

Abstract
The complete genome sequence of the nematode Caenorhabditis elegans provides an excellent basis for studying the distribution and evolution of protein families in higher eukaryotes. Three fundamental questions are as follows: How many paralog clusters exist in one species, how many of these are shared with other species, and how many proteins can be assigned a functional counterpart in other species? We have addressed these questions in a detailed study of predicted membrane proteins in C. elegans and their mammalian homologs. All worm proteins predicted to contain at least two transmembrane segments were clustered on the basis of sequence similarity. This resulted in 189 groups with two or more sequences, containing, in total, 2647 worm proteins. Hidden Markov models (HMMs) were created for each family, and were used to retrieve mammalian homologs from the SWISSPROT, TREMBL, and VTS databases. About one-half of these clusters had mammalian homologs. Putative worm-mammalian orthologs were extracted by use of nine different phylogenetic methods and BLAST. Eight clusters initially thought to be worm-specific were assigned mammalian homologs after searching EST and genomic sequences. A compilation of 174 orthology assignments made with high confidence is presented. [Tables describing transmembrane protein families and orthology assignments are available from ftp.cgr.ki.se/pub/data/worm.]
The first multicellular organism with a completely sequenced genome is the roundworm Caenorhabditis elegans (Consortium 1998). The genome comprises ∼100 million basepairs, and is predicted to encode ∼19,000 proteins. Previous analysis has revealed that approximately two-thirds of the proteins can be assigned a tentative biochemical function on the basis of sequence homology to proteins of known function. A majority of the proteins have homologs within the worm genome (paralogs). The fraction of proteins with a detectable paralog has been estimated by two different methods to within 66% and 95% (Sonnhammer and Durbin 1997; Teichmann and Chothia 2000). It was also noted that a large fraction of the paralog clusters do not match proteins in other species. Sonnhammer and Durbin (1997) presented a dozen of the largest such cases and showed that some clusters could be assigned a tentative function by use of hidden Markov models (HMMs). Multiple alignment-based homology search methods such as HMMs and profiles are considered more sensitive than single-sequence methods (Eddy 1996, 1998; Park et al. 1998). Examples of HMM-based protein family databases are Pfam (Bateman et al. 2000) and SMART (Schultz et al. 2000).
Our aim was to classify all membrane proteins in C. elegans by grouping them in clusters of paralogs, and annotating them according to sequence homology to proteins with a known function and orthology to mammalian proteins. The membrane proteins are attractive for several reasons. They form a rather well-defined group of an easily predicted class of proteins. Membrane proteins contain many interesting receptors and signaling proteins that are particularly important in multicellular organisms and cannot be studied in bacteria or in yeast. Membrane proteins are also challenging for many sequence comparison programs because of high degeneracy in hydrophobic membrane domains. Because of their special environment, membrane proteins, overall, are thought to be less constrained in sequence than water-soluble domains, and therefore evolve more rapidly.
Our main interest was concentrated on the G-protein-coupled receptor (GPCR) family (Kolakowski 1994; Horn et al. 1998). GPCRs are represented in the Pfam database by five different families. These models and/or simple pairwise comparisons are often used to discover novel GPCR genes (Bargmann 1998; Robertson 1998, 2000). However, the use of only models of well-established families reduces the chance of finding any new genes that could have the same function, but lack obvious sequence similarity. Alternative methods, not dependent on sequence similarity to known GPCRs could be useful to discover distinct novel families. For example, several new GPCR sequences have been found from Drosophila genome by modeling the pattern of their transmembrane domains (Clyne et al. 1999). We use a similar approach to find and characterize membrane proteins from C. elegans. Briefly, we cluster all predicted membrane proteins from the C. elegans proteome into families, generate an HMM for each family, and use these HMMs to find related mammalian genes.
After this classification of worm membrane proteins and their mammalian homologs, it is also important to find orthologs, as they are likely to be functional counterparts. By definition, orthologs are genes that have a common ancestor and are separated by a speciation event (Fitch 1970; Hillis et al. 1996). After the speciation, one or both orthologs may be duplicated and form paralogous gene families. Paralogs often undergo functional differentiation, and are therefore less likely to be functional counterparts in different species. Distinguishing orthologs from paralogs in genomic studies is often not straightforward. Furthermore, when analyzing partially sequenced genomes, one cannot rule out the possibility of incorrect ortholog assignments to paralogs if the true ortholog has not yet been sequenced.
