-mediated DNA damage response in murine cerebellar neurons. J Neurosci. 2006;26:7767–7774. doi: 10.1523/JNEUROSCI.2055-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A. 1996;93:13084–13089. doi: 10.1073/pnas.93.23.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, Ersoy F, Foroud T, Jaspers NG, Lange K, Lathrop GM, Leppert M, Nakamura Y, O'Connell P, Paterson M, Salser W, Sanal O, Silver J, Sparkes RS, Susi E, et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22–23. Nature. 1988;336:577–580. doi: 10.1038/336577a0. [DOI] [PubMed] [Google Scholar]
- Gatti RA, Nieberg R, Boder E. Uterine tumors in ataxia-telangiectasia. Gynecol Oncol. 1989;32:257–260. doi: 10.1016/s0090-8258(89)80045-9. [DOI] [PubMed] [Google Scholar]
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science. 1998;280:1089–1091. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006;25:3504–3514. doi: 10.1038/sj.emboj.7601231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuljis RO, Xu Y, Aguila MC, Baltimore D. Degeneration of neurons, synapses, and neuropil and glial activation in a murine Atm knockout model of ataxia-telangiectasia. Proc Natl Acad Sci U S A. 1997;94:12688–12693. doi: 10.1073/pnas.94.23.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- Li A, Swift M. Mutations at the ataxia-telangiectasia locus and clinical phenotypes of A-T patients. Am J Med Genet. 2000;92:170–177. doi: 10.1002/(sici)1096-8628(20000529)92:3<170::aid-ajmg3>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Li J, Han YR, Plummer MR, Herrup K. Cytoplasmic ATM in neurons modulates synaptic function. Curr Biol. 2009;19:2091–2096. doi: 10.1016/j.cub.2009.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval N, Platzer M, Rosenthal A, Dörk T, Bendix R, Skawran B, Stuhrmann M, Wegner RD, Sperling K, Banin S, Shiloh Y, Baumer A, Bernthaler U, Sennefelder H, Brohm M, Weber BH, Schindler D. Characterization of ATM gene mutations in 66 ataxia telangiectasia families. Hum Mol Genet. 1999;8:69–79. doi: 10.1093/hmg/8.1.69. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Sedgwick RP, Boder E. Progressive ataxia in childhood with particular reference to ataxia-telangiectasia. Neurology. 1960;10:705–715. doi: 10.1212/wnl.10.7.705. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–2422. doi: 10.1101/gad.10.19.2411. [DOI] [PubMed] [Google Scholar]
- Yang Y, Herrup K. Loss of neuronal cell cycle control in ataxia-telangiectasia: a unified disease mechanism. J Neurosci. 2005;25:2522–2529. doi: 10.1523/JNEUROSCI.4946-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]