

Abstract
(N)-Methanocarba nucleosides containing bicyclo[3.1.0]hexane replacement of the ribose ring previously demonstrated selectivity as A3 adenosine receptor (AR) agonists (5′-uronamides) or antagonists (5′-truncated). Here, these two series were modified in parallel at the adenine C2 position. N6-3-Chlorobenzyl-5′-N-methyluronamides derivatives with functionalized 2-alkynyl chains of varying length terminating in a reactive carboxylate, ester, or amine group were full, potent human A3AR agonists. Flexibility of chain substitution allowed the conjugation with a fluorescent cyanine dye (Cy5) and biotin, resulting in binding Ki values of 17 and 36 nM, respectively. The distal end of the chain was predicted by homology modeling to bind at the A3AR extracellular regions. Corresponding l-nucleosides were nearly inactive in AR binding. In the 5′-truncated nucleoside series, 2-Cl analogues were more potent at A3AR than 2-H and 2-F, functional efficacy in adenylate cyclase inhibition varied, and introduction of a 2-alkynyl chain greatly reduced affinity. SAR parallels between the two series lost stringency at distal positions. The most potent and selective novel compounds were amine congener 15 (Ki = 2.1 nM) and truncated partial agonist 22 (Ki = 4.9 nM).
Introduction
Selective agonist and antagonist ligands of the A3 adenosine receptor (ARa), a G-protein-coupled receptor (GPCR), have been reported.1 A3AR antagonists are proposed for the treatment of cancer, inflammation, glaucoma, and asthma.2–5 A3AR agonists are already in clinical trials for liver cancer, rheumatoid arthritis, psoriasis, and dry eye disease,6,7 with additional envisioned applications for bowel inflammation, ischemia, and other autoimmune inflammatory disorders.8–10
Selective A3AR antagonists encompass both non-purine heterocyclic antagonists1,11 and nucleoside12,13 antagonists. Nucleoside-derived A3AR antagonists have the advantage of a species-independent pharmacological profile;4 i.e., they tend to maintain A3AR selectivity in both human and murine species, while most of the non-purine antagonists are selective for the human (h) but not rat (r) A3AR. Modification of the ribose moiety of adenosine derivatives, and to a lesser extent N6 and C2 substituents, has been shown to reduce agonist efficacy and thus be useful for converting A3AR agonists into antagonists.12,13
Both the binding and selectivity of adenosine derivatives as prototypical A3AR agonists can be substantially increased by replacing the flexible ribose scaffold with a rigid bicyclo[3.1.0] hexane ring system resulting in a class of carbocyclic nucleosides related to 1 (Chart 1).14–16 These (N)-methanocarba adenosine agonists have been found to possess high potency and enhanced selectivity for the A3AR,11 and an agonist of this class (MRS3558, 1a) has been shown to display antiarthritic activity and protection in a model of lung ischemia.17,18 The N6-3-iodobenzyl derivative 1b (MRS1898) was recently radioiodinated to form an A3AR agonist radioligand that can be used selectively in murine and in hA3AR binding studies.19
Chart 1.
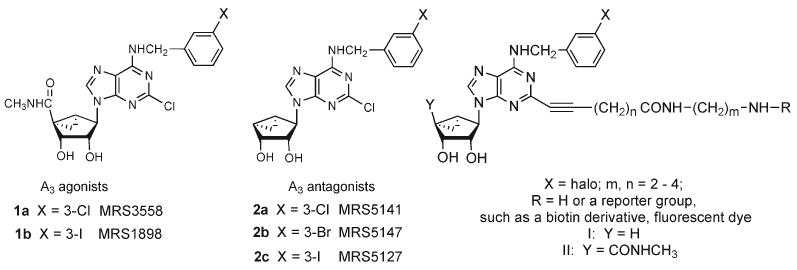
(N)-Methanocarba Derivatives of Adenosine as A3AR-Selective Agonists (1a, 1b) and Antagonists (2a, 2b)a
aGeneral formulas I and II represent two parallel structural series included in the present study. The agonist corresponding to X = Cl, n = 3, m = 2, R = H in series II was already reported.21
We describe in this work two new series of selective A3AR ligands that extend recent reports, i.e., agonists and antagonists of the (N)-methanocarba family, derived through largely parallel modifications at the adenine C2 position.20,21 Thus, the new agonists contained a 5′-N-methyluronamide group, and this amide group was removed entirely in the truncated series. The principle of lowering the relative efficacy of full agonists by removing a flexible 5′-amide group to produce, in some cases, hA3AR antagonists was shown for 4′-thionucleosides by Jeong et al. and for (N)-methanocarba analogues by Melman et al. (i.e., 2a–c).13,20 The stereospecificity of binding to the ARs was also probed with the preparation of corresponding l-nucleosides. In both series, the conformationally restricted (N)-methanocarba scaffold was retained, and most of the analogues contained a substituted 6-(benzylamino) moiety. The most structurally diverse modifications were made at the adenine C2 position, with the addition of extended alkynyl chains terminating in a carboxylic, ester, or amino group, following a functionalized congener approach.22,23 This terminal functionalized group was remote from the minimal pharmacophore required for recognition at the primary binding site of the receptor. The intended application of the functional groups is for covalent coupling to various other moieties, including spectroscopic reporter groups, without losing biological activity.
Results
Chemical Synthesis
In this study, we have elaborated and extended functionalization at the C2 position of previously reported (N)-methanocarba nucleoside derivatives, which were shown to be selective A3AR agonists15,16,21 and antagonists.20 The series of 5′-N-methyluronamides (Chart 1, II) are similar to the reported agonists 1, and the series of truncated nucleosides (I) are similar to the reported antagonists 2. Here, these two series were modified in parallel at the adenine C2 position.
The synthetic route to the 5′-N-methyluronamide (N)-methanocarba derivatives 5–16 is shown in Scheme 1A. The synthesis of the 2′,3′-protected intermediate 27, containing an N6-(3-chlorobenzyl) group, from l-ribose was previously reported.24 This 2-iodo intermediate was then subjected to Sonogashira coupling25 with the corresponding alkyne esters. The products 28–30 of varying alkynyl chain length were deprotected to liberate the 2′,3′-hydroxyl groups to provide nucleosides 8–10, followed by hydrolysis of the methyl ester to afford agonists 5–7. Alternatively, compounds 28–30 were aminolyzed using the appropriate alkyldiamine in excess, and the product amines were deprotected to yield the amine functionalized agonists 11–16 of varying chain length.
Scheme 1a.

a (A) (i) HC≡C(CH2)nCOOMe, Pd(PPh3)2Cl2, CuI, Et3N, DMF, room temp; (ii) 10% TFA, MeOH, 70 °C; (iii) appropriate alkyldiamine, MeOH, room temp; (iv) KOH, MeOH, room temp. (B) (v) For 17a: biotin, HATU, DIEA, DMF, room temp. For 17b: biotin-ε-aminocaproyl N-succinimidyl ester, Et3N, DMF, room temp. (vi) N-succinimidyl ester of chain-derivatized Cy5 cyanine dye, bicarbonate buffer, DMF.
The amino and carboxylic derivatives contained chemically functionalized, extended alkynyl chains to serve as functionalized congeners for conjugation to other biologically active moieties or to carriers. 23,26 Several representative biologically active conjugates for the detection and characterization of the A3AR were prepared as shown in Scheme 1B. Biotin conjugates of varying length 17a and 17b were synthesized from the amine 11, which was the analogue having the shortest chain in the homologous series of amine congeners. The biotin conjugate 17a was prepared by a HATU coupling with biotin, and the extended chain analogue 17b was prepared from the corresponding biotin-ε-aminocaproyl active ester. The fluorescent derivative 18 similarly was prepared by treating 11 with the active ester of a chain-derivatized Cy5 cyanine dye.27
Selected l-enantiomers 19–21 of the 5′-N-methyluronamide methanocarba agonist series were also prepared, as shown in Scheme 2. The isopropylidene intermediate 31 was prepared from d-ribose as previously reported. A Mitsunobu condensation21 with 2-iodo-6-chloropurine provided 32, which was first converted to the N6-(3-chlorobenzyl) derivative 33 and then treated with excess methylamine to yield 34. This 2-iodo intermediate was subjected to a Sonogashira coupling25 with the corresponding alkyne esters to provide compounds 35–37 of varying alkynyl chain length. Esters 35 and 37 were aminolyzed with ethylenediamine followed by hydrolysis of the acetonide group with 10% TFA to provide compounds 20 and 21. In the case of compound 36, the 2′,3′-isopropylidene group was first deprotected, and hydrolysis of the ester afforded the carboxylic acid derivative 19.
Scheme 2 a.
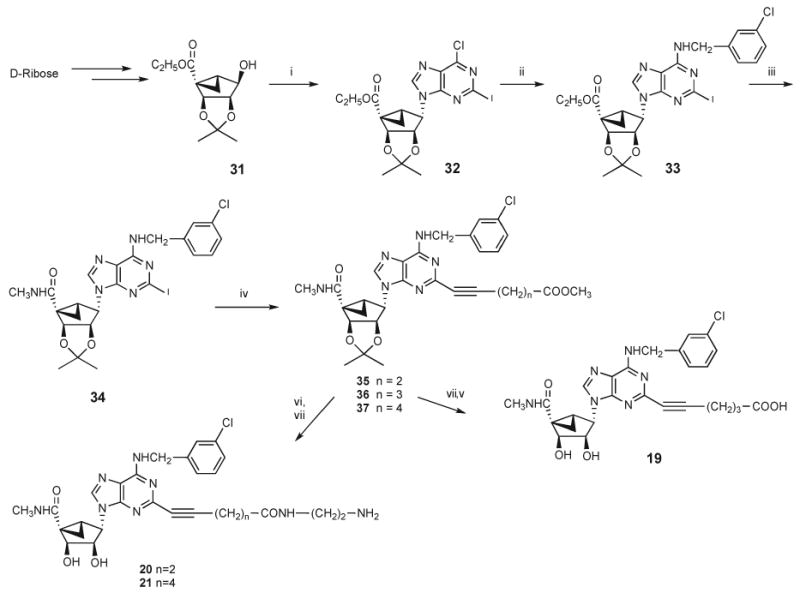
a (i) 2-Iodo-6-chloropurine, Ph3P, DIAD, THF, room temp; (ii) 3-chlorobenzylamine, Et3N, MeOH, room temp; (iii) 10% methylamine, MeOH, room temp; (iv) HC≡C(CH2)nCOOMe, Pd(PPh3)2Cl2, Cul, Et3N, DMF, room temp; (v) KOH, MeOH; (vi) ethylenediamine, MeOH, room temp; (vii) 10% TFA, MeOH, 70 °C.