Traditionally, phylogenetic trees have been constructed to detect orthologous proteins (Chervitz et al. 1998; Yuan et al. 1998). As a quick alternative, the BLAST program (Altschul et al. 1997) has often been used to find probable orthologs between different species (Tatusov et al. 1997; Mushegian et al. 1998; Makarova et al. 1999; Wheelan et al. 1999). In this case, all sequences from one species (or clade) are compared with all sequences from other species (or clade). The sequences that are most similar to each other in both comparisons (best hits) are considered putative orthologs. Available ortholog databases include COGs (Tatusov et al. 2000), a comprehensive collection of bacterial and yeast orthologs, and HOVERGEN (Duret et al. 1994) with mammalian orthologs and phylogenetic trees.
All orthology detection methods have a substantial error rate. To reduce the risk of incorrect orthology assignments and increase confidence in the prediction, we have used nine different phylogenetic methods and BLAST, and only accepted assignments in which most methods agree. This procedure resulted in 174 putative worm–mammal orthology assignments. We provide a detailed description of these assignments that can serve as a basis for preparing model experiments in C. elegans to investigate the function of human genes.
RESULTS
Prediction of Membrane Proteins in C. elegans Reveals a Clear Bias Toward 7 TM Proteins
To find transmembrane proteins in the C. elegans genome, we ran all proteins from the Wormpep98 database through the TMHMM program (Sonnhammer et al. 1998). This program predicts transmembrane regions in proteins with a HMM trained on known membrane proteins. The distribution of proteins with different numbers of predicted transmembrane regions is shown in Figure 1.
Figure 1.

The number of predicted transmembrane domains in all proteins of the wormpep98 database. The high peak of 7 transmembrane proteins corresponds to large G-protein-coupled receptor families. On the right axis, the number of proteins in each category can be read as percentages of the entire C. elegans proteome, assuming 20,000 proteins in total.
In total, 6167 proteins were predicted to contain one or more transmembrane regions. However, no method exists to distinguish reliably signal peptides from TM regions, thus, many of the proteins with a single N-terminal TM segment are probably secreted soluble proteins. However, as most worm proteins are predicted from genomic DNA, the N terminus is unverified, and using an N-terminal criterion for recognizing signal peptides is unreliable. TMHMM has a false prediction rate of ∼1% in soluble proteins, and about the same rate of false negatives. The false negative rate for TM segments in multi-spanning proteins is much higher, ∼10%–15% (A. Krogh, pers. comm.).
As seen in Figure 1, there is a large peak at 6–7 predicted transmembrane regions. This is in agreement with the known overrepresentation of 7-transmembrane GPCR proteins in C. elegans, which has been described before (Troemel et al. 1995; Sonnhammer and Durbin 1997; Robertson 1998; Troemel 1999; Robertson 2000).
Because our main interest is to study integral membrane proteins, and to avoid non-membrane proteins, we only considered the 3854 proteins with two or more predicted TM regions for cluster analysis.
Clustering of the Predicted Membrane Proteins
Many of the membrane proteins belong to paralogous families — groups of proteins with similar sequences and functions. We collected such families by all-versus-all BLAST searches, followed by single linkage clustering of homologous domains. For detailed descriptions of clustering, see the Methods section. Multiple alignments for each cluster were created and corrected manually, and incorrectly clustered sequences were removed. A HMM was created from each multiple alignment. This HMM was used to search for additional members in the Wormpep98 database. If one protein matched several HMM models, it was assigned to the group/HMM with the lowest E-value. The length of the HMMs (i.e., of the conserved domain) was nearly always between 100 and 1000 states, with an average of 385 states (see Fig. 2A for a distribution of the conserved domain lengths).
Figure 2.

(A) Distribution of the length of HMMs used in this study. This distribution illustrates the typical length of conserved domains in worm membrane proteins. (B) Paralogous family sizes and distribution of proteins between these families. The X-axis is drawn in logarithmic scale. (Open bars) Cluster distribution by size; (gray bars) the number of proteins belonging to given size of families.
After creating nonoverlapping models for each cluster, we searched for additional members of those families from the wormpep98 using HMMPFAM with a cutoff E-value of 1e-2. Forty-six proteins that had less than two TM regions predicted initially were added to the clusters by this procedure. This resulted in 189 clusters with more than two members, containing 2744 domains, which belong to 2630 unique proteins, as 114 domains belong to multi-domain proteins, many of which appear to be fused gene predictions. A total of 1270 of the predicted membrane sequences were not included in clusters because they did not have any well-conserved paralogs.
