

Abstract
In insects, initiation of metamorphosis requires a surge in the production of the steroid hormone 20-hydroxyecdysone from the prothoracic gland, the primary endocrine organ of juvenile larvae. Here, we show that blocking TGFβ/Activin signaling, specifically in the Drosophila prothoracic gland, results in developmental arrest prior to metamorphosis. The terminal, giant third instar larval phenotype results from a failure to induce the large rise in ecdysteroid titer that triggers metamorphosis. We further demonstrate that activin signaling regulates competence of the prothoracic gland to receive PTTH and insulin signals, and that these two pathways act at the mRNA and post-transcriptional levels, respectively, to control ecdysone biosynthetic enzyme expression. This dual regulatory circuitry may provide a cross-check mechanism to ensure that both developmental and nutritional inputs are synchronized before initiating the final genetic program leading to reproductive adult development. As steroid hormone production in C. elegans and mammals is also influenced by TGFβ/Activin signaling, this family of secreted factors may play a general role in regulating developmental transitions across phyla.
Keywords: TGFβ, Activin, Ecdysone, Insulin, PTTH, Prothoracic gland
INTRODUCTION
In many multicellular organisms, developmental transitions are necessary for proper progression from the juvenile stage to adulthood. In humans, for example, puberty initiates a series of physical, morphological and physiological changes that culminate in an adult capable of reproduction. Likewise, insects, particularly those in the holometabolous group, undergo a process known as metamorphosis that transforms sexually immature individuals into breeding adults. Both puberty and insect metamorphosis rely on steroid hormones that systemically coordinate gene expression changes that change the physiology and morphology of the whole animal. (Mauras et al., 1996; Sisk and Foster, 2004; Gilbert et al., 1997; Gilbert et al., 2002).
During larval and pupal stages of insect development, the prothoracic gland (PG) is the major endocrine organ in which biosynthesis of the insect steroid hormone ecdysone (E) takes place. The production of E in the PG requires the action of the enzymes encoded by the Halloween gene family (Gilbert et al., 2002; Gilbert, 2004). With the exception of neverland (nvd) (Yoshiyama et al., 2006) and shroud (sro) (Niwa et al., 2010), all the Halloween genes identified so far including spook (spo), spookier (spok), phantom (phm), disembodied (dib) and shadow (sad) code for cytochrome P450 proteins that sequentially convert cholesterol to E (Warren et al., 2002; Warren et al., 2004). Once made and released from the PG, E is rapidly converted in peripheral tissues to 20-hydroxyecdysone (20E), the active derivative of E, by Shade, another P450 enzyme (Petryk et al., 2003). In Drosophila and other holometabolous insects, a large increase in the titer of E/20E generated at the end of the third-instar stage triggers metamorphosis by inducing the expression of the downstream target genes (Andres and Thummel, 1992; Baehrecke, 1996).
Elucidating the mechanisms that modulate and time the production of the E/20E titer is crucial to understanding several aspects of developmental timing in insects. In the past few decades, a great deal of work has centered on understanding the function of various neuropeptides in modulating the timing and size of the E/20E pulses during development (for a review, see Marchal et al., 2010). Key among these is prothoracicotropic hormone (PTTH), a brain-derived factor that can trigger a surge in the production and secretion of E from the PG (Gilbert et al., 1997; Gilbert et al., 2002). Recently, Torso, a receptor tyrosine kinase that activates the Ras/Raf/ERK pathway during embryonic terminal development in response to Trunk (Li, 2005; Perrimon et al., 1995), a distant relative of PTTH, has been identified as the PTTH receptor (Rewitz et al., 2009). Knockdown of Ras, Raf or ERK in the PG gives rise to a developmental delay phenotype similar to that caused by PTTH-neuron ablation, suggesting that this MAPK pathway is also used to transduce the PTTH signal in the PG (Rewitz et al., 2009). Consistent with this view, over-activation of Ras or Raf in the PG accelerates larval development caused by increased E production (Caldwell et al., 2005). One potential mechanism for how the PTTH pathway promotes E biosynthesis is by stimulating transcription of E biosynthetic enzymes, as PTTH neuron ablation leads to marked reduction in the steady-state mRNA levels of several E biosynthetic enzymes prior to metamorphosis (McBrayer et al., 2007).
In addition to PTTH signaling, the highly conserved insulin pathway has also been implicated in controlling E synthesis (Colombani et al., 2005; Mirth et al., 2005; Caldwell et al., 2005). In Drosophila, there are seven insulin-like peptides (DILPs) that activate a single insulin-like receptor (InR) (Mirth and Riddiford, 2007; Wu and Brown, 2006). The most intensively studied DILPs, DILP2, DILP3 and DILP5, are expressed in two clusters of seven neurosecretory cells in the brain known as insulin-producing cells (IPCs) (Ikeya et al., 2002). PG-specific expression of a dominant-negative Pi3K, a downstream effector of InR, or PTEN, a phosphatase that antagonizes Pi3K (Goberdhan et al., 1999), results in low ecdysteroid titers (Colombani et al., 2005; Mirth et al., 2005), slow development (Caldwell et al., 2005) and increased body size. By contrast, PG-specific expression of either wild-type or activated Pi3K, or hyperactivation of insulin signaling in IPCs reduces body size and advances the onset of metamorphosis, probably because of precocious ecdysone synthesis (Colombani et al., 2005; Caldwell et al., 2005; Walkiewicz and Stern, 2009). These observations suggest that insulin signaling influences both growth rate and metamorphic timing by acting on the PG to affect E synthesis.