Scheme 3 shows the new synthetic route used to prepare the truncated (N)-methanocarba derivatives 22–26. While the previous study of truncated (N)-methanocarba nucleosides as antagonists of the hA3AR depended on a low yielding decarboxyation step, an alternative simple synthetic approach was designed. The protected cyclopentenone 38 was stereoselectively reduced to the corresponding alcohol 39,28 which was then subjected to cyclopropanation to yield the glycosyl donor 40 for the truncated nucleosides and therefore converged on the previously reported route.20 Compound 40 was subjected to a Mitsunobu condensation20 with the appropriate C2 substituted 6-chloropurine derivative to give the 2-H (41) and 2-halopurine (42, 43) derivatives. The 2-1 derivative 44 was prepared in a similar manner and was substituted with 3-chlorobenzylamine at the 6 position to give 45, which serves as a precursor for 2-alkynyl derivatives in the truncated series. This route afforded the isopropylidene-protected truncated nucleosides 46–49, which were subsequently deprotected to provide compounds 22–25 for biological testing.
Scheme 3a.
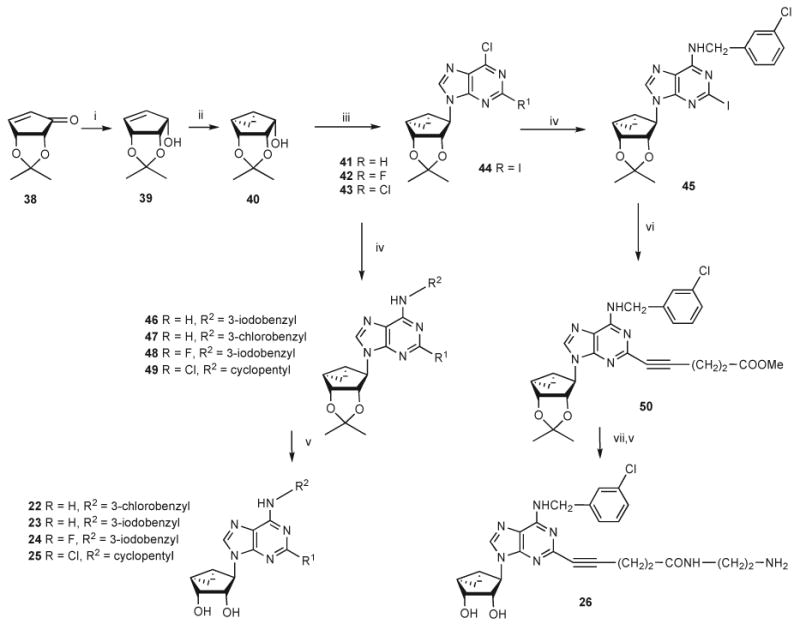
a (i) NaBH4, CeCl3 ·7H2O, MeOH, 0 °C; (ii) Et2Zn, CH2I2, CH2Cl2, room temp; (iii) 2-substituted-6-chloropurine, Ph3P, DIAD, THF, room temp; (iv) 3-iodo- or 3-chlorobenzylamine or cyclopentylamine, Et3N, MeOH, room temp; (v) 10% TFA, MeOH, 70 °C; (vi) HC≡C(CH2)2COOMe, Pd(PPh3)2Cl2, CuI, Et3N, DMF, room temp; (vii) ethylenediamine, MeOH, room temp.
A truncated 2-iodo intermediate 45 was subjected to Sonogashira coupling25 with methyl pentynate to give compound 50, which upon amination with ethylenediamine and subsequent acid hydrolysis provided the truncated nucleoside 26 containing an amine functionalized chain.
Pharmacological Activity
Several previously reported 2-chloro-(N)-methanocarba agonists (1a, 1b, 3, 4, 6, 9, and 12) and antagonists (2a–c) were used for comparison in the biological assays (Table 1). Binding assays at three hAR subtypes were carried out using standard radioligands29–31 and membrane preparations from Chinese hamster ovary (CHO) cells (A1 and A3) or human embryonic kidney (HEK293) cells (A2A) stably expressing a hAR subtype (Table 1).32,33 Since activity within the class of (N)-methanocarba nucleosides was previously noted to be very weak or absent at the hA2BAR,16 we did not include this receptor in the screening protocol. Functional data were determined in an assay of adenylate cyclase (A3AR-induced inhibition).34 Functional data for selected compounds in a functional assay consisting of A3AR-induced stimulation of guanine nucleotide binding are reported in Table 1.35
Table 1. Potency of a Series of (N)-Methanocarba Adenosine Derivatives at Three Subtypes of Human ARs and the Functional Efficacy at the A3AR.
 | ||||||
|---|---|---|---|---|---|---|
| compd | structure | affinity (Ki, nM) or % inhibitiona | % efficacy,b A3 | |||
| R1 | R2 | A1 | A2A | A3 | ||
| Series A | ||||||
| 1ac | Cl | 3-Cl-Bn | 260 ± 60h | 2300 ±100 | 0.29 ± 0.04 | 103 ± 7 |
| 1b | Cl | 3-I-Bn | 136 ± 22d,h | 784 ± 97d | 1.5 ± 0.2c | 100c |
| 3c | H | 3-I-Bn | 700 ± 270h | 6200 ± 100 | 2.4 ± 0.5 | 100 |
| 4c | Cl | cyclopentyl | 18.3 ± 6.3h | 3250 ± 300 | 3.7 ± 0.9 | 101 |
| 5 | C≡C(CH2)2COOH | 3-Cl-Bn | (17 ± 4%) | (24 ± 12%) | 61.1 ± 35.8 | 106 ±18 |
| 6c,e | C≡C(CH2)3COOH | 3-Cl-Bn | 14,900 ± 3500h | (43%) | 2.38 ± 0.56 | ND |
| 7 | C≡C(CH2)4COOH | 3-Cl-Bn | (11 ± 5%) | (38 ± 2%) | 12.4 ± 1.8 | 74.4 ± 5.1 |
| 8 | C≡C(CH2)2COOCH3 | 3-Cl-Bn | (41 ± 0%) | (43 ± 5%) | 5.5 ± 0.3 | 90.0 ± 6.2 |
| 9c | C≡C(CH2)3COOCH3 | 3-Cl-Bn | 482 ± 23h | (49%) | 1.17 ± 0.27 | ND |
| 10 | C≡C(CH2)4COOCH3 | 3-Cl-Bn | (20 ± 7%) | 3540 ± 1370 | 11.1 ± 2.3 | 85.0 ± 10.2 |
| 11 | C≡C(CH2)2CONH(CH2)2NH2 | 3-Cl-Bn | 2320 ± 520 | 550 ± 83 | 2.5 ± 0.5 | 96.9 ± 6.8 |
| 12c | C≡C(CH2)3CONH(CH2)2NH2 | 3-Cl-Bn | 454 ± 44h | (81%) | 2.17 ± 0.51 | ND |
| 13 | C≡C(CH2)4CONH(CH2)2NH2 | 3-Cl-Bn | (47 ± 3%) | 277 ± 33 | 3.4 ± 0.7 | 100 ± 2 |
| 14 | C≡C(CH2)2CONH(CH2)3NH2 | 3-Cl-Bn | (31 ± 9%) | 979 ± 181 | 3.1 ± 0.4 | 97.9 ± 18.4 |
| 15 | C≡C(CH2)2CONH(CH2)4NH2 | 3-Cl-Bn | (30 ± 2%) | 766 ± 109 | 2.1 ± 0.4 | 102 ± 13 |
| 16 | C≡C(CH2)2CO[NH(CH2)2]2NH2 | 3-Cl-Bn | (33 ± 2%) | 890 ± 110 | 15.4 ± 4.4 | 87.6 ± 21.2 |
| 17a | C≡C(CH2)2CONH(CH2)2NH-biotin | 3-Cl-Bn | (1 ± 1%) | (51 ± 2%) | 36.4 ± 5.6 | 84.5 ± 12.0 |
| 17b | C≡C(CH2)2CONH(CH2)2NH-CO(CH2)5NH-biotin | 3-Cl-Bn | (12 ± 4%) | (47 ± 11%) | 57.7 ± 16.2 | 107 ± 18 |
| 18 | C≡C(CH2)2CONH(CH2)2NH-CO-(CH2)5Cy5g | 3-Cl-Bn | (36 ± 3%) | 4730 ± 1020 | 17.2 ± 3.1 | 94.4 ± 9.6 |
| Series B | ||||||
| 19 | C≡C(CH2)3COOH | 3-Cl-Bn | (0%) | (9 ± 2%) | 5270 ± 1090 | ND |
| 20 | C≡C(CH2)2CONH(CH2)2NH2 | 3-Cl-Bn | (33 ± 7%)h | (8 ± 1%) | (30 ± 1%) | 25.6 ± 10.9 |
| 21 | C≡C(CH2)4CONH(CH2)2NH2 | 3-Cl-Bn | (36 ± 5%)h | (5 ± 1%) | 4630 ± 1060 | 52.0 ± 9.4 |
| Series C | ||||||
| 2ad | Cl | 3-Cl-Bn | 3070 ± 1500h | 4510 ± 910 | 1.06 ± 0.36 | 2.9 ± 3.7f |
| 2bd,e | Cl | 3-Br-Bn | 1760 ± 1010h | 1600 ± 480 | 0.73 ± 0.30 | 46 ± 4,5.8 ± 0.8f |
| 2cd,e | Cl | 3-I-Bn | 3040 ± 610h | 1080 ± 310 | 1.44 ± 0.60 | 44 ± 6, 1.0 ± 3.2f |
| 22 | H | 3-Cl-Bn | 1600 ± 150 | 4520 ± 830 | 4.9 ± 0.7 | 58.3 ± 8.0 |
| 23 | H | 3-I-Bn | 839 ± 68 | 2330 ± 680 | 11.0 ± 2.2 | 80.8 ± 19.0 |
| 24 | F | 3-I-Bn | 513 ± 4 | 4000 ± 780 | 10.7 ± 0.9 | 19.0 ± 3.0 |
| 25 | Cl | cyclopentyl | 109 ± 16 | 1640 ± 360 | 120 ± 31 | 95.1 ± 4.6 |
| 26 | C≡C(CH2)2CONH(CH2)2NH2 | 3-Cl-Bn | (15 ± 2%) | (35 ± 6%) | 404 ± 67 | 0.9 ± 8.5 |
All experiments were done on CHO or HEK293 (A2A only) cells stably expressing one of four subtypes of human ARs. The binding affinity for A1, A2A, and A3ARs was expressed as Ki values (n = 3–5) and was determined by using agonist radioligands ([3H]51, [3H]54, or [125I]53, respectively), unless otherwise noted. A percent in parentheses refers to inhibition of radioligand binding at 10 μM.
Unless otherwise noted, the efficacy at the human A3AR was determined by inhibition of forskolin-stimulated cAMP production in AR-transfected CHO cells. At a concentration of 10 μM, in comparison to the maximal effect of 51 (= 100%) at 10 μM. Data are expressed as mean ± standard error (n = 3). ND, not determined.
Values from Melman et al.20
A3AR functional assay consisted of stimulation of [35S]GTPγS binding at 10 μM, expressed as a percentage of the full effect induced by 10 μM 51 (100 ± 5%).