The resulting clusters, with two or more members and two or more TM regions, cover 13.8% of all proteins in the wormpep98 database. The four largest clusters are G-protein-coupled receptor (GPCR) families, the largest family containing 624 members. The largest non-GPCR cluster was the acetylcholine and GABA receptor family with 80 members. The distribution of cluster sizes and of proteins in different cluster sizes is shown in Figure 2B. About one-third of all TM proteins are found in large clusters with >65 members, about one-third in medium sized clusters with 2–64 members, and the remainder are singletons.
Finding Mammalian Homologs
After clustering the paralogous families, we had a representative set of HMMs for membrane proteins in C. elegans. We used these HMMs to search for mammalian homologs in a dataset consisting of the 71,822 mammalian entries in the SWISSPROT, TREMBL, and VTS databases. Of these sequences, 3608 had an HMMPFAM match at an E-value of 1e-2 or better to 106 HMMs. These homologs were then used to (1) collect worm-mammal ortholog candidates; (2) assign a potential function to paralog families in the worm genome; and (3) define worm-specific protein families, that is, the families without obvious mammalian homologs.
The clusters of paralogs annotated with a median number of predicted transmembrane domains, and a number of worm and mammalian homologs, are described in Table 1. The clusters are divided by their general function. The most abundant functional categories were characterized as GPCR proteins (24 clusters, 1529 worm proteins) and ion channels or various transporters (50 clusters, 558 worm proteins). There were four GPCR families with mammalian homologs. The remaining 20 GPCR clusters did not have clearly significant mammalian homologs, but were assigned putative GPCRs on the basis of weak similarity to well-characterized GPCR proteins. These GPCR families cover 86% of the proteins with six to eight predicted TM regions. A total of 63 non-GPCR families (401 worm proteins) did not have any mammalian homolog in protein databases. As shown below, subsequent searches in EST databases and human genomic sequences revealed mammalian homologs for some of these seemingly worm-specific families.
Table 1.
Description of Clustered Worm Protein Families
 
|
The columns WORM and MAMMALIAN show the number of Caenorhabditis elegans and mammalian homologs found in used protein databases with each HMM-model (with HMMPFAM cutoff level E < 1e-2). The column MEDIAN TM shows median number of predicted transmembrane domains in all matching worm proteins. The number of mammalian–worm ortholog assignments within each cluster is shown in column ORTHOLOGS. The description line shows description of mammalian homologs. Each orthology assignment is described in detail in Table 2, available as supplementary information at http://www.genome.org. Families are organized into groups by their general biochemical function. Fifty-six families for which mammalian homologs were not found are not shown here. Mammalian homologs found in unfinished genomic DNA are marked “phase1” or “phase2”. This table can also be downloaded from ftp.cgr.ki.se/pub/data/worm.
Finding Orthologs
As a next step, we tried to find orthologous membrane proteins between C. elegans and human. Orthologs are likely to have the same functions and similar biological roles. Thus, these orthologs might be an invaluable source of clarifying the function of uncharacterized human genes. The study of many gene functions in the worm is significantly simpler than in mammalian model organisms (e.g., transgenic mice).
If two orthologs had a common ancestor and directly diverged by speciation only, they should be easy to pick up as the most similar sequences in a two-way all-versus-all sequence similarity comparison. However, complicating factors such as subsequent gene duplication and different divergence rates make the simple two-way sequence comparison technique unreliable. Therefore, we also use phylogenetic methods to reconstruct the evolution of these sequences more reliably.
If no duplication has occurred since the speciation, the two genes form a one-to-one relationship. If subsequent duplications have occurred, one-to-many or many-to-many types of orthology was assigned.
The phylogenetic trees were analyzed to identify orthologous sequences between C. elegans and human proteins. All worm sequences from a given cluster and all found mammalian homologs were aligned and used to calculate phylogenetic trees. Other non-human homologs were included into the phylogenetic tree to improve the chance of finding correct orthologs if the human sequence is still undiscovered. Ten such pairs of orthologs were found with mouse or rat sequences.
The phylogenetic trees can be calculated in different ways and the results are highly dependent on the chosen method and parameters used. We used several different programs and different models of evolution to calculate different trees that were analyzed for orthologs. Overall, nine different combinations of programs, methods, and evolutionary models were used (see Methods). Assignments were made only if a majority of programs supported the orthology with high confidence value.