The ability of nutrient signals to regulate developmental transitions is not restricted to insects. Pubertal onset in humans is intimately linked to nutritional status, with precocious puberty becoming more prevalent in well-fed western societies (Kaplowitz et al., 2001). Likewise, in C. elegans the formation of dauer, an arrested larval state that allows survival through times of stress, is induced by nutrient deprivation. Recently, dafachronic acids have been identified as the steroid hormones controlling dauer diapause (Motola et al., 2006). The production of dafachronic acids involves G protein-coupled receptors that act as environmental sensors to control the production of TGFβ-type and insulin peptides that, either directly or indirectly, regulate hormone biosynthesis (for a review, see Fielenbach and Antebi, 2008).
The involvement of TGFβ-type factors in regulating this important developmental transition in C. elegans and the conservation of the pathway in regulating many developmental and physiological processes suggest that TGFβ signals might be general regulators of steroid hormone production. In Drosophila, the Activin/TGFβ family comprises the ligands Actβ, Dawdle (Daw) and Myoglianin (Myo), while the BMP family ligands are represented by Decapentaplegic, Glass bottom boat and Screw. The family assignment of the ligand Maverick (Mav) is unclear based on either sequence or signaling properties (Nguyen et al., 2000). Both ligand families probably use a common set of type II receptors, coded for by punt and wishful thinking, whereas pathway specificity is mediated by the type I receptor encoded by baboon (babo), in the case of Activins and either thickveins or saxophone, in the case of BMPs. Downstream signal transduction is mediated by the R-Smads, dSmad2 (Smox – FlyBase) for Activin ligands or Mad for BMPs, whereas Medea serves as a common co-Smad that complexes with the dSmad2 or Mad to regulate target gene transcription.
Here, we demonstrate that the TGFβ/Activin pathway plays a pivotal role in regulating developmental transitions in Drosophila. We found that knockdown of various Activin pathway components in the PG causes developmental arrest at the last larval stage owing to the absence of the E/20E peak required for eliciting metamorphosis. The defect in producing the E/20E peak is caused, in part, by loss of PTTH and insulin signal reception in the PG, which primarily affect transcription and translation of E biosynthetic enzymes, respectively. We conclude that Activin signaling is required in the PG to maintain competence of the endocrine gland to respond to the metamorphic stimulation by PTTH and insulin signaling pathways, and that each of these pathways regulates E biosynthesis by distinct molecular mechanisms.
MATERIALS AND METHODS
Drosophila strains and husbandry
All Drosophila stocks and crosses were maintained on the standard cornmeal food at 25°C. The strains used include daw-Gal4 (Zhu et al., 2008), Actβ-Gal4 (Ting et al., 2007), UAS-babo(CA) (Brummel et al., 1999), phm-Gal4 (Ono et al., 2006) and UAS-RhebEP50.084 (Chang and Neufeld, 2009). UAS-dSmad2 RNAi (#14609 and #105687), UAS-babo RNAi (#3825 and #853), UAS-punt RNAi (#848 and #37279) and UAS-medea RNAi (#19688 and #19689) were obtained from the Vienna Drosophila RNAi center. Additional RNAi babo, mad, punt and dSmad2 lines were generated for this study (details available upon request). UAS-nuGFP (#4775), UAS-Akt (#8191) and UAS-InR (#8262) were obtained from the Bloomington Drosophila Stock Center. UAS-Daw and UAS-Actβ were generated in a y,w1118 background by using standard methods. UAS-RasV12 and tub>PH-GFP are kind gifts from Dr Neufeld (UMN, USA).
Staging larvae
Females were allowed to lay eggs on apple juice agar plates for 6 hours. Larvae were synchronized at the L2-to-L3 transition. The newly molted third instar larvae were collected, transferred into vials containing standard cornmeal food, and allowed to develop to the desired time points. The larvae were reared inside an insulated moist chamber at 25°C under a 12 hour light/dark cycle.
20E feeding and whole-body ecdysteroid titer determination
The 20E feeding experiment was performed as previously described (McBrayer et al., 2007). For ecdysteroid determination, 50 larvae per genotype per time point were collected and frozen at −80°C. The procedures for ecdysteroid extraction and determination by radioimmunoassay can be found elsewhere (Warren et al., 2006).
In situ hybridization
For examining endogenous gene expression, in situ hybridization was carried out as described by Chávez et al. (Chávez et al., 2000). cDNAs were used to generate both the antisense and sense probes according to standard methods.