Structure given in Scheme 1B.
A1AR binding determined using [3H]52.
Compounds 5–15 in the (N)-methanocarba 5′-N-methyluronamide series (series A), which had functionalized 2-alkynyl chains terminating in a carboxylic acid, ester, or amino group, were potent A3AR ligands. These derivatives having charged, polar, and chemically reactive groups at the end of a chain were intended for structural probing of distal regions of the ligand binding site of the receptor and for the design of specialized high-affinity ligands, such as fluorescent probes.
The optimal chain length for A3AR affinity among carboxylic acid derivatives occurred with the previously reported compound 6,21 which had three methylenes in the chain. The corresponding ester derivative 921 was also the most potent in the series of homologous esters 8–10. A shorter chain carboxylic acid derivative 5 was considerably less potent than other members of the series in binding to the hA3AR.
The carboxylic acid derivatives were then extended by amide linkage to diaminoalkyl groups. In the series of resulting terminal amino derivatives 11–13, the length of the carboxyl alkyl chain was varied, and in the series of 11 and 14 and 15 the length of the diaminoalkyl chain was varied. While 12 displayed an atypically high A 1AR affinity (Ki = 454 nM) and 13 an atypically high A2AAR affinity (Ki = 270 nM), most of the members of these series were highly selective for the A3AR. The Ki values at the hA3AR of the primary amino derivatives 11–15 of varying chain length were in the range of 2–3 nM. The most potent and selective novel member of this series, an amine derivative 15 (Figure 1, Table 1), was 360 and > 5000-fold selective in binding to the hA3AR in comparison to hA1 and A2AARs, respectively. Compound 16, containing two free amino groups in the chain, one primary and another secondary, was slightly reduced in affinity at the A3AR with a Ki value of 15.4 nM.
Figure 1.
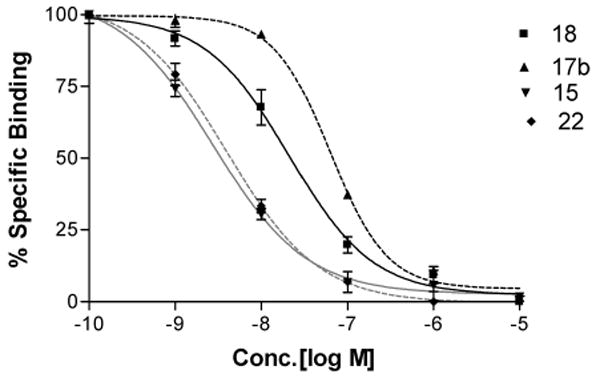
Inhibition of radioligand binding by (N)-methanocarba nucleoside analogues in membranes of CHO cells expressing the human A3AR. Inhibition curves are shown for the agonist analogues containing a 5′-N-methyluronamide group and compounds 15 (amine functionalized congener), 17b (biotinylated probe), and 18 (fluoresecent probe) and for the truncated partial agonist 22. All of the analogues shown were highly selective for the A3AR in comparison to the A1 and A2aARs.
As functionalized congeners, these long chain derivatives were intended for conjugation to carrier moieties for receptor detection and characterization, in a manner that did not prevent receptor recognition.22,23 A biotin conjugate 17b derived from amine 11 in the series of 5′-N-methyluronamide derivatives displayed a Ki value at the hA3AR of 58 nM (Figure 1) and selectivity of > 170-fold and 66-fold in comparison to the A1 and A2AAR, respectively. A shorter analogue 17a lacking the ε-aminocaproyl spacer chain was slightly more potent in hA3AR binding with a Ki value of 36 nM. A fluorescent conjugate 18 containing the sulfonated carbocyanine dye Cy527 was potent and selective in A3AR binding. The fluorescent excitation and emission maxima of 18 in aqueous medium, 646 and 660 nm, respectively, were characteristic of Cy5 dye conjugates.
Various compounds were examined for the ability to inhibit the production of adenosine 3′,5′-cyclic phosphate (cAMP) in membranes of CHO cells expressing the hA3AR (Table 1).34 Inhibition by 10 μM NECA (51, 5′-N-ethylcarboxamidoadenosine) was set at 100% relative efficacy. The (N)-methanocarba 5′-N-methyluronamide derivatives (series A) were consistently full agonists. Full concentration response curves for compounds 17a and 18 provided IC50 values in hA3AR-mediated inhibition of adenylate cyclase of 2.50 ± 0.42 and 1.09 ± 0.28 nM, respectively (Figure 2A). Thus, this series provided many new potent and selective A3AR agonists, and alkynyl chain derivatization at the C2 position appears to be an effective strategy for covalent attaching sterically bulky groups while maintaining A3AR recognition and activation.
Figure 2.

Functional agonism by the tested in an assay of adenylate cyclase in membranes of CHO cells expressing the hA3AR. Each experiment was repeated three times. (A) Activity of the 5′-N-methyluronamide (N)-methanocarba analogues 17a and 18. The EC50 values of 17a and 18 were 2.50 ±0.42 and 1.09 ±0.28 nM, respectively. (B) Activity of the truncated (N)-methanocarba analogues 2b and 2c at the hA3AR. The full agonist 51 was included for comparison (representing 100% efficacy). The percent relative efficacy of 2b and 2c was 46 ± 4% and 44 ± 6%, respectively. The EC50 values of 2b and 2c were 4.2± 0.6 and 12± 1 nM, respectively.
A series of l-nucleosides was prepared as enantiomers of members of the potent agonist functionalized congeners (series B). Compounds 19, 20, and 21 were the enantiomers of the potent A3AR agonists 5, 11, and 13, respectively. We confirmed that at submicromolar concentrations no substantial AR binding activity was seen with these l-enantiomers, confirming stereoselectivity of binding at the ARs. At 10 μM, 19–21 were weak or inactive in binding at the A1 and A2AARs, and at the A3AR they tended to be slightly more potent in binding. These l-nucleosides induced partial activation of the A3AR at 10 μM, as confirmed in the cAMP functional assay in which compounds 20 and 21 activated to the degree of 26% and 52%, respectively. Nevertheless, these l-nucleosides can be used at nanomolar concentrations in pharmacological studies as inactive control compounds of similar physicochemical properties, by analogy to the use of inactive enantiomers of other GPCRs.36 At these concentrations, interactions with ARs were lacking. In the truncated 4′-thionucleoside class of A3AR antagonists, stereospecificity of binding was similarly demonstrated.45
The effects of modification at the C2 position were explored in the truncated series C. In the 5′-N-methyluronamide derivatives (series A), even the presence of a long chain at the C2 position did not interfere with the binding and activation of the A3AR. However, in the series of nucleosides that were truncated at the 4′-carbon, variability in both binding affinity and efficacy at the hA3AR was observed, depending on the nature of the C2 substitution. Compounds 22–24, lacking a functionalized chain at the C2 position, were potent and selective A3AR ligands with binding Ki values of 5–11 nM. Removal of the 2-C1 substitution in N6-benzyl-type 4′-truncated derivatives (e.g., 22, 23) or its replacement with 2-F (e.g., 24) maintained A3AR selectivity but reduced the A3 AR affinity by 5- to 10-fold. Replacement of 2-C1 with 2-H either had no effect (cf. 2a and 22) on affinity at the A2AAR or moderately reduced it (cf. 2c and 23), and affinity at the A1AR was moderately increased. The most A3AR selective truncated compound was the 2-unsubstituted N6-(3-chlorobenzyl) derivative 22 with 330-fold and 940-fold selectivity in comparison to the A1 and A2AAR, respectively.
An amine functionalized congener 26 in the truncated series, with a Ki value of 403 nM, proved to be weaker in binding to the A3AR than those analogues with less sterically bulky substitution at the C2 position. However, it nevertheless displayed moderate selectivity of > 20-fold in comparison to both the A1 and A2AARs. This derivative was analogous to compound 11 in the 5′-N-methyluronamide series, which was 160-fold more potent in binding to the A3AR.
A 5′-truncated N6-cyclopentyl derivative 25, by analogy to compound 4,37 was intended to display enhanced affinity at both A1 and A3ARs but not at the A2AAR. However, the loss of the 5′-N-methyluronamide group reduced affinity at A1 and A3ARS by 6- and 32-fold, respectively. Thus, N6-benzyl substitution of the (N)-methanocarba nucleosides preserved the AR selectivity profile upon truncation better than an N6-cyclopentyl group.
Selected compounds in the present series were tested in binding at the rA3AR. The Ki values (nM) were determined to be the following: 6, 66.5 ± 3.9; 15, 8.68 ± 0.85; 18, 9.62 ± 1.50; 22, 20.3 ± 2.0. Thus, the high A3AR affinity was maintained in a murine species.
The A3AR efficacy in the cAMP functional assay varied greatly in this series of truncated (N)-methanocarba derivatives. Compounds 2b and 2c, which were reported as A3AR antagonists based on an assay of [35S]GTPγS binding,20 were partial agonists in the assay of adenylate cyclase (Figure 2B). Both partial (e.g., 22) and nearly full agonism (e.g., 23 and 25) were observed for the novel truncated nucleosides in the cyclase assay. Replacement of the 2-Cl of the truncated analogues 2b with hydrogen in 22 resulted in a 58% relative efficacy, which was comparable to 46% for 2b. The 2-H 23 and 2-F 24 N6-(3-iodobenzyl) analogues displayed relative efficacy of 81% and 19%, respectively. Compound 25 inhibited the production of cAMP via the hA3AR to the same extent as 51 and was therefore a full agonist as judged by adenylate cyclase inhibition.
A homology model of the hA3AR based on the A2AAR X-ray structure38,39 was used to study the putative interactions between the chain-functionalized agonist 15 and the A3AR. The binding mode of 15 obtained after InducedFit docking revealed the following binding mode of the ligand (Figure 3). The 2′- and 3′-hydroxyl groups were located in proximity to Ser271 (7.42) and His272 (7.43) and could form H-bonds with these residues. The oxygen atom of the 5′-N-methylcarboxamido moiety was involved in H-bonding with the side chain hydroxyl group of Thr94 (3.36). Both N6-amino group and the N7 atom of the adenine ring formed H-bonds with Asn250 (6.55). A π−π interaction was observed between the adenine ring of 15 and Phe168 (EL2). The ligand–receptor interactions observed in the present model were in good agreement with the data of site-directed mutagenesis and with our previously published models of ARs, including the studies of AR agonists docked to the A2AAR crystal structure.38,40 In the model obtained, the 3-chlorobenzyl ring was located in the hydrophobic pocket formed by Val169 (EL2). Met172(EL2), Met174(EL2), Met177(5.38), and Ile253(EL3).
Figure 3.