Table 2 lists the proteins involved in 174 putative human-worm orthology assignments that were identified with the described procedure. The list is grouped by general functions of the mammalian proteins. From the orthologs in the table, ∼30% were one-to-one relationships, 40% were one-to-many or many-to-one, and 30% were many-to-many orthologous relationships. With the completion of the human genome sequence, the fraction of one-to-many and many-to-many orthologs is likely to increase. The different types of orthologous relationships are illustrated in Figure 3.
Table 2.
Sample of Orthology Assignments between Caenorhabditis elegans and Human Membrane Proteins
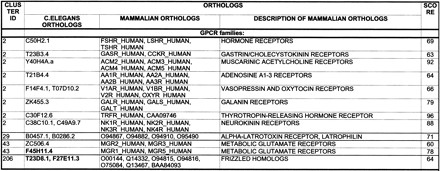 |
Non-human orthologs are shown only in groups where the corresponding human gene is missing. The names starting with letters AAC, AAD, AAF, BAA, CAA or CAB are coding sequences from EMBL database, not indexed in TREMBL. They can be retrieved by searching “proteinID” field in “EMBL_features” database. The C. elegans orthologs for which worm EST sequences are known are shown in bold. The description column shows brief information from the SWISSPROT or TREMBL description field (uppercase) and from other sources (lowercase). The last column shows the reliability index (average bootstrap value) for orthologous pairs. Families are organized into groups by their general biochemical function. Functional categories not shown here include Enzymes, Transporters and ion channels, Miscellaneous receptors and adhesion molecules, Exact biochemical function unknown, and Matches to mammalian or human genomic sequences. The complete table is available as supplementary information at http://www.genome.org.
Figure 3.

Examples of orthology. A part of the tree of the cluster 15 is shown. The tree is calculated with CLUSTALW, with 1000 bootstrap replicates. The bottom pair of orthologs illustrates one-to-one type of orthology. The top group shows many-to-many type of orthology in which two worm proteins are orthologous to two human proteins (and two mouse proteins). Sequences with a dot in the name are worm proteins.
Orthologs in EST and Genomic Sequences to Worm-Specific Families
In the previous sections, we used worm-centric gene clusters and corresponding HMMs to find worm–mammalian homologs and orthologs. This method is efficient for finding reliable orthologous relationships between proteins from distant species. The HMMs can also be used for searching DNA databases for new, uncharacterized human genes. We used the program ESTWISE to search the human UNIGENE and EMBL EST databases with the remaining worm-specific HMMs. The matching ESTs were assembled and the homology was verified with the DOTTER program (Sonnhammer and Durbin 1995). The sequences were translated and aligned together with the C. elegans homologs to build phylogenetic trees, which were analyzed for orthologous relationships. As a result, we detected five putative orthologs in clusters that did not have any known mammalian homologs previously. We also searched against all currently available human genomic sequences (∼90% of the genome) and detected fragments of homologs in three more families. These results are listed at the bottom of Tables 1 and 2, but as they are based on less reliable data, we did not include them in the set of 174 high-confidence assignments. An example with alignment and a phylogenetic tree of two assembled human EST sequences and several previously thought worm-specific proteins is shown in Figure 4.
Figure 4.
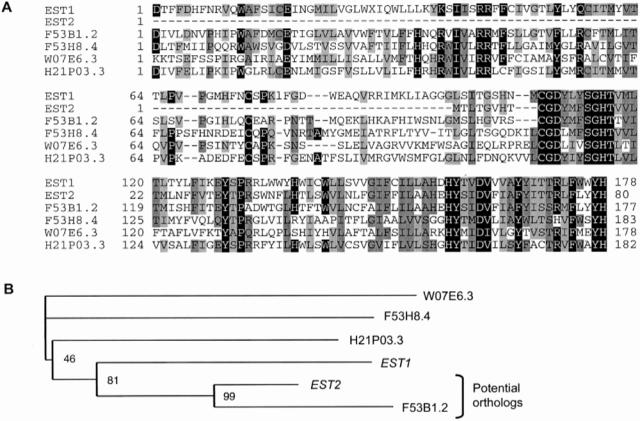
Human EST sequences matching apparently worm-specific families. An ESTWISE search with a worm-specific HMM model (cluster 173) identified at least two new mammalian genes, one of which seems orthologous with a C. elegans gene. (A) Multiple alignment of the translated and assembled mammalian ESTs and their worm homologs. EST1 was assembled from mammalian EST sequences AI113283, HSM011691, and HSZZ18477. EST2 was assembled from mammalian EST sequences AI060175, MM1135871, and AA559273. (B) Phylogenetic tree of the same family, calculated with CLUSTALW.
Phylogenetic Methods or Two-Way BLAST?