Immunofluorescence
Brain-ring gland complexes were dissected in pre-chilled PBS, fixed in 3.7% formaldehyde in PBS for 20 minutes at room temperature and then washed 3× in PBS with 0.1% Triton X-100 (PBT). The primary antibodies used in this study include: mouse anti-Coracle, 1:200 (Hybridoma, C566.9); rabbit anti-Phm, 1:1000 (Parvy et al., 2005); rabbit anti-Dib, 1:1000 (Parvy et al., 2005); rabbit anti-Sad, 1:1000 (raised against the peptide sequence CIRVQEDQRRPHDEA); guinea pig anti-Spo/Spok, 1:100 (raised against peptide sequence CDWSQLQQKRRNLARRH); mouse monoclonal anti-MAPK activated, 1:10,000 (Sigma, M8159); and rabbit anti-dFOXO, 1:1000 (Puig et al., 2003). Guinea pig anti-DIMM antibody (1:500) is a kind gift from P. Taghert (WUSTL, MO, USA). Fluorescent-conjugated secondary antibodies (ALEXA FLUOR) were purchased from Invitrogen and were used at a 1:500 dilution. DNA was stained by DAPI (1 μg/ml, Sigma) in PBT for 5 minutes. All primary and secondary antibodies were diluted in PBT. Primary antibody incubation was carried out at 4°C overnight and secondary antibody incubation was performed at room temperature for 2 hours. DNA was stained by DAPI. Images were acquired using a Zeiss LSM710 laser scanning confocal microscope.
qRT-PCR
For each sample, 10 ring gland-brain complexes were dissected and stored in TRIzol Reagent (Invitrogen). Total RNA was isolated by using RNeasy mini kit and RNase-Free DNase Set (QIAGEN). Reverse transcription was carried out by using ThermoScript RT PCR System (Invitrogen). qRT-PCR was performed on Roche LightCycler 480 System (Roche) using LightCycler DNA Master SYBR Green I (Roche). The Drosophila ribosomal protein L23 (rpL23) was used as a reference gene to normalize gene expression levels. Three independent biological samples for each genotype at each specific time point were collected for statistical analysis. The qRT-PCR primers used are listed in Table S2 in the supplementary material.
Western blotting
Western blotting was carried out according to standard protocols. The primary antibodies include mouse anti-α-tubulin, 1:5000 (Sigma, # T9026), rabbit anti-PHM, 1:1000 (Parvy et al., 2005), rabbit anti-DIB, 1:1000 (Parvy et al., 2005), rabbit anti-Sad, 1:1000 (raised against the peptide sequence CIRVQEDQRRPHDEA), guinea pig anti-Spok, 1:5,000 [raised against the peptide sequence that matches the Spok protein (FBgn0086917) from amino acid 356 to 485]. The secondary antibodies used are IRDye 700 and 800 (1:10,000), and blots were scanned with the Odyssey Infrared Imaging System (LI-COR Biosciences).
Statistical analysis
Student's t-test was used to calculate the statistical significance of the observed differences in the protein expression levels and gene expression levels. Error bars represent s.d.
RESULTS
Loss and gain of Activin signaling in PG causes developmental arrest
The finding that TGFβ signaling is involved in regulating dauer formation in C. elegans prompted us to ask whether this signaling pathway plays a similar regulatory role in controlling developmental transitions in Drosophila. Previously, we noted that babo mutant larvae are developmentally delayed and exhibit ~70% larval lethality (Brummel et al., 1999). This led us to examine whether perturbation of the Activin/Babo pathway specifically in the ring gland, a composite endocrine organ that secretes hormones essential for development (Jones and Jones, 2007), produces any developmental abnormalities. To this end, we used Akh (CC, corpus cardiacum), Phm (PG) and Aug21 (CA, corpus allatum) Gal4 drivers to specially overexpress or knockdown various Activin signaling components in different regions of the ring gland. Overexpression of an constitutively active form of Babo (Babo*), the type I receptor for Activin-type ligands (Brummel et al., 1999) or knockdown of Smad2, the primary transcriptional transducer of Drosophila Activin signaling (Brummel et al., 1999) in either the CA or the CC produced no obvious developmental phenotypes (data not shown). By contrast, overexpression of Babo* or the ligand Actβ in the PG resulted in larvae that wander at a relatively small size (Fig. 1A) with mouth hooks exhibiting second instar characteristics (Fig. 1C versus 1D), indicating that they have failed to progress to the third instar stage. After wandering for about 2 days, these second instar larvae formed stage precocious puparia that failed to survive to pharate adults (Fig. 1B), perhaps because they did not achieve minimal viable weight (see Fig. S1A in the supplementary material). Growth analysis shows that these second instar precocious larvae suffered from delayed development and took ~6.5 days after egg laying (AEL) to reach 50% pupariation, compared with the typical 5-day period in wild-type larvae (see Fig. S1B in the supplementary material).
Fig. 1.