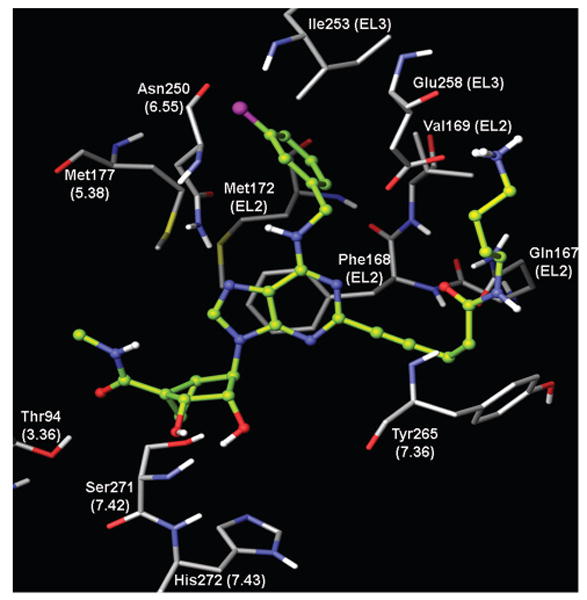
Binding mode of amine congener 15 in the 5′-N-methyluronamide series obtained after InducedFit docking to the A3AR homology model built based on the crystal structure of the A2AAR.
The long chain substituent at the C2 position of 15 was oriented toward the extracellular part of the receptor, which is a prerequisite in functionalized congeners.23 A similar orientation of the 2-propynyl fragment of 15 was previously suggested for C2 substituted agonists of the A2B receptor.41,42 The carbonyl oxygen of the amide group of the C2 substituent of 15 was H-bonded to the side chain NH2-group of Gln 167 (EL2), and consequently the NH-group of the C2 substituent of 15 was oriented toward the hydroxyl group of Tyr265 (7.36) (Figure 3). The terminal amine of the ligand interacted with negatively charged Glu258 (EL3), and it was also H-bonded to the backbone oxygen atom of Val259 (EL3).
Discussion
In the present work and in a previous study,20 parallel patterns of SAR in receptor binding were observed in the two series of 5′-CONHCH3 (full agonist) and truncated (N)-methanocarba nucleosides, in cases with substituted N6-benzyl groups and small substituents present at the adenine C2 position. This parallel behavior was not maintained in a derivative bearing a chain at the C2 position.
The replacement of the adenine 2-Cl with H in the series of substituted N6-benzyl-(N)-methanocarba agonists containing the 5′-CONHCH3 group was previously shown to have only minor effects on AR binding.16 Specifically, it moderately reduced affinity at the A1 and A2AARs without a significant reduction at the A3AR (cf. 1a and 3). In the present 5′-N-methyluronamide series with more elaborate modification at the C2 position, the preservation of potent and selective binding to the hA3AR was demonstrated.
The flexibility of chain substitution at C2 allowed the conjugation with sterically bulky moieties. This distal region of the nucleosides was predicted by homology modeling to be at A3AR extracellular regions and consequently free from the steric constraints present at the pharmacophore binding region. Thus, this series of highly potent and selective A3AR agonist functionalized congeners is suitable for conjugation to prosthetic groups for radiolabeling, fluorescent detection, avidin complexation, and polymeric coupling with retention of biological activity. The functionalized congener approach was already applied to 1,4-dihydropyridine derivatives as antagonists of the hA3AR.26
Specifically, agonists in this series were conjugated with biotin and a fluorescent dye with retention of high A3AR affinity and selectivity. The fluorescent conjugate 18 was a highly potent, full agonist with a Ki values at the hA3AR and the rA3AR of 17.2 and 9.6 nM, respectively. The fluorescent dye Cy5, which was incorporated covalently in 18, has favorable properties for use in biological systems including near-infrared imaging, with excitation and emission maxima at 649 and 670 nm, respectively, and with a relatively high quantum yield of 0.28.27 Another fluorescent agonist of the hA3AR having lower affinity than 18 was used in fluorescence correlation spectroscopy to demonstrate that the receptor exists in heterogeneous microdomains of individual living cells.43
In our previous study of truncated (N)-methanocarba nucleosides,20 the docking of two representative structures in a rhodopsin-based homology model of the hA3AR indicated that both (N)-methanocarba agonists and nucleoside-derived antagonists were able to bind in a very similar mode in the receptor binding site, which is common to other A3AR-selective nucleosides, such as the prototypical agonist 2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine (Cl-IB-MECA).20 We have shown that the SAR parallels between these two series were less stringent with respect to distal modification, e.g., when a long functionalized chain was attached at the C2 position. Thus, in the truncated series the attachment of a functionalized amino chain at the C2 position greatly reduced A3AR affinity. Therefore, among the 2-alkynyl nucleosides in this study, only those containing a 5′-N-methyluronamide group, i.e., full agonists, were suitable as functionalized congeners.
Truncation of the 5′-CONHCH3 moiety of A3AR agonists in the (N)-methanocarba class was again demonstrated as a means of reducing the relative efficacy of A3AR-selective agonists to produce partial agonism, as indicated in the cyclase assay. A previous study of 2-triazolo derivatives of adenosine demonstrated modulation of the relative efficacy at the A3AR by structural changes at the 2 position.12c In the truncated series, the remaining efficacy at the hA3AR, as was evident in a cAMP functional assay, ranged from small (e.g., 24 and 26) to substantial (e.g., 23).
We compared selected compounds in the ability to stimulate the A3AR using multiple functional criteria. When stimulation of [35S]GTPγS binding was used as a criterion of receptor activation, truncated (N)-methanocarba nucleosides (e.g., 2a–c) reported by Melman et al.20 did not induce a substantial activation of the A3AR, with < 10% of the full agonism. However, in the current study using a different functional assay that measured inhibition of the production of cAMP to detect antagonism, considerable activity in the truncated series was seen. The efficacy in the cyclase assay generally exceeded the efficacy in [35S]GTPγ S binding. Other truncated nucleosides in the 4′-thio series were A3 AR antagonists in both [35S]GTPγS and cAMP assays.44 Recently, truncated nucleosides in the 4′-oxo series were demonstrated to be A3 AR antagonists in the cAMP assay.45
In the truncated series, removal of the 2-Cl substitution or replacement with 2-F in 24 moderately reduced the A3AR affinity but retained high selectivity. The introduction of fluorine in 24 was intended for possible radiofluorination as a means of preparing tracers for in vivo PET imaging of the A3AR.47 One member of the previously reported truncated (N)-methanocarba nucleoside series 2b, (1R,2R,3S,4R,5S)-4-(6-(3-bromobenzylamino)-2-chloro-9H-purin-9-yl)bicyclo-[3.1.0]hexane-2,3-diol (MRS5147), was recently labeled with the short-lived positron-emitter 76Br to form a selective hA3AR radioligand of high affinity for use in positron emission tomography (PET). This ligand was intended for imaging of the A3 AR in murine as well as primate species, because high affinity binding was maintained at the rA3AR. Binding of these truncated (N)-methanocarba nucleosides at the A3AR was competitive,20 and therefore, the related derivatives in this study should be useful as receptor probes.
In conclusion, two highly potent and selective series of nucleoside ligands containing the (N)-methanocarba ring system have been synthesized and characterized as A3AR agonists, partial agonists, and antagonists. Furthermore, in the 5′-N-methyluronamide series but not in the truncated series, the functionalized congener approach was found to maintain potency and selectivity at the hA3AR and therefore provides an entry into high affinity molecular probes. Thus, the binding and activation profiles of the truncated analogues indicated the biological interdependence of substitution at the C2, 5′, and N6 positions. Potent agonist ligands containing biotin and fluorescent dye moieties should be useful in future pharmacological studies of the localization, association, regulation, and internalization of the A3AR and might provide an alternative to the use of radioligands for this receptor.
Experimental Section
Chemical Synthesis. Materials and Instrumentation
d-Ri-bose, l-ribose, and other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). Alcohol derivative 31 was prepared as reported.201 H NMR spectra were obtained with a Varian Gemini 300 spectrometer using CDCl3 and CD3OD as solvents. Chemical shifts are expressed in δ values (ppm) with tetramethylsilane (δ 0.00) for CDCl3 and water (δ 3.30) for CD3OD. TLC analysis was carried out on aluminum sheets precoated with silica gel F254 (0.2 mm) from Aldrich. All target compounds are ≥95% pure as determined by HPLC. HPLC mobile phases consisted of the following: for system A, linear gradient solvent system of CH3CN/triethyl ammonium acetate from 5/95 to 60/40 in 20 min and flow rate of 1.0 mL/min; for ystem B, linear gradient solvent system of CH3CN/tetrabutyl ammonium phosphate from 20/80 to 60/40 in 20 min and flow rate of 1.0 mL/min. Low-resolution mass spectrometry was performed with a JEOL SX102 spectrometer with 6 kV Xe atoms following desorption from a glycerol matrix or on an Agilent LC/MS 1100 MSD, with a Waters (Milford, MA) Atlantis C18 column. High resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine, unless otherwise noted. Observed mass accuracies are those expected on the basis of known performance of the instrument as well as trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy.
(1′S,2′R,3′S,4′S,5′S)-4′-[6-(3-Chlorobenzylamino)-2-(4-methoxycarbonyl-l-butynyl)-9-yl]-2′,3′-0-isopropylidenebicyclo[3.1.0]-hexane-1′-carboxylic Acid N′-Methylamide (28)
To a solution of compound 27 (52 mg, 0.087 mmol) in anhydrous DMF (1.5 mL), PdCl2(PPh3)2 (12 mg, 0.017 mmol), CuI (2 mg, 0.010 mmol), methyl-ω-pentynate (39 mg, 0.347 mmol), and then triethylamine (0.12 mL, 0.86 mmol) were added. The reaction mixture was stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH = 30:1) to give compound 28 (40 mg, 78%) as a foamy syrup. 1H NMR (CD3OD, 300 MHz) δ 8.16 (s, 1H), 7.42–7.44 (m, 4H), 5.81 (d, J =6.7 Hz, 1H), 5.02 (s, 1H), 4.82 (m, 2H), 3.78 (s, 3H), 2.85 (s, 3H), 2.72–281 (m, 5H), 2.09–2.14 (m, 1H), 1.55 (s, 3H), 1.41 (t, J=5.1 Hz, 1H), 1.29 (s, 3H), 0.82–0.96 (m, 1H). HRMS calculated for C29H32ClN6O5 (M + H)+: 579.2105: found 579.2123.
(1′S,2′R,3′S,4′S,5′S)-4′-[6-(3-Chlorobenzylamino)-2-(4-(β-aminoethyl aminocarbonyl)-1-butynyl)-9-yl]-2′,3′-dihydroxybicyclo [3.1.0]hexane-1′-carboxylic Acid N-Methylamide (11)
To a solution of compound 28 (20 mg, 0.034 mmol) in methanol (0.3 mL), ethylenediamine (1.5 mL) was added. The mixture was stirred overnight at room temperature. Solvent was evaporated, and the residue was roughly purified on flash silica gel column chromatography. The aminated product was dissolved in methanol (1.5 mL) and 10% trifluoroacetic acid (1.5 mL) and heated at 70 °C for 15 h. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH/NH4OH = 10:1:0.1) to give compound 11 (15 mg, 79%). 1H NMR (CD3OD, 300 MHz) δ 8.09 (s, 1H), 7.39 (s, 1H), 7.27–7.32 (m, 3H), 4.99 (d, J = 6.6 Hz, 1H), 4.78– 4.91 (m, 2H), 3.97 (d, J = 6.6 Hz, 1H), 3.33–3.36 (m, 4H), 2.87 (s, 3H), 2.75–2.80 (m, 4H), 2.53 (t, J = 7.2 Hz, 2H), 2.59–3.41 (m, 1H), 1.88 (t, J = 4.5 Hz, 1H), 1.34–1.43 (m, 1H). HRMS calculated for C27H31ClN6O2Na (M + Na)+: 589.2068; found 589.2054.