As mentioned above, we used the consensus of nine different phylogenetic methods to assign orthology. This approach is relatively reliable, but is labor intensive and time consuming, particularly if compared with the two-way all-versus-all BLAST method. Ortholog detection with the BLAST program is very fast and can be automated easily, but it has several drawbacks. We were interested in comparing the performance of phylogenetic methods with the commonly used two-way BLAST method.
In our hands, two-way BLAST detects 168 ortholog pairs of the final 174 orthologs (true positives). In addition, BLAST detects 34 pairs that were not confirmed by phylogenetic methods. We took a closer look at cases in which the BLAST results were different from tree-based methods. For 17 of these cases, we were able to find a reason for the BLAST failure; hence, we believe they are false positives. Another 17 cases remained unresolved, some of them could be real orthologs that were missed by the phylogenetic methods because of errors in multiple alignment or low confidence. The following reasons were found to be responsible for the errors in two-way BLAST: (1) If an alignment contains a large insertion, BLAST reports two segments and the overall significance is based on the strongest match only; (2) BLAST uses gap penalties, but phylogenetic methods do not; (3) BLAST assumes a constant molecular clock, which is sometimes not correct (see Fig. 5); and (4) BLAST does not separate orthologs from paralogs, but this can be achieved by rooting the tree with an outgroup.
Figure 5.

Example of different orthologs detected by phylogenetic or BLAST-based methods (CLUSTER 150). The tree was calculated by the neighbor-joining method. BLAST detects the pair with the shortest distance, whereas phylogenetic methods can compensate for unequal rates of divergence and make a more correct grouping.
The number of different reasons observed for two-way BLAST errors is shown in Table 3. The main problem with two-way BLAST is the relatively high rate of false positive hits — only a few true assignments were missed. Note that this is only true when BLAST is run as described in Methods; default BLAST parameters will generate many more errors. The BLAST method can thus be used rather safely to generate ortholog candidates, which then need to be verified by phylogenetic methods. Phylogenetic methods may also produce false positives by so-called long branch attraction of distant outliers. This is a known artifact of distance-based phylogenetic methods. These false positives can be revealed by adding a proper outgroup or by comparing pairwise similarity scores of the potentially orthologous sequences (see Methods). We detected two cases of long branch attraction in our initial list of orthologs.
Table 3.
Causes of Incorrect Orthology Assignments by Two-Way BLAST
| Reason | 2 segments | Gap penalties | Constant molecular clock | No rooting with outgroup | Total |
|---|---|---|---|---|---|
| False negatives | 2 | 4 | 0 | 0 | 6 |
| False positives | 3 | 4 | 5 | 5 | 17 |
See text for details.
We conclude that although the BLAST-based method is fast and works relatively well, phylogenetic methods are still necessary to reliably assign orthology suggested by BLAST. However, the best end result is gained when results from phylogenetic methods are confirmed also by the BLAST-based method.
DISCUSSION
This work describes a number of steps taken to arrive at a comprehensive list of orthologous membrane proteins between C. elegans and mammals. Because the mammalian gene set is not yet complete, these assignments are tentative, but they have been subjected to rigorous testing and manual analysis. The steps taken include prediction of membrane proteins in C. elegans, classifying these proteins into families, finding mammalian homologs with HMM searches, and a detailed analysis of orthologous relationships.
The classification of predicted membrane proteins also provided a good overview of the distribution and evolution of these families in C. elegans. As expected, a number of worm families did not have any mammalian homologs in the protein databases. However, for five of these families, mammalian homologs were found after searching EST databases with HMMs. Homologs for three additional families were found from human genomic sequences. Thus, our HMM models are an excellent platform for novel gene discovery. It is possible that homologs will be found for some of the remaining 75 worm-specific families once the human genome is completely finished and assembled.
Nineteen of the seventy-five worm-specific families are likely to be GPCRs. This is in good agreement with the primitive structure of the worm nervous system (Mombaerts 1999).
The list of orthologs is an important result of our research. Orthologs are likely to have the same biological function. Thus, the list of potential orthologs is an excellent starting point for experimental studies in C. elegans. The prediction of orthologs is typically done in a relatively quick and dirty way, by use of only pairwise-similar detection programs. We present a more careful approach using phylogenetic methods, which is more consistent with the definition of orthology (Fitch 1970). The study shows that the orthologs predicted by BLAST are often different from tree-based orthologs. Moreover, different phylogenetic methods can produce highly different results. Thus, we strongly suggest using several different methods to assign orthology.