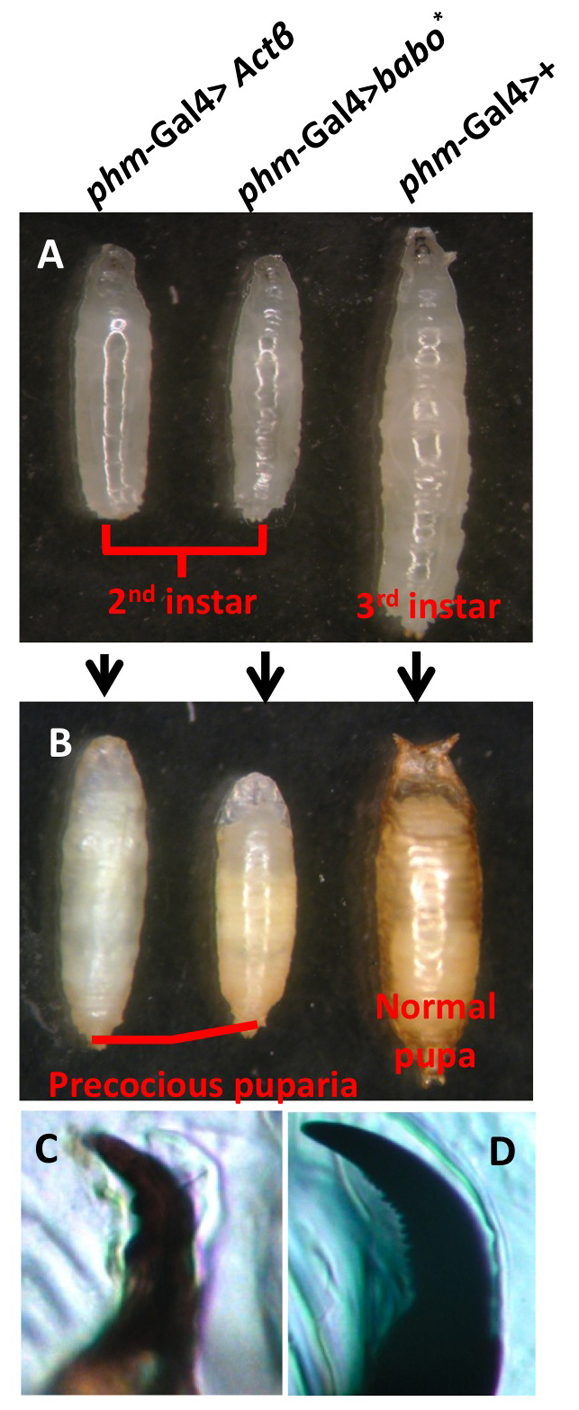
Overactivation of Activin signaling in the PG leads to stage precocious pupariation. (A) Ectopic expression of the ligand Actβ (left) or a constitutively active receptor Babo* (middle) in the PG results in small wandering larvae compared with a 3rd instar control wandering larva (right). (B) The small wandering larvae in A (left and middle) formed stage precocious pupae. A wild-type pupa is shown on the right. (C,D) The mouth hook of the small wandering larvae from A exhibit 2nd instar characteristics (two or three large teeth) (C) compared with the mouth hooks of a typical 3rd instar control larva (D), which have many smaller serrated teeth.
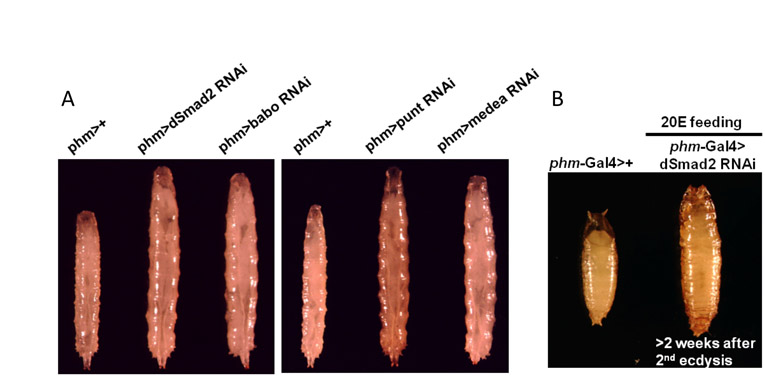
Although these gain-of-function experiments demonstrate that excess Activin signaling in the PG can alter developmental progression through larval stages, we sought to determine whether Activin signaling is required in the PG for normal development. Accordingly, we employed RNAi to specifically knockdown dSmad2 expression in the PG using a variety of Gal4 drivers. As dSmad2 is the sole downstream mediator of canonical Activin signaling, its knockdown should remove contributions of all Activin-type ligands, including Actβ, Daw, Myo and perhaps Mav. Interestingly, larvae in which dSmad2 is knocked down in the PG (thereafter referred to as dSmad2 RNAi larvae) failed to initiate metamorphosis and arrested at the third instar stage for more than 2 weeks, during which time they continued to feed and grow to a very large size (Fig. 2A). Knockdown of dSmad2 was also able to suppress the stage precocious pupariation of activated Babo, indicating that the gain-of-function phenotype requires dSmad2 (data not shown). To eliminate possible off-target effects of the RNAi technology, we used several UAS-dSmad2 RNAi constructs that target different regions of the dSmad2 gene. All of these constructs individually gave rise to the same non-pupariating phenotype (see Table S1 in the supplementary material). To show that the non-pupariating phenotype is specific to loss of Activin signaling rather than to other unrelated cellular functions of dSmad2, we knocked down the type I receptor, Babo, a type II receptor, Punt (Childs et al., 1993), and a transcriptional co-factor for dSmad2, Medea (Das et al., 1998). Again, using several RNAi lines for each gene, we observed the same non-pupariating phenotype (see Fig. S2A and Table S1 in the supplementary material). However, when the transcriptional transducer of the BMP pathway, Mad, is knocked down in PG cells, no effect on developmental timing is observed, even though the same Mad RNAi lines were capable of generating strong BMP loss-of-function phenotypes when expressed in other tissues. In addition, simultaneous knockdown of both dSmad2 and Mad in the PG produced no stronger phenotype than dSmad2 knockdown alone (data not shown).