(1′S,2′R,3′S,4′S,5′S)-4′-[6-(3-Chlorobenzylamino)-2-(4-methoxycarbonyl)-1 -butyn yl)-9-yl]- 2′,3′ -dihydroxybicyclo [3.1.0] hexane-1′-carboxylic Acid N-Methylamide (8)
A solution of compound 28 (67 mg, 0.115 mmol) in methanol (3 mL) and 10% triflromethanesulfonic acid (2 mL) was heated at 70 °C overnight. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH = 20:1) to give compound 8 (46 mg, 75%). 1H NMR (CD3OD, 300 MHz) δ 8.13 (s, 1H), 7.40 (s, 1H), 7.22–7.45 (m, 3H), 5.10 (d, J = 6.3 Hz, 1H), 4.80–4.86 (m, 2H), 4.03 (d, J = 6.6 Hz, 1H), 3.63 (s, 3H), 2.86 (s, 3H), 2.76–2.85 (m, 2H), 2.46–2.58 (m, 2H), 2.05–2.10 (m, 1H), 1.77–1.83 (m, 1H), 1.35–1.40 (m, 1H), 0.82–0.96 (m, 1H). HRMS calculated for C26H28ClN6O5 (M + H)+: 539.1731; found 539.1743.
(1′S,2′R,3′S,4′S,5′S)-4′-[6-(3-Chlorobenzylamino)-2-(4-hydroxycarbonyl)-l-butynyl)-9-yl]-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic Acid N′-Methylamide (5)
To a solution of ester 8 (30 mg, 0.055 mmol) in methanol (1.5 mL), 1 M solution of potassium hydroxide (1 mL) was added. The mixture was stirred at room temperature overnight. The reaction mixture was neutralized with acetic acid, and solvent was evaporated, and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH = 10:1) to give compound 5 (23 mg, 80%). 1H NMR (CD3OD, 300 MHz) δ 8.06 (s, 1H), 7.38 (s, 1H), 7.24–7.35 (m, 3H), 5.14 (d, J= 5.8 Hz, 1H), 4.79–4.86 (m, 2H), 4.03 (d, J = 6.3 Hz, 1H), 2.87 (s, 3H), 2.42–2.80 (m, 4H), 2.04– 2.32 (m, 1H), 1.82 (t, J =4.8 Hz, 1H), 1.36–1.40 (m, 1H), 0.84– 0.97 (m, 1H). HRMS calculated for C25H26ClN6O5 (M + H)+: 525.9562; found 525.9583.
(1′S,2′R,3′S,4′S,5′S)-4′-[6-(3-Chiorobenzylamino)-2-(N-biotinyl(β-aminoethylaminocarbonyl)-1-butynyl)-9-yl]-2′, 3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic Acid N-Methylamide (17a)
To a solution of compound 11 (4 mg, 0.007 mmol) in dry DMF (0.5 mL), biotin (1.89 mg, 0.0077 mmol), HATU (3.2 mg, 0.0084 mmol), and DIEA (1.6 μL, 0.009 mmol) were added. The mixture was stirred at room temperature overnight. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH/NH4OH =5:1:0.1) to give compound 17a (3.5 mg, 62%). 1H NMR (CD3OD, 300 MHz) δ 8.14 (s, 1H), 7.43 (s, 1H), 7.29–7.39 (m, 3H), 5.07 (d, J = 6.9 Hz, 1H), 4.78–4.86 (m, 2H), 4.48–4.52 (m, 1H), 4.28–4.32 (m, 1H), 4.04 (d, J =6.3 Hz, 1H), 3.70–3.82 (m, 2H), 3.24–3.31 (m, 1H), 3.08–3.23 (m, 1H), 2.91 (s, 3H), 2.70–2.83 (m, 4H), 2.56 (t, J =7.5 Hz, 2H), 2.04–2.15 (m, 2H), 1.89 (t, J =5.1 Hz, 1H), 1.33–1.70 (m, 10H), 0.84–1.02 (m, 1H). HRMS calculated for C37H46ClN10O6S (M + H)+: 793.3011; found 793.3030.
(1′S,2′R,3′S,4′S,5′S)4′-[6-(3-Chlorobenzylamino)-2-(N-biotinyl{5-aminopentanyl}(β-aminoethylaminocarbonyl)-1-butynyl)-9-yl]-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic Acid N-Methylamide (17b)
To a solution of compound 11 (3.4 mg, 0.0059 mmol) in DMF (0.5 mL), sulfo-NHS-LC-biotin (10 mg, 0.017 mmol, Pierce) and 10 μL of triethyl amine were added. The mixture was stirred overnight at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH/NH4OH = 7:1:0.1) to give compound 17b (2.9 mg, 55%). 1H NMR (CD3OD, 300 MHz) δ 7.99 (s, 1H), 7.29 (s, 1H), 7.19–7.32 (m, 3H), 4.93 (d, J = 5.4 Hz, 1H), 4.70–4.80 (m, 2H), 4.36–4.42 (m, 1H), 4.16–4.22 (m, lH), 3.88 (d,J = 6.9 Hz, 1H), 3.42–3.62 (m, 2H), 3.03–3.15 (m, 4H), 2.89 (s, 3H), 2.56–2.86 (m, 6H), 2.41 (t, J = 6.6 Hz, 1H), 1.96–2.13 (m, 5H), 1.15–1.16 (m, 14H), 0.74–0.88 (m, 1H). C43H57C1N11O7S (M+H)+: 906.3852; found 906.3878.
(1′S,2′R,3′S,4′S,5′S)-4′-[6-(3-Chlorobenzylamino)-2-(N-cyaiune(β-aminoethylaminocarbonyl)-1-butynyl)-9-yl]-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic Acid N-Methylamide (18)
To a solution of compound 11 (1.69 mg, 0.0029 mmol) in DMF (0.3 mL), Cy5 fluorescent dye (2.36 mg, 0.0029 mmol) and bicarbonate buffer (60 μL) were added. The mixture was stirred at room temperature overnight. The reaction mixture was covered with aluminum foil in order to protect from light. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH/NH4OH = 3:1:0.1) to give compound 18 (3.2 mg, 89%) as a dark-blue syrup. 1H NMR (CD3OD, 300 MHz) δ 8.21–8.42 (m, 1H), 8.08 (s, 1H), 7.85–7.93 (m, 2H), 7.29–7.46 (m, 6H), 6.28–6.45 (m, 1H), 5.07 (d, J = 6.9 Hz, 1H), 4.84–4.86 (m, 2H), 3.97–4.20 (m, 3H), 3.53 (t,J = 6.0Hz, 2H), 3.12 (t,J = 6.0Hz, 2H), 2.89 (s, 3H), 2.49 (t,J =7.5 Hz, 2H), 2.52–2.64 (m, 2H), 2.01–2.17 (m, 2H), 1.56–1.89 (m, 9H), 1.22–1.49 (m, 10H), 0.86–0.96 (m, 1H). HRMS calculated for C60H68ClIN10 O11S2 (M +): 1203.4199; found 1203.4175.
(1′R,2′S,3′R,4′R,5′R)-4′-(6-Chloro-2-iodopurin-9-yl)-2′,3′-isopropylidenebicyclo[3.1.0]hexane-1′-carboxylic Acid Ethyl Ester (32)
To a solution of triphenylphosphine (0.357 g, 1.361 mmol) and 6-chloro-2-iodopurine (0.286 g, 1.019 mmol) in dry THF (4 mL), DIAD (0.26 mL, 1.359 mmol) was added. The mixture was stirred at room temperature for 10 min. A solution of compound 31 (0.165 g, 0.681 mmol) in THF (2 mL) was added to the reaction mixture, and the mixture was further stirred overnight at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 3:1) to give the product 32 (0.2 g, 58%) as a foam. 1H NMR (CDCl3, 300 MHz) δ 1.31–1.37 (m, 4H), 1.56 (s,6H), 1.77–1.80 (m, 1H), 2.10–2.22 (m, 1H), 4.19–4.27 (m, 2H), 4.77 (d, J = 14.2 Hz, 1H), 4.91 (s, 1H), 5.83 (d, J = 15.3 Hz, 1H),7.97(s, 1H). HRMS calculated for C17H19ClIN4O4 (M + H)+: 505.0061; found 505.0073.
(1′R,2′S,3′R,4′R,5′R)-4′-[6-(3-Chlorobenzylamino)-2-iodopurin-9- yl]-2′,3′-isopropylidenebicyclo[3.1.0]he xane-1′-carboxylic Acid Ethyl Ester (33)
To a solution of compound 32 (0.273 g, 0.54 mmol) in anhydrous methanol (5 mL), triethylamine (1 mL, 7.569 mmol) and 3-chlorobenzylamine (0.33 mL, 2.697 mmol) were added. The mixture was stirred overnight at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 1:1) to give the product 33 (0.255 g, 78%) as a foamy solid. 1H NMR (CDCl3, 300 MHz) δ 7.57 (s, 1H), 7.20–7.25 (m, 4H), 6.03–6.15 (m, 1H), 5.83 (d, J = 4.45 Hz, 1H), 5.28 (s, 1H), 4.65– 4.88 (m, 3H), 4.11–4.22 (m, 2H), 2.18–2.23 (m, 1H), 1.22–1.80 (m, 7H), 1.10–1.21 (m, 4H). HRMS calculated for C24H26ClI-N5O4 (M + H)+: 610.0639; found 610.0649
(1′R,2′S,3′R,4′R,5′R)-4′-[6-(3-Chlorobenzylamino)-2-iodopurin-9-yl]-2′-,3′-isopropylidenebicyclo[3.1.0]hexane-1′-carboxylic Acid-N-Methylamide (34)
To a solution of compound 33 (0.258 g, 0.423 mmol) in methanol (5 mL), 40% methylamine (4 mL) was added. The mixture was stirred at room temperature for 2 days. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH = 40:1) to give the product 34 (0.165 g, 66%) as a foam. 1H NMR(CDCl3, 300 MHz) δ 7.58 (s, 1H), 7.25–7.37 (m, 4H), 6.52 (bs, 1H), 5.65 (d, J = 6.9 Hz, 1H), 4.73–4.83 (m, 3H), 2.97 (s, 3H), 2.04–2.07 (m,1H), 1.62–1.82 (m, 1H), 1.55 (s, 3H), 1.22–1.33 (m, 5H). HRMS calculated for C23H25ClIN6O3 (M + H)+: 595.0643: found 595.0649.