Naturally, all of the identified ortholog pairs and groups are preliminary, because not all of the human proteins are yet available through protein or EST databases. The list can be finally confirmed only after the completion of the human genome sequence.
METHODS
Data
The Wormpep98 database was used as the full proteome sequence of the C. elegans that is based on the official version wormpep17 (http://www.sanger.ac.uk/Projects/C_elegans/wormpep/) which contains 19,099 protein sequences. The data in this database is of somewhat low quality and is known to contain some errors due to wrong gene predictions. Thus, we were cautious when interpreting any alignment data involving worm proteins from wormpep98 database. The mammalian proteins were retrieved from a nonredundant set of SWISSPROT + TREMBL + SWISSNEW + TREMBLNEW from October 11, 1999 (Baker et al. 2000). This set of mammalian proteins contained 52,838 proteins, 20,181 of which were of human origin. Additionally, 19,093 hypothetical human proteins were added to mammalian dataset from the VTS database, version 7 (N. Miyajima, Kazusa DNA Research Institute, Kisarazu, Japan). Overall, 71,882 mammalian proteins were used in this study. UNIGENE database (Schuler 1997) version 105, or later, and EMBL EST database (Baker et al. 2000) were used to search for additional orthologs.
Prediction of Transmembrane Regions
Transmembrane regions were predicted with the HMM-based program TMHMM (Sonnhammer et al. 1998). The default settings for this program are rather conservative and may miss weak transmembrane domains. The underestimation of the number of transmembrane segments is more frequent than overestimation.
Clustering of C. elegans Paralogs
The clustering was done in several steps. First, only the proteins with 6–8 predicted transmembrane domains were clustered. Subsequently, the remaining proteins with 2–48 predicted transmembrane domains were clustered. The clustering of both datasets was based on similarities detected by an all-to-all search with the BLAST2 program (Altschul et al. 1997). BLAST search and clustering was performed in several rounds, with decreasing stringency (E-value cutoff from 1e-50 to 1e-5). The sequence similarities detected by BLAST were clustered together by single-linkage clustering.
The domain boundaries were not used in the clustering step. Nevertheless, only the actual matching domains of sequences were retrieved and used in multiple alignment. This helped to manually eliminate wrongly clustered sequences after multiple alignment. Multiple alignment was done with the program CLUSTALW, version 1.4 (Thompson et al. 1994) and edited with the alignment viewer BELVU (E. Sonnhammer, unpubl.). The manual editing involved pruning unaligned parts, removing poorly aligned sequences, correcting of alignment errors around gaps, and an occasional realignment with ClustalW.
HMM Construction
The HMM models were constructed from manually edited multiple alignments. The HMMBUILD and HMMCALIBRATE programs from the HMMER2.1.1 (S. Eddy, unpubl.) package. Both global (default) and local (-f option) models were created with HMMBUILD, but in most searches, only the default model was used. For EST database search, the local HMM models that allow partial matches to the HMM were used. The HMM files and seed alignments are available upon request.
Homology Searches
HMMPFAM programs from the HMMER package were used to find both worm and mammalian homologs. In all of our searches, we used the E-value cutoff 1e-2. Wormpep98 database to search C. elegans homologs and a dataset of 52,838 mammalian protein sequences was used to find mammalian homologs (see above). Many of the proteins matched several different HMMs. In this case, the sequences (domains) were assigned to the HMM/group with the best matching E-value.
Ortholog Detection with Phylogenetic Methods
Multiple alignments for the tree calculation were constructed from each group of homologs by the program HMMALIGN from the HMMER package. Gappy columns and gappy sequences (>50% gaps) were removed from the alignment before calculating the phylogenetic tree. Sequences >99% identical to any other sequence were also removed from alignment.
Different phylogenetic programs give different tree topology according to the method used and the model of evolution assumed. There is no overall consensus among biologists as to which phylogenetic method best reflects the evolution proteins, particularly in the case of membrane proteins. Thus, instead of choosing one arbitrary method, we used several different programswith different options. The programs PHYLOWIN with observed distance, Poisson correction and PAM distance (Galtier et al. 1996), ClustalW with observed distance and Kimura correction (Thompson et al. 1994), PHYLIP with PAM distance (Felsenstein 1993), PUZZLE with BLOSUM62 matrix (Strimmer and von Haeseler 1996), ProtML with Jones matrix (Adachi and Hasegawa 1996), and PAUP with parsimony criterion (Swofford 1998) were used to build distance-based phylogenetic trees for ortholog detection. For most programs, the bootstrap technique was used to estimate reliability of the given branching order. A total of 100–500 bootstrap tests were run on trees to assess the significance of the branching order. Only bootstrap values >50% were considered positive. In PUZZLE, the reliability is shown as the number of puzzling attempts that support a given branch. The ProtML method does not use bootstrapping, but the significance of branching order can be estimated directly from the standard error of branch lengths. The list of orthologs was made on a consistency principle — the ortholog was marked only if the majority of nine phylogenetic methods used supported given pairing of orthologs. The final reliability index (the score in Table 2) for the orthologous branching is calculated as the average support of the nine different methods. The different methods seem sufficiently uncorrelated to justify this simple procedure. In the 76 cases in which one or more methods did not support the consensus branching, 37 different binary patterns of support among the 9 methods were observed. The most frequent pattern was observed 15 times (all methods but ProtML was supported). The pattern of support thus varies considerably and is not fixed on a particular set of methods.
In smaller families, the location of root and pairing of orthologs is not obvious. In these cases, all similar sequences from SWISSPROT–TREMBL were used to root the phylogenetic tree. Another weakness of distance-based phylogenetic methods is a possibility of the long-branch attraction. In this case, putative orthologs are not truly related and are paired together only because of a common dissimilarity to other sequences. Long-branch attraction was checked and eliminated by checking the similarity of paired sequences in BELVU. If sequences had a negative similarity score, they were removed from the list of orthologs.
Ortholog Detection with Two-Way BLAST
Exactly the same worm and mammalian sequence domains were used for phylogenetic methods and BLAST searches. Both worm–mammalian and mammalian–worm BLAST searches were run without SEG filtering. The sequences that were best hits to each other in both directions were marked as orthologs. The cutoff value for ortholog pairs was set at 50 bits (corresponds approximately to E-value 1e-5 if recalculated to current size of SWISSPROT + TREMBL).
DNA Database Searches with HMMs
The program ESTWISEDB version 2.1.19c from the WISE2 package (E. Birney, unpubl.; http://www.sanger.ac.uk/Software/Wise2/) was used for searching DNA databases. This program can directly compare HMM with DNA sequences. The human UNIGENE database (ftp://ncbi.nlm.nih.gov/repository/unigene/), version 104 or higher, was searched directly with ESTWISEDB. The EST database was too large to search directly, so the filtering program BLASTWISE was used. The BLASTWISE procedure is as follows: (1) Twenty-five protein sequences are generated from the HMM (with the program HMMEMIT from the HMMER2.1.1 package); (2) these 25 protein sequences are used to search the EST database with the program TBLASTN and with cutoff E-value 10 for potential homologs; and (3) matching EST sequences are retrieved and used in the final ESTWISEDB similarity search against the original HMM (local match model).
Searching Human Genomic Sequences
All available human genomic sequences were downloaded 2000–07–25 from Genbank (ftp://ncbi.nlm.nih.gov/refseq/H_sapiens/Contigs/). These were searched with TBLASTN using the consensus sequence from each worm cluster alignment as query sequence.
Acknowledgments
This work was supported by a grant from Pharmacia Corporation (Kalamazoo, Michigan).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL erik.sonnhammer@cgr.ki.se; FAX 46-8-337983.
Article published online before print: Genome Res., 10.1101/gr.149100.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.149100.
REFERENCES
- Adachi J, Hasegawa M. Computer Science Monographs No. 27. Tokyo: Institute of statistical mathematics; 1996. MOLPHY: Programs for molecular phylogenetics. [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker W, van den Broek A, Camon E, Hingamp P, Sterk P, Stoesser G, Tuli MA. The EMBL nucleotide sequence database. Nucleic Acids Res. 2000;28:19–23. doi: 10.1093/nar/28.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caenorhabditis elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2360–2365. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Chervitz SA, Aravind L, Sherlock G, Ball CA, Koonin EV, Dwight SS, Harris MA, Dolinski K, Mohr S, Smith T, et al. Comparison of the complete protein sets of worm and yeast: Orthology and divergence. Science. 1998;282:2022–2028. doi: 10.1126/science.282.5396.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyne PJ, Warr CG, Freeman MR, Lessing D, Kim J, Carlson JR. A novel family of divergent seven-transmembrane proteins: Candidate odorant receptors in Drosophila. Neuron. 1999;22:327–338. doi: 10.1016/s0896-6273(00)81093-4. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D, Gouy M. HOVERGEN: A database of homologous vertebrate genes. Nucleic Acids Res. 1994;22:2360–2365. doi: 10.1093/nar/22.12.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Hidden Markov models. Curr Opin Struct Biol. 1996;6:361–365. doi: 10.1016/s0959-440x(96)80056-x. [DOI] [PubMed] [Google Scholar]
- ————— Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP: Phylogeny inference package. (Version 3.2) Cladistics. 1989;S:164–166. [Google Scholar]
- Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: Two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- Hillis DM, Moritz C, Mable BK. Molecular systematics. Sunderland, MA: Sinauer Associates; 1996. [Google Scholar]
- Horn F, Weare J, Beukers MW, Horsch S, Bairoch A, Chen W, Edvardsen O, Campagne F, Vriend G. GPCRDB: An information system for G protein-coupled receptors. Nucleic Acids Res. 1998;26:275–279. doi: 10.1093/nar/26.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski LF., Jr GCRDbA G-protein-coupled receptor database. Receptors Channels. 1994;2:1–7. [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Galperin MY, Grishin NV, Tatusov RL, Wolf YI, Koonin EV. Comparative genomics of the Archaea (Euryarchaeota): Evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 1999;9:608–28. [PubMed] [Google Scholar]
- Mombaerts P. Seven-transmembrane proteins as odorant and chemosensory receptors. Science. 1999;286:707–711. doi: 10.1126/science.286.5440.707. [DOI] [PubMed] [Google Scholar]
- Mushegian AR, Garey JR, Martin J, Liu LX. Large-scale taxonomic profiling of eukaryotic model organisms: A comparison of orthologous proteins encoded by the human, fly, nematode, and yeast genomes. Genome Res. 1998;8:590–598. doi: 10.1101/gr.8.6.590. [DOI] [PubMed] [Google Scholar]
- Park J, Karplus K, Barrett C, Hughey R, Haussler D, Hubbard T, Chothia C. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J Mol Biol. 1998;284:1201–1210. doi: 10.1006/jmbi.1998.2221. [DOI] [PubMed] [Google Scholar]
- Robertson HM. Two large families of chemoreceptor genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae reveal extensive gene duplication, diversification, movement, and intron loss. Genome Res. 1998;8:449–463. doi: 10.1101/gr.8.5.449. [DOI] [PubMed] [Google Scholar]
- ————— The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 2000;10:192–203. doi: 10.1101/gr.10.2.192. [DOI] [PubMed] [Google Scholar]
- Schuler GD. Pieces of the puzzle: Expressed sequence tags and the catalog of human genes. J Mol Med. 1997;75:694–698. doi: 10.1007/s001090050155. [DOI] [PubMed] [Google Scholar]
- Schultz J, Copley RR, Doerks T, Ponting CP, Bork P. SMART: A web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 2000;28:231–234. doi: 10.1093/nar/28.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:GC1–GC10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- ————— Analysis of protein domain families in Caenorhabditis elegans. Genomics. 1997;46:200–216. doi: 10.1006/geno.1997.4989. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Intell Systems Mol Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- Strimmer K, von Haeseler A. Quartet-puzzling: A quartet maximum-likelihood method for reconstructing tree topologies. Mol Biol Evol. 1996;13:964–969. [Google Scholar]
- Swofford DL. PAUP: Phylogenetic analysis using persimony and other methods, version 4. New York, NY: Sinauer Press; 1998. [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann SA, Chothia C. Immunoglobulin superfamily proteins in Caenorhabditis elegans. J Mol Biol. 2000;296:1367–1383. doi: 10.1006/jmbi.1999.3497. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER. Chemosensory signaling in C. elegans. BioEssays. 1999;21:1011–1020. doi: 10.1002/(SICI)1521-1878(199912)22:1<1011::AID-BIES5>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Troemel ER, Chou JH, Dwyer ND, Colbert HA, Bargmann CI. Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell. 1995;83:207–218. doi: 10.1016/0092-8674(95)90162-0. [DOI] [PubMed] [Google Scholar]
- Wheelan SJ, Boguski MS, Duret L, Makalowski W. Human and nematode orthologs–lessons from the analysis of 1800 human genes and the proteome of Caenorhabditis elegans. Gene. 1999;238:163–170. doi: 10.1016/s0378-1119(99)00298-x. [DOI] [PubMed] [Google Scholar]
- Yuan YP, Eulenstein O, Vingron M, Bork P. Towards detection of orthologues in sequence databases. Bioinformatics. 1998;14:285–289. doi: 10.1093/bioinformatics/14.3.285. [DOI] [PubMed] [Google Scholar]