Fig. 2.

Loss of Activin signaling in the PG leads to a failure to initiate metamorphosis. (A) Knockdown of dSmad2 (left panel) results in larval developmental arrest and giant larvae compared with control (right panel). (B) Immunostaining for Coracle in both the control and dSmad2 RNAi larvae. DAPI is in blue. (C,C′) DAPI staining of RGBCs of the control and dSmad2 RNAi larvae. (D,D′) DAPI staining of wing imaginal discs of the control and dSmad2 RNAi larvae. (E) The whole larva ecdysteroid titer of the control and dSmad2 RNAi larvae. Data are from a single determination using 50 larvae for each data point. (F) 20E feeding at 48 hours after the second ecdysis rescues the dSmad2 RNAi larvae to the pupa stage.
The above data suggest that Activin signaling mediated by Babo, Punt, dSmad2 and Medea in the PG is essential for proper developmental timing and for the initiation of metamorphosis.
Loss of Activin signaling leads to low 20E levels due to downregulation of E biosynthetic enzymes
Next, we explored the underlying mechanism of how TGFβ/Activin signaling affects metamorphosis. As TGFβ signaling has been shown to regulate cell fate during development (Larsson and Karlsson, 2005; Mondal et al., 2004; ten Dijke et al., 2003), we first examined whether Activin signaling is required for specifying PG cell fate. Both the PG morphology and cell number appear to be normal in the dSmad2 RNAi larvae when compared with the control, at least when visualized by immunostaining for Coracle, a septate junction marker (Fehon et al., 1994) (Fig. 2B,B′) and DAPI staining (Fig. 2C,C′). In addition, the fact that Phm and Sad are expressed at near normal levels in the dSmad2 RNAi PG (see Fig. S3 in the supplementary material), and the fact that there is still detectable but low expression of several other ecdysone biosynthetic enzymes (see below), indicates that loss of TGFβ signaling does not generally interfere with the assignment of endocrine properties to the PG cells. Although we did not observe any major morphological defects in the PG, we noticed that dSmad2 RNAi larvae have smaller brains and imaginal discs (Fig. 2C,C′,D,D′), despite the fact that the larvae are abnormally large. These phenotypes have been previously reported in without children mutants, which have low ecdysteroid levels (Wismar et al., 2000). As a high titer of 20E is essential for metamorphosis to occur, we examined whether dSmad2 RNAi larvae exhibit a decreased 20E titer, and found that, indeed, they do not generate a 20E peak during the prolonged third instar stage. Although control larvae that normally undergo pupariation ~48 hours after ecdysis into the third instar stage have a sharp rise in total ecdysteroid content, dSmad2 RNAi larvae failed to mount an ecdysone surge even after 4 days in the third instar stage (Fig. 2E). Feeding 20E to dSmad2 RNAi larvae at 48 hours post second ecdysis, a stage where larvae mount a ecdysteroid titer under wild-type conditions, enables 45% of the larvae (n=54) to undergo metamorphosis (Fig. 2F), but the pupae formed failed to progress to the pharate stage. The dSmad2 RNAi third instar larvae that were aged for more than 2 weeks can also initiate pupariation in the presence of dietary 20E (see Fig. S2B in the supplementary material), although to a lesser extent (20%, n=10). We note that many of the mitotic tissues, including brain, imaginal discs and parts of the gut show pronounced degeneration in these aged individuals likely accounting for the reduced pupation rate when feed 20E. These results confirm that 20E deficits underlie the failure to elicit metamorphosis. Based on these observations, we conclude that the Activin signaling pathway, mediated by dSmad2, modulates E metabolism in the PG.
One explanation for impaired 20E production in dSmad2 RNAi larvae could be that the E biosynthetic enzymes themselves are not properly expressed in the PG. To test this idea, we used immunostaining to visualize expression of various E biosynthetic enzymes in the PGs of dSmad2 RNAi larvae. Intriguingly, we found that staining for Spok and Dib, two P450 enzymes required in the PG for the synthesis of E, is reduced (Fig. 3A,A′). Curiously, a rare individual PG cell (Fig. 3A, arrow) will exhibit significant staining, probably because of cell heterogeneity in phm>Gal4 expression.
Fig. 3.