(1′R,2′S,3′R,4′R,5′R)-4′-[6-(3-Chlorobenzylamino)-2-(4-hydroxycarbonyl)-1-butynyl)-9-yl]-2′,3′-dihydroxybicyclo[3.1.0]hexane-1′-carboxylic Acid N-Methylamide (19)
To a solution of compound 34 (32 mg, 0.053 mmol) in anhydrous DMF (1.0 mL), PdCl2(PPh3)2 (7 mg, 0.01 mmol), CuI (2 mg, 0.01 mmol), methyl-ω-hexynate (27 mg, 0.214 mmol), and then triethylamine (0.014 mL, 0.10 mmol) were added. The reaction mixture was stirred at room temperature overnight. Solvent was evaporated under vacuum, and the residue was roughly purified on flash silica gel column chromatography. To a solution of the product in methanol (1 mL), 10% TFA (1 mL) was added, and the mixture was heated at 70 °C for 7 h. Solvent was evaporated under vacuum, the crude reaction mixture was further dissolved in methanol (1 mL), and 1 M solution of KOH (1 mL) was added, and the mixture was stirred at room temperature overnight. Reaction mixture was neutralized with acetic acid, solvent was evaporated under vacuum, and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH = 20:1) to give compound 19 (18 mg, 62%) as a colorless powder. 1H NMR (CD3OD, 300 MHz) δ 7.97 (s, 1H), 7.30 (s, 1H), 7.14– 7.22 (m, 3H), 4.97 (d, J = 6.7 Hz, 1H), 4.73 (brs, 3H), 3.90 (d, J = 6.6 Hz, 1H), 2.75 (s, 3H), 2.42 (t, J = 7.2 Hz, 2H), 2.28 (t, J = 7.2 Hz, 2H), 1.96 (m, 1H), 1.83 (m, 3H), 1.75 (m, 1H), 1.25 (m, 1H). HRMS calculated for C26H28ClN6O5 (M + H)+: 539.1731: found 539.1724.
(1R,2R,3S,4S,5S)-2,3-O-Isopropylidene-2,3,4-trihydroxybicyclo[3.1.0]hexane (40)
To a solution of compound 39 (0.773 g, 4.95 mmol) in dry CH2Cl2 (25 mL), Et2Zn (19.8 mL, 1 M solution in hexane) was added at 0 °C. The mixture was stirred for 15 min. After 15min, CH2I2 (3.2 mL, 39.57 mmol) was added at the same conditions and then the reaction mixture was stirred at room temperature overnight. After completion of reaction, it was quenched with saturated ammonium chloride solution and the aqueous layer was extracted with CH2Cl2. The combined organic layer was dried (Na2SO4), filtered, and evaporated. The residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 3:1) to give compound 40 (0.710 g, 85 %) as a colorless syrup. 1H NMR (CDCl3, 300 MHz) δ 4.88 (t, J = 6.3 Hz, 1H), 4.46 –4.54 (m, 2H), 2.37 (d, J = 9.3 Hz, 1H), 1.83 – 1.86 (m, 1H), 1.61–1.66 (m, 1H), 1.55 (s,3H), 1.29 (s,3H), 0.95– 0.99 (m, 1H), 0.62–0.68 (m, 1H). HRMS calculated for C9H15O3 (M + H)+: 171.0943; found 171.0954.
(1′R,2′R,3′S,4′S,5′S)-4′-(6-Chloro-2-iodopurine)-2′,3′-O-isopropylidene-2′,3′-dihydroxybicyclo[3.1.0]hexane (44)
To a solution of triphenylphosphine (132 mg, 0.5 mmol) and 6-chloro-2-iodopurine (141 mg, 0.50 mmol) in dry THF (2 mL), DIAD (0.1 mL, 0.50 mmol) was added. The mixture was stirred at room temperature for 10 min. A solution of compound 31 (43 mg, 0.252 mmol) in THF (2 mL) was added to the reaction mixture, and the mixture was further stirred overnight at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 3:1) to give the product 44 (65 mg, 61%) as a foam. 1H NMR (CDCl3, 300 MHz) d 8.07 (s, 1H), 5.37 (t, J = 2.1 Hz, 1H), 4.67 (d, J = 5.7Hz, 1H), 2.13–2.18 (m, 1H), 1.65–1.69 (m, 2H), 1.27 (s, 3H), 1.26 (s, 3H), 0.96–1.01 (m, 2H). HRMS calculated for C14H15ClIN4O2 (M + H)+: 432.9927; found 432.9928.
(1′R,2′R,3′S,4′S,5′S)-4′-[6-(3-Chlorobenzylamino)-2-iodopurine]-2′,3′- O-isopropylidene-2′,3′-dihydroxybicyclo[3.1.0]hexane (45)
To a solution of compound 44 (0.105 g, 0.243 mmol) in anhydrous methanol (5 mL), triethylamine (0.4 mL, 3.38 mmol) and 3-chlorobenzylamine (0.15 mL, 1.21 mmol) were added. The mixture was stirred overnight at room temperature. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (hexane/ethyl acetate = 1:1) to give the product 45 (0.096 g, 74%) as a foam. 1H NMR (CDCl3, 300 MHz) δ 7.63 (s, 1H), 7.37 (s, 1H), 7.27–7.26 (m, 3H), 6.25 (bs, 1H), 5.34 (t, J = 5.7Hz, 1H), 4.94 (s,2H), 4.64 (d, J =6.9Hz, 1H), 2.06–2.09 (m, 1H), 1.61–1.69 (m,2H), 1.54(s,3H), 1.26(s,3H), 0.88–0.96 (m, 2H). HRMS calculated for C21H22ClIN5O2 (M + H)+: 538.0497; found 538.0507.
(1′R,2′R,3′S,4′ S,5′ S)-4′-[6-(3-Iodobenzylamino)purine]-2′,3′-dihydroxybicyclo[3.1.0]hexane (23)
A solution of compound 46 (32 mg, 0.063 mmol) in MeOH (2 mL) and 10% trifluoroacetic acid (1.5 mL) was heated at 70 °C overnight. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH =30:1) to give compound 23 (23 mg, 79%). 1H NMR (CD3OD, 300 MHz) δ 8.34 (s, 1H), 8.32 (s, 1H), 7.8 (s, 1H), 7.64 (d,J = 8.1 Hz, 1H), 7.42 (d, J =7.5 Hz, 1H), 7.13 (t, J= 7.8 Hz, 1H), 4.90 (s, 2H), 4.72 (t, J= 6.3 Hz, 2H), 3.96 (d, J = 6.6 Hz, 1H), 1.96–2.04 (m, 1H), 1.67–1.72 (m, 1H), 1.30–1.34 (m, 1H), 0.75–0.83 (m, 1H). HRMS calculated for C18H19IlN5O2 (M +H)+: 464.0596; found 464.0584.
(1′R,2′R,3′S,4′S,5′S)-4′-[6-(3-Iodobenzylamino)-2-fluoro-purine]-2′,3′-dihydroxybicyclo[3.1.0]hexane (24)
To a solution of compound 42 (10 mg, 0.03 mmol) in anhydrous methanol (0.5 mL), triethylamine (0.06 mL, 0.42 mmol) and 3-iodobenzylamine (0.02 mL, 0.15 mmol) were added. The mixture was stirred overnight at room temperature. Solvent was evaporated, and the residue was roughly purified on flash silica gel column chromatography. The product was dissolved in methanol (1 mL) and 10% TFA (0.5 mL), and the reaction mixture was heated at 70 °C for 6 h. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (hexane/ ethyl acetae = 1:1) to give compound 24 (8 mg, 55%). 1H NMR (CD3OD, 300 MHz) δ 8.13 (s, 1H), 7.78 (s, 1H), 7.63 (dd, J = 7.8 Hz, 1H), 7.41 (d, J = 7.8 Hz, 1H), 7.11 (t, J = 7.8 Hz, 1H), 4.76 (s, 1H), 4.71–4.76 (m, 2H), 3.92–3.96 (m, 2H), 1.96–2.01 (m, 1H), 1.64–1.67 (m, 1H), 0.73–0.91 (m, 2H). HRMS calculated for C18H18FIN5O2 (M + H)+: 482.0473; found 482.0489.
(1′R,2′R,3′S,4′S,5′S)-4′-[6-(3-Chlorobenzylamino)-2-(4-methoxycarbonyl-1-butynyl)-9-yl]-2′,3′-O-isopropylidenebicyclo[3.1.0]-hexane (50)
To a solution of compound 45 (43 mg, 0.08 mmol) in anhydrous DMF (1.5mL),PdCl2(PPh3)2 (11 mg,0.015mmol), CuI (1.5 mg, 0.007 mmol), methyl-ω-pentynate (35 mg, 0.31 mmol), and then triethylamine (0.11 mL, 0.80 mmol) were added. The reaction mixture was stirred at room temperature overnight. Solvent was evaporated under vacuum and the residue was purified on flash silica gel column chromatography (hexane/ ethyl acetate = 1:1) to give compound 50 (30 mg, 74%) as a foamy syrup. 1H NMR (CDCl3, 300 MHz) 7.98 (s, 1H), 7.32– 7.21 (m, 4H), 6.19 (bs, 1H), 5.31 (t, J = 5.24, 1H), 5.12 (s, 2H), 4.60 (d, J= 7.2 Hz, 1H), 3.78 (s, 3H), 2.69 –2.84 (m, 4H), 2.06– 2.14 (m, 1H), 1.62–1.72 (m, 1H), 1.56 (s,3H), 1.25 (s,3H), 0.88– 1.04 (m, 2H). HRMS calculated for C27H29ClN5O2 (M+ H)+: 522.9953; found 522.9967.
(1′ R,2′ R,3′S,4′S,5′S)-4′ -[6-(3-Chlorobenzylamino)-2-(4-β-aminoethylaminocarbonyl)-1-butynyl)-9-yl]-2′,3′-dihydroxybicyclo [3.1.0]hexane (26)
To a solution of compound 50 (25 mg, 0.047 mmol) in methanol (0.4 mL), ethylenediamine (1.5 mL) was added. The mixture was stirred overnight at room temperature. Solvent was evaporated, and the residue was roughly purified on flash silica gel column chromatography. The aminated product was dissolved in methanol (1.5 mL) and 10% trifluoroacetic acid (1.0 mL), and the mixture was heated at 70 °C for 15 h. Solvent was evaporated and the residue was purified on flash silica gel column chromatography (CH2Cl2/MeOH = 5:1) to give compound 26 (14 mg, 58%). 1H NMR (CD3OD, 300 MHz) δ 8.12 (s, 1H), 7.25–7.39 (m, 4H), 5.97 (s, 1H), 4.70 (t, J = 6.3 Hz, 1H), 3.95 (d, J = 6.6 Hz, 1H), 3.77 (t, J = 6.3 Hz, 2H), 2.94 (m, 2H), 2.52 (t, J = 7.8 Hz, 2H), 1.97–2.01 (m, 1H), 1.68–1.72 (m, 2H), 1.30–1.35 (m, 2H), 0.89–0.91 (m, 2H), 0.78–0.82 (m, 1H). HRMS calculated for C25H29ClN7O3 (M + H)+: 510.2000; found 510.2020.
UV–Visible Spectra of Biotinyl (17) and Fluorescent (18) Probes and Fluorescence Spectrum of 18
UV–visible spectra were measured on an HP diode array spectrophotometer, model no. 8452A. The concentration of each was 40 μM in pH 7.5 aqueous buffer (50 mM Tris-HCl containing 10 mM MgCl2). The parameters for the compounds are as follows: 17a, λ 276 nm (ε = 9890 L/(mol·cm)); 17b, λ 276 nm (ε = 7400 L/(mol·cm)); 18, λ 274 nm (ε = 16 100 L/(mol·cm)), λ 650 nm (ε = 44900 L/(mol·cm)). Fluorescence spectra were measured on a PerkinElmer LS503: excitation λmax = 646 nm, emission λmax = 660 nm.
Receptor Binding and Functional Assays
[3H]2-Chloro-N6-cyclopentyladenosine (52, [3H]CCPA, 42.6 Ci/mmol) was a custom synthesis product (Perkin-Elmer). [125I]N6-(4-Amino-3-iodobenzyl)adenosine-5′-N′-methyluronamide (53, [125I]I-AB-MECA, 2200 Ci/mmol) and [3H](2-[p-(2-carboxyethyl)-phenylethylamino]-5′-N′-ethylcarboxamidoadenosine) (54, [3H] CGS21680, 40.5 Ci/mmol) were purchased from Perkin-Elmer Life and Analytical Sciences (Boston, MA). Test compounds were prepared as 5 mM stock solutions in DMSO and stored frozen.
Cell Culture and Membrane Preparation
CHO cells stably expressing the recombinant hA1, hA3, and rA3Rs and HEK-293 cells stably expressing the hA2AAR were cultured in Dulbecco's modified Eagle medium (DMEM) and F12 (1:1) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100μg/mL streptomycin, and 2 μmol/mL glutamine. In addition, 500 μg/mL Geneticin was added to the A2A media, while 800 μg/mL hygromycin was added to the A1 and A3 media. After being harvested, cells were homogenized and suspended in PBS. Cells were then centrifuged at 240g for 5 min, and the pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.5) containing 10 mM MgCl2. The suspension was homogenized and was then ultracentrifuged at 14,330 g for 30 min at 4 °C. The resultant pellets were resuspended in Tris buffer and incubated with adenosine deaminase (3 units/mL) for 30 min at 37 °C. The suspension was homogenized with an electric homogenizer for 10 s, pipetted into 1 mL vials, and then stored at −80 °C until the binding experiments. The protein concentration was measured using the BCA protein assay kit from Pierce Biotechnology, Inc. (Rockford, IL).49
Binding Assays
Into each tube in the binding assay was added 50 μL of increasing concentrations of the test ligand in Tris-HCl buffer (50 mM, pH 7.5) containing 10 mM MgCl2, 50 μL of the appropriate agonist radioligand, and finally 100 μL of membrane suspension. For the A1AR (22 μg of protein/tube) the radioligand used was [3H]51 (final concentration of 3.5 nM) or [3H]52 (final concentration of 1.0 nM). For the A2AAR (20 μg/tube) the radioligand used was [3H]53 (10 nM). For the A3AR (21 μg/tube) the radioligand used was [125I]54 (0.34 nM). Nonspecific binding was determined using a final concentration of 10 μM 51 diluted with the buffer. The mixtures were incubated at 25 °C for 60 min in a shaking water bath. Binding reactions were terminated by filtration through Brandel GF/B filters under reduced pressure using a M-24 cell harvester (Brandel, Gaithersburg, MD). Filters were washed three times with 3 mL of 50 mM ice-cold Tris-HCl buffer (pH 7.5). Filters for A1 and A2AAR binding were placed in scintillation vials containing 5 mL of Hydrofluor scintillation buffer and counted using a Perkin-Elmer liquid scintillation analyzer (Tri-Carb 2810TR). Filters for A3AR binding were counted using a Packard Cobra II γ-counter. The Ki values were determined using GraphPad Prism for all assays.
cAMP Accumulation Assay
Intracellular cAMP levels were measured with a competitive protein binding method.34 CHO cells that expressed the recombinant hA3AR were harvested by trypsinization. After centrifugation and resuspended in medium, cells were planted in 24-well plates in 1.0 mL medium. After 24 h, the medium was removed and cells were washed three times with 1 mL of DMEM, containing 50 mM HEPES, pH 7.4. Cells were then treated with the agonist 51 or test compound in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL). After 45 min forskolin (10 μM) was added to the medium, and incubation was continued for an additional 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20 °C. For determination of cAMP production, 100 μL of the HCl solution was used in the Sigma direct cAMP enzyme immunoassay following the instructions provided with the kit. The results were interpreted using a Bio-Tek Instruments ELx808 Ultra microplate reader at 405 nm.
Molecular Modeling
The Prime program of the Schrodinger package48 was utilized to build a new molecular model of the hA3AR. The recently reported model of the complex of the nonselective AR agonist 51 docked to the crystal structure of the A2AAR was used as a template for modeling of the A3AR.38 The sequence alignment of the A2A and A3ARs was performed with Prime, taking into account positions of highly conserved amino acid residues. The default values of all Prime parameters were used. The position of full agonist 51 inside the A2A receptor was taken into account during the modeling. Thus, the resulting model of the A3AR contained 51 prepositioned inside the receptor. The Glide program of the Schrodinger package was used to redock 51 to the A3AR model obtained. The receptor grid generation was performed for the box with a center in the centroid of 51 in its initial position. The size of the box was determined automatically. The extra precision mode (XP) of Glide was used for the docking. The ligand scaling factor was set to 1.0. The geometry of the ligand binding site of the complex of A3AR with 51 docked was optimized. The binding site was defined as 51 and all amino acid residues located within 5 Å from 51. All A3AR residues located within 2 Å from the binding site were used as a shell. The following parameters of energy minimization were used: OPLS2005 force field; water was used as an implicit solvent; a maximum of 5000 iterations of the Polak–Ribier conjugate gradient minimization method was used with a convergence threshold of 0.01 kJ·mol−1 ·A−1. Another reference nucleoside, 5′-N′-methylcarboxamidoadenosine, was also docked in this hA3AR homology model (Supporting Information).
The A3AR model obtained was utilized to study a binding mode of amine congener 15 using InducedFit docking implemented in the Schrödinger package. The grid generation was performed for the box with a side of 32 Å with a center in the centroid of 51. The default values were used for other parameters.
Supplementary Material
Acknowledgments
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations. This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Dilip Tosh thanks Can-Fite Biopharma for financial support.
Footnotes
Contributed to mark the 100th anniversary of the Division of Medicinal Chemistry of the American Chemical Society.
Abbreviations: AR, adenosine receptor; cAMP, adenosine 3′,5′-cyclic phosphate; CCPA, 2-chloro-N6-cyclopentyladenosine; CHO, Chinese hamster ovary; Cl-IB-MECA, 2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; DMEM, Dulbecco's modified Eagle's medium; EDTA, ethylenediaminetetraacetic acid; GPCR, G-protein-coupled receptor; HATU, 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate methanaminium; HEK, human embryonic kidney; I-AB-MECA, 2-[p-(2-carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamidoadenosine; NECA, 5′-N-ethylcarboxamidoadenosine; DIPEA, diisopropylethylamine; DCM, dichloromethane; DMF, N,N-dimethylformamide; HEPES,4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; HRMS, high resolution mass spectroscopy; PET, positron emission tomography; TEA, triethylamine; TLC, thin layer chromatography.
Supporting Information Available: Synthetic procedures for compounds 30, 13–16, 10, 7, 35, 37, 20, 21, 41, 42, 46, 47, 49, 22, and 25 and their characterization; UV–visible spectra of 17a, 17b, and 18; docking view of a reference agonist in the hA3AR homology model. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jacobson KA, Klutz AM, Tosh DK, Ivanov AA, Preti D, Baraldi PG. Medicinal Chemistry of the A3 Adenosine Receptor: Agonists, Antagonists, and Receptor Engineering In HEP Adenosine Receptors in Health and Disease; Handbook of Experimental Pharmacology. Vol. 193. Springer; New York: 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gessi S, Merighi S, Varani K, Cattabriga E, Benini A, Mirandola P, Leung E, Mac Lennan S, Feo C, Baraldi S, Borea PA. Adenosine receptors in colon carcinoma tissues and colon tumoral cell lines: focus on the A3 adenosine subtype. J Cell Physiol. 2007;211:826–836. doi: 10.1002/jcp.20994. [DOI] [PubMed] [Google Scholar]
- 3.Yamano K, Inoue M, Masaki S, Saki M, Ichimura M, Satoh M. Generation of adenosine A3 receptor functionally humanized mice for the evaluation of the human antagonists. Biochem Pharmacol. 2006;71:294–306. doi: 10.1016/j.bcp.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Curr Eye Res. 2005;30:747–754. doi: 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hua X, Chason KD, Fredholm BB, Deshpande DA, Penn RB, Tilley SL. Adenosine induces airway hyperresponsiveness through activation of A3 receptors on mast cells. J Allergy Clin Immunol. 2008;122:107–113. doi: 10.1016/j.jaci.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fishman P, Jacobson KA, Ochaion A, Cohen S, Bar-Yehuda S. The anti-cancer effect of A3 adenosine receptor agonists: a novel, targeted therapy. Immunol, Endocr Metab Agents Med Chem. 2007;7:298–303. doi: 10.2174/187152207781369878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J Rheumatol. 2008;35:41–48. [PubMed] [Google Scholar]
- 8.Zheng J, Wang R, Zambraski E, Wu D, Jacobson KA, Liang BT. A novel protective action of adenosine A3 receptors: attenuation of skeletal muscle ischemia and reperfusion injury. Am J Physiol: Heart Circ Physiol. 2007;293:3685–3691. doi: 10.1152/ajpheart.00819.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan TC, Ge ZD, Tampo A, Mio Y, Bienengraeber MW, Tracey WR, Gross GJ, Kwok WM, Auchampach JA. The A3 adenosine receptor agonist CP-532,903 [N6-(2,5-dichlorobenzyl)-3′-aminoadenosine-5′-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel. J Pharmacol Exp Ther. 2008;324:234–243. doi: 10.1124/jpet.107.127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guzman J, Yu JG, Suntres Z, Bozarov A, Cooke H, Javed N, Auer H, Palatini J, Hassanain HH, Cardounel AJ, Javed A, Grants I, Wunderlich JE, Christofi FL. ADOA3R as a therapeutic target in experimental colitis: proof by validated high-density oligonucleotide microarray analysis. Inflammatory Bowel Dis. 2006;12:766–789. doi: 10.1097/00054725-200608000-00014. [DOI] [PubMed] [Google Scholar]
- 11.Moro S, Deflorian F, Bacilieri M, Spalluto G. Novel strategies for the design of new potent and selective human A3 receptor antagonists: an update. Curr Med Chem. 2006;13:639–645. doi: 10.2174/092986706776055670. [DOI] [PubMed] [Google Scholar]
- 12.(a) Gao ZG, Kim SK, Biadatti T, Chen W, Lee K, Barak D, Kim SG, Johnson CR, Jacobson KA. Structural determinants of A3 adenosine receptor activation: nucleoside ligands at the agonist/antagonist boundary. J Med Chem. 2002;45:4471–4484. doi: 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao ZG, Joshi BV, Klutz AM, Kim SK, Lee HW, Kim HO, Jeong LS, Jacobson KA. Conversion of A3 adenosine receptor agonists into selective antagonists by modification of the 5′-ribofuran-uronamide moiety. Bioorg Med Chem Lett. 2006;16:596–601. doi: 10.1016/j.bmcl.2005.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cosyn L, Palaniappan KK, Kim SK, Duong HT, Gao ZG, Jacobson KA. Van Calenbergh, S. 2-Triazole-substituted adenosines: a new class of selective A3 adenosine receptor agonists, partial agonists, and antagonists. J Med Chem. 2006;49:7373–7383. doi: 10.1021/jm0608208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeong LS, Choe SA, Gunaga P, Kim HO, Lee HW, Lee SK, Tosh DK, Patel A, Palaniappan KK, Gao ZG, Jacobson KA, Moon HR. Discovery of a new nucleoside template for human A3 adenosine receptor ligands: d-4′-thioadenosine derivatives without 4′-hydroxymethyl group as highly potent and selective antagonists. J Med Chem. 2007;50:3159–3162. doi: 10.1021/jm070259t. [DOI] [PubMed] [Google Scholar]
- 14.Jacobson KA, Ji Xd, Li AH, Melman N, Siddiqui MA, Shin KJ, Marquez VE, Ravi RG. Methanocarba analogues of purine nucleosides as potent and selective adenosine receptor agonists. J Med Chem. 2000;43:2196–2203. doi: 10.1021/jm9905965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee K, Ravi RG, Ji Xd, Marquez VE, Jacobson KA. Ring-constrained (N)-methanocarba-nucleosides as adenosine receptor agonists: independent 5′-uronamide and 2′-deoxy modifications. Bioorg Med Chem Lett. 2001;11:1333–1337. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tchilibon S, Joshi BV, Kim SK, Duong HT, Gao ZG, Jacobson KA. (N)-Methanocarba 2, N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J Med Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochaion A, Bar-Yehuda S, Cohen S, Amital H, Jacobson KA, Joshi BV, Gao ZG, Barer F, Patoka R, Del Valle L, Perez-Liz G, Fishman P. The A3 adenosine receptor agonist CF502 inhibits the PI3K, PKB/Akt and NF-κB signaling pathway in synoviocytes from rheumatoid arthritis patients and in adjuvant induced arthritis rats. Biochem Pharmacol. 2008;76:482–494. doi: 10.1016/j.bcp.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matot I, Weininger CF, Zeira E, Galun E, Joshi BV, Jacobson KA. A3 adenosine receptors and mitogen activated protein kinases in lung injury following in-vivo reperfusion. Crit Care. 2006;10:R65. doi: 10.1186/cc4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao ZG, Teng B, Wu H, Joshi BV, Griffiths GL, Jacobson KA. Synthesis and pharmacological characterization of [125I]MRS1898, a high affinity, selective radioligand for the rat A3 adenosine receptor. Purinergic Signalling. 2009;5:31–37. doi: 10.1007/s11302-008-9107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melman A, Wang B, Joshi BV, Gao ZG, de Castro S, Heller CL, Kim SK, Jeong LS, Jacobson KA. Selective A3 adenosine receptor antagonists derived from nucleosides containing a bicyclo[3.1.0]hexane ring system. Bioorg Med Chem. 2008;16:8546–8556. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg Med Chem Lett. 2008;18:2813–2819. doi: 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson KA, Ukena D, Padgett W, Kirk KL, Daly JW. Molecular probes for extracellular adenosine receptors. Biochem Pharmacol. 1987;36:1697–1707. doi: 10.1016/0006-2952(87)90056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobson KA. Functionalized congener approach to the design of ligands for G protein-coupled receptors (GPCRs) Bioconjugate Chem. 2009 doi: 10.1021/bc9000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi BV, Melman A, Mackman RL, Jacobson KA. Synthesis of ethyl (1S,2R,3S,4S,5S)-2,3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from l-ribose: a versatile chiral synthon for preparation of adenosine and P2 receptor ligands. Nucleosides, Nucleotides Nucleic Acids. 2008;27:279–291. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chinchilla R, Najera C. The Sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem Rev. 2007;107:874–922. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 26.Li AH, Chang L, Ji Xd, Melman N, Jacobson KA. Functionalized congeners of 1,4-dihydropyridines as antagonist molecular probes for A3 adenosine receptors. Bioconjugate Chem. 1999;10:667–677. doi: 10.1021/bc9900136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS. Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjugate Chem. 1993;4:105–111. doi: 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- 28.Choi WJ, Park JG, Yoo SJ, Kim HP, Moon HR, Chun MW, Jung YH, Jeong LS. Syntheses of d- and l-cyclopentenone derivatives using ring-closing metathesis: versatile intermediates for the synthesis of d- and l-carbocyclic nucleosides. J Org Chem. 2001;66:6490–6494. doi: 10.1021/jo015733w. [DOI] [PubMed] [Google Scholar]
- 29.Klotz KN, Lohse MJ, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-[3H]cyclopentyladenosine ([3H]-CCPA), a high affinity agonist radioligand for A1 adenosine receptors. Naunyn Schmiedeberg's Arch Pharmacol. 1989;340:679–683. doi: 10.1007/BF00717744. [DOI] [PubMed] [Google Scholar]
- 30.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J Pharmacol Exp Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 31.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-aminobenzyl-5′-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 32.Englert M, Quitterer U, Klotz KN. Effector coupling of stably transfected human A3 adenosine receptors in CHO cells. Biochem Pharmacol. 2002;64:61–65. doi: 10.1016/s0006-2952(02)01071-7. [DOI] [PubMed] [Google Scholar]
- 33.Jacobson KA, Park KS, Jiang Jl, Kim YC, Olah ME, Stiles GL, Ji Xd. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology. 1997;36:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 35.Lorenzen A, Lang H, Schwabe U. Activation of various subtypes of G-protein alpha subunits by partial agonists of the adenosine A1 receptor. Biochem Pharmacol. 1998;56:1287–1293. doi: 10.1016/s0006-2952(98)00207-x. [DOI] [PubMed] [Google Scholar]
- 36.Pakdeechote P, Dunn WR, Ralevic V. Cannabinoids inhibit noradrenergic and purinergic sympathetic cotransmission in the rat isolated mesenteric arterial bed. Br J Pharmacol. 2007;152:725–733. doi: 10.1038/sj.bjp.0707397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobson KA, Gao ZG, Tchilibon S, Duong HT, Joshi BV, Sonin D, Liang BT. Semirational design of (N)-methanocarba nucleosides as dual acting A1 and A3 adenosine receptor agonists: novel prototypes for cardioprotection. J Med Chem. 2005;48:8103–8107. doi: 10.1021/jm050726b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivanov AA, Barak D, Jacobson KA. Evaluation of homology modeling of G-protein-coupled receptors in light of the A2A adenosine receptor crystallographic structure. J Med Chem. 2009;52:3284–3292. doi: 10.1021/jm801533x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. The 2.6 ångstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Costanzi S, Ivanov AA, Tikhonova IG, Jacobson KA. Structure and function of G protein-coupled receptors studied using sequence analysis, molecular modeling and receptor engineering: adenosine receptors. Front Drug Des Discovery. 2007;3:63–79. [Google Scholar]
- 41.Ivanov AA, Palyulin VA, Zefirov NS. Computer aided comparative analysis of the binding modes of the adenosine receptor agonists for all known subtypes of adenosine receptors. J Mol Graphics Modell. 2007;25:740–754. doi: 10.1016/j.jmgm.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 42.Ivanov AA, Wang B, Klutz AM, Chen VL, Gao ZG, Jacobson KA. Probing distal regions of the A2B adenosine receptor by quantitative structure–activity relationship modeling of known and novel agonists. J Med Chem. 2008;51:2088–2099. doi: 10.1021/jm701442d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cordeaux Y, Briddon SJ, Alexander SP, Kellam B, Hill SJ. Agonist occupied A3 adenosine receptors exist within heterogeneous microdomains of individual living cells. FASEB J. 2008;22:850–860. doi: 10.1096/fj.07-8180com. [DOI] [PubMed] [Google Scholar]
- 44.Jeong LS, Pal S, Choe SA, Choi WJ, Jacobson KA, Gao ZG, Klutz AM, Hou X, Kim HO, Lee HW, Lee SK, Tosh DK, Moon HR. Structure activity relationships of truncated d- and l-4′-thioadenosine derivatives as species-independent A3 adenosine receptor antagonists. J Med Chem. 2008;51:6609–6613. doi: 10.1021/jm8008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pal S, Choi WJ, Choe SA, Heller CL, Gao ZG, Chinn M, Jacobson KA, Hou X, Lee SK, Kim HO, Jeong LS. Structure–activity relationships of truncated adenosine derivatives as highly potent and selective human A3 adenosine receptor antagonists. Bioorg Med Chem. 2009;17:3733–3738. doi: 10.1016/j.bmc.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wadsak W, Mien LK, Shanab K, Ettlinger DE, Haeusler D, Sindelar K, Lanzenberger RR, Spreitzer H, Viernstein H, Keppler BK, Dudczak R, Kletter K, Mitterhauser M. Preparation and first evaluation of [F-18]FE@SUPPY: a new PET tracer for the adenosine A3 receptor. Nucl Med Biol. 2008;35:61–66. doi: 10.1016/j.nucmedbio.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 47.Kiesewetter DO, Lang L, Ma Y, Bhattacharjee AK, Gao ZG, Joshi BV, Melman A, Castro S, Jacobson KA. Synthesis and characterization of [76Br]-labeled high affinity A3 adenosine receptor ligands for positron emission tomography. Nucl Med Biol. 2009;36:3–10. doi: 10.1016/j.nucmedbio.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mohamadi FN, Richards GJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel an integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J Comput Chem. 1990;11:440–467. [Google Scholar]
- 49.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 50.Walsh MK, Wang X, Weimer BC. Optimizing the immobilization of single-stranded DNA onto glass beads. J Biochem Biophys Methods. 2001;47:221–231. doi: 10.1016/s0165-022x(00)00146-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.