Activin signaling regulates expression of the Halloween genes in the PG. (A,A′) Immunostaining of Dib (A) and Spok (A′) in the PG of wild-type and the dSmad2 RNAi larvae. (B,B′) Western blot analysis on the expression of Dib and Spok in the control and dSmad2 RNAi larvae. A total of four RGBCs were used in each lane. Tubulin serves as a loading control. (C,C′) Quantitative analysis of the Western blot results from B and B′. The Dib or Spok protein level is normalized to the tubulin level in the same sample. The normalized Dib or Spok level in the control larvae at D0 is set as 1. Three replicate blots were used for quantification. Data are mean±s.d. (D) qPCR analysis on the transcript levels of the Halloween genes in dSmad2 RNAi larvae. Data are mean±s.d. *P<0.05, **P<0.005.
To quantify this downregulation, we performed Western blot analysis on brain-ring gland complexes (BRGCs) to measure the levels of the Dib and Spok proteins (Fig. 3B,B′). Densitometry shows that BRGCs from dSmad2 RNAi larvae have extremely low levels of Dib, ranging from a fivefold reduction at the beginning of the third instar to an 11-fold loss at the onset of metamorphosis when compared with the well-staged control larvae (Fig. 3C). Spok also shows significant reduction in dSmad2 RNAi larvae (Fig. 3C′) compared with wild type. Conversely, Phm and Sad protein levels are unaffected (see Fig. S3 in the supplementary material).
Since TGFβ signaling is known to regulate gene transcription, we examined whether downregulation of Dib and Spok at the protein level is a consequence of reduced transcription. Using qRT-PCR analysis of the RNA samples isolated from BRGCs in dSmad2 RNAi and the control larvae, we found that the steady-state levels of dib, spok and nvd transcripts are significantly decreased (Fig. 3D), whereas, consistent with the protein expression data, the transcript levels of phm and sad are unaltered (Fig. 3D). These results identified the E biosynthetic genes dib, spok and nvd as being potential regulatory targets of Activin signaling in the PG, and suggest that the reduced expression of these genes probably contributes to the dSmad2 RNAi phenotype.
PTTH signaling is impaired by blocking Activin signaling
In Lepidoptera and Drosophila, the rise of the E/20E titer at the onset of metamorphosis is thought to be triggered in part by the neuropeptide PTTH (Ishizaki and Suzuki, 1994; McBrayer et al., 2007; Smith and Gilbert, 1989). Therefore, we investigated the possibility that loss of Activin signaling might indirectly affect E production by modifying PTTH signaling. First, we examined whether the ligand PTTH is expressed normally in dSmad2 RNAi larvae and found that the ptth transcript is expressed in a pair of neurons in each brain hemisphere at a level comparable with wild type (see Fig. S4A,B in the supplementary material). Next, we investigated whether the downstream components of the PTTH pathway are affected. Recently, Torso, a receptor tyrosine kinase, has been shown to function as a PTTH receptor and signal through the Ras-Raf-Erk cascade (Rewitz et al., 2009). When we examined the expression of torso in dSmad2 RNAi larvae, the level of torso mRNA was markedly reduced, as demonstrated by both in situ hybridization and qRT-PCR analysis (Fig. 4A,B). Furthermore, phosphorylated Erk (pErk), an indicator of PTTH signal activation, also shows strong downregulation (Fig. 4C′), consistent with a block of the PTTH signaling pathway at the receptor level.
Fig. 4.

Loss of Activin signaling reduces PTTH signal reception and Halloween gene mRNA levels. (A,A′) In situ hybridization of the torso transcript in a control and a dSmad2 RNAi PG. (B) qPCR analysis of the torso transcript levels in control and dSmad2 RNAi larvae. Data are mean±s.d. *P<0.05. (C,C′) Immunostaining of pErk (red) in the control and the dSmad2 RNAi. DAPI is shown in blue. All the images were taken with the same exposure time. (D) Expression of RasV12 in the PG rescues the dSmad2 RNAi larvae to the pharate stage. (E,E′) Immunostaining of Dib and Spok in the PG of the phm>dSmad2 RNAi; Rasv12 larvae. (F) Comparison of the transcript levels of the Halloween genes in the control, dSmad2 RNAi and phm>dSmad2 RNAi; Rasv12 larvae. Data are mean±s.d.
If loss of PTTH signaling is responsible for the lack of E in dSmad2 RNAi larvae, we rationalized that reactivating the PTTH pathway might partially restore E biosynthetic enzyme expression and rescue the pupariation defect. To this end, we expressed in the PG a constitutively active form of Ras (RasV12), an essential component in PTTH signaling that acts downstream of Torso (Rewitz et al., 2009). Similar to a previous report (Caldwell et al., 2005), we found that expression of RasV12 alone in the PG accelerated larval development and gave rise to very small third instar pharate pupae (Fig. 4D) that failed to eclose, When RasV12 was expressed in the PG of dSmad2 RNAi larvae (phm-Gal4> dSmad2 RNAi; RasV12), the larvae successfully initiated metamorphosis and died at the pharate stage, similar to the phm-Gal4> RasV12 animals (Fig. 4D). We reasoned that this rescue was probably a consequence of restored E biosynthetic enzyme expression. At the protein level, Spok and Dib, which are barely detectable in dSmad2 RNAi larvae, are strongly expressed in the PG of phm-Gal4> dSmad2 RNAi; RasV12 RNAi larvae (Fig. 4E). This increase at the protein level is probably a result of increased transcription because the amount of the nvd, spok and dib mRNA increased to almost normal or even more than the wild-type levels upon RasV12 expression (Fig. 4F). Surprisingly, transcription of phm and sad, which are not affected by dSmad2 RNAi, also exhibit dramatic upregulation (Fig. 4F), confirming that PTTH signaling regulates the expression of phm and sad, as suggested previously (McBrayer et al., 2007). The expression of molting defective (mld), which encodes a transcription factor required for spok expression (Neubueser et al., 2005; Ono et al., 2006), is not influenced by RasV12 expression (data not shown), suggesting that the upregulation of mRNA is not seen for all PG-expressed genes when the PTTH signal pathway is hyperactivated. Overall, these results indicate that loss of Activin signaling in the PG impairs the expression of the PTTH receptor Torso and thus indirectly reduces E levels by rendering PG cells incompetent to respond to PTTH stimulation.
Insulin signaling is disturbed by loss of Activin signaling
Although loss of torso expression in dSmad2 RNAi larvae is consistent with the observed development timing defect, it is important to recognize that ablation of PTTH-producing neurons or knockdown of torso expression in the PGs only delays development during the third instar stage (McBrayer et al., 2007; Rewitz et al., 2009) and does not arrest it in the way that the knockdown of Activin signaling components does. We previously speculated that a second prothoracicotropic signal exists that is responsible for producing an eventual rise in the E titer in larvae lacking PTTH signal that leads to metamorphosis (McBrayer et al., 2007). The more severe developmental arrest phenotype exhibited by dSmad2 RNAi larvae could be accounted for by a loss of competence to respond to the hypothesized alternative prothoracicotropic signal. We speculated that insulin-like factors might be good candidates as genetic manipulations of insulin signaling in the PG have revealed that insulin signaling has an impact on 20E titers (Colombani et al., 2005; Mirth et al., 2005). We first sought to determine whether insulin signaling in the PG is disturbed by loss of Activin signaling. We used a pleckstrin homology domain-GFP (PH-GFP) fusion protein, which serves as an indicator of insulin signal activation (Britton et al., 2002). Although none of the control PG cells show cytoplasmic localization of the PH-GFP protein, we observed that this protein is diffusely distributed throughout the cytoplasm of many dSmad2 RNAi PG cells (see Fig. S5A in the supplementary material), indicating that insulin signaling is probably downregulated when Activin signaling is blocked. Consistently, we also found that dFOXO is present in the nucleus of the dSmad2 RNAi PGs, further suggesting reduced insulin signaling (Puig et al., 2003), whereas this nuclear localization pattern was not observed in the control PGs (see Fig. S5B in the supplementary material). Analysis of the mRNA levels of the insulin signaling components revealed that, whereas Pi3K and Akt show mild but significant reduction, InR exhibits a dramatic decrease in the BRGC of the dSmad2 RNAi larvae (Fig. 5A). Although downregulation in both the PG and the brain could account for the observed transcriptional reduction of InR, expressing InR or its downstream mediator Akt specifically in the PG rescued the ability of the dSmad2 RNAi larvae to undergo the larval-to-pupal transition and form viable adults (Fig. 5B). These results strongly imply that loss of InR in the PG contributes to the negative effect of dSmad2 RNAi on 20E production.
Fig. 5.

Loss of Activin signaling in the PG reduces insulin receptor expression and lowers Halloween gene protein levels. (A) The transcript levels of InR, Pi3K, Akt, TOR and S6k in the BPGC of the control and the dSmad2 RNAi larvae. Data are mean±s.d. (B) Expression of either a UAS-InR or a UAS-Akt transgene in the PG rescues the dSmad2 RNAi larvae to the adult stage. (C) Comparison of the transcript levels of the Halloween genes in the control, dSmad2 RNAi and phm>dSmad2 RNAi; InR larvae. No statistical differences are found between dSmad2 RNAi larvae and phm>dSmad2 RNAi; InR larvae. Data are mean±s.d. (D,D′) Western blot analysis on Dib and Spok proteins levels in the control, dSmad2 RNAi and phm>dSmad2 RNAi; InR larvae. (E,E′) Quantitation. The levels of Dib and Spok are normalized to Tubulin and are relative to the phm>+ animals. Data are mean±s.d. *P<0.05, **P<0.005.
To determine whether the rescue by upregulating insulin signaling was a result of enhanced expression of the E biosynthetic enzymes, we analyzed the mRNA levels of nvd, dib and spok, and the protein levels of Dib and Spok. Although upregulation of insulin signaling has no effect on the steady-state mRNA levels of these E enzymes (Fig. 5C), there is a considerable elevation of the amount of the Dib and Spok protein upon insulin signaling stimulation in dSmad2 RNAi larvae (Fig. 5D,D′). Altogether, the above results suggest that insulin signaling is compromised in dSmad2 RNAi larvae and that its primary effect is to regulate post-transcriptionally the levels of several E biosynthetic enzymes.
DISCUSSION
Developmental transitions are carefully timed processes that enable organisms to adapt and respond to various environmental and developmental cues so that reproduction occurs under conditions most advantageous for species survival. Previous work in a number of holometabolous insects, including Drosophila, has highlighted the importance of the PTTH and insulin signaling pathways in stimulating 20E production in the PGs to trigger metamorphosis (Colombani et al., 2005; Kawakami et al., 1990; McBrayer et al., 2007; Mirth et al., 2005; Sauman and Reppert, 1996; Walkiewicz and Stern, 2009). We demonstrate here that PG competence to respond to these two essential metamorphic stimuli in Drosophila is crucially dependant on TGFβ/Activin signaling, which controls production of the primary receptors for these two pathways. The ability of activins to act as either direct or indirect permissive signals in the PG is interesting as competence factors have long been thought to play a central role in modulating cellular responses to hormonal signals (Broadus et al., 1999) and one generally recognized mechanism through which they function is by regulating the expression of receptors for a variety of signals (Shi et al., 1996). In the mammalian ovary, Activin has been shown to induce the expression of the receptor for follicle stimulating hormone (FSH) in rat granulosa cells (Hasegawa et al., 1988; Xiao et al., 1992), contributing to the stage-specific response of the developing follicles to FSH stimulation (Knight and Glister, 2001). It is also interesting to note in this regard that in Bombyx, the prothoracic gland, has been shown to be refractory to PTTH signals at certain stages (Ciancio et al., 1986) and that Torso levels fluctuate dramatically during the 5th instar stage (Rewitz et al., 2009), potentially accounting for the lack of PTTH response at certain times. At present we do not know whether Torso or InR levels in the PG fluctuate during normal Drosophila larval development, nor do we know whether activins play a role in regulating Torso and InR levels in response to some specific timing or nutritional cue. It is possible that activins simply impart constitutive expression of these receptors in the PG as part of a general developmental program responsible for endowing the PG cells with their steroidogenic capacity. As the general morphological features of the PG cells are not perturbed by Activin signal knockdown, even when using the phm-Gal4 driver, which is activated in the early embryo, we do not suspect that activins are required for specification of the gland itself. However, this conclusion must be tempered by the fact that RNAi knockdown is probably not complete and the phm-Gal4 driver may not turn on until after gland formation is largely finished. To fully rule out a role of activins in gland specification, null germline clones for babo or dSmad2 need to be analyzed, and at this time it is not clear that any such alleles exist for either gene (M.B.O., unpublished).
The lack of true null mutations in both babo and dSmad2 may also account for the stronger phenotype produced by PG-specific knockdown of babo or dSmad2 compared with the reported babo genetic loss-of-function phenotype. Our previous studies examining the phenotype of babo zygotic mutants revealed a 2-3 day developmental delay in puparium formation, but up to 30% of the mutant larvae do initiate metamorphosis (Brummel et al., 1999). By contrast, elimination of activin signal reception only in the PG produces a stronger phenotype where virtually 100% of the knockdown larvae arrest development without puparium formation. Although residual Babo function is a possible explanation for this difference, we point out that another explanation exists that highlights the difficulties associated with interpreting tissue-specific knockdown experiments. It is theoretically possible to produce a stronger whole animal phenotype by tissue-specific knockdown than by a genetic null mutation, owing to potential compensatory changes in other mutant tissues in the genetically null animal. This is especially true when the factor potentially regulates multiple systemic signals in different tissues. For example, the ligand Actβ is produced in IPCs of the brain (M.B.O., unpublished). If Actβ negatively regulates insulin production or secretion, then, in a babo genetic null background, upregulation of insulin signal upon loss of Actβ signaling in IPCs may compensate for reduced insulin reception capacity in the PGs. This compensatory mechanism may enable a percentage of the babo mutant larvae to undergo puparium formation. By contrast, knockdown of activin signal reception in the PG alone, as in the case of phm>babo RNAi animals, would probably not lead to this compensatory response and thus results in a stronger developmental arrest phenotype. Consistent with this view, we find that using a ubiquitous driver, such as daughterless-Gal4, to knockdown dSmad2 in all tissues, including the PG, does not lead to developmental arrest, whereas using a PG specific driver does. These potential complications in evaluating phenotypic differences obtained using genetic mutations and tissue-specific knockdown methods should be borne in mind as they are likely to be observed more frequently with the increasing use of tissue-specific knockdown analyses in numerous model organisms.
The issue of which activin-like ligand(s) are responsible for providing the competence signal and whether they are regulated by particular developmental or nutritional cues is also important to answer but is problematic because of redundancy concerns. We suspect that Actβ probably plays a role as it is expressed in numerous neurosecretory cells including the IPCs (M.B.O., unpublished), which innervate the heart tube (Rulifson et al., 2002) and thereby probably provide systemic delivery of this ligand to many tissues. In addition, overexpression of this ligand in the PG causes stage precocious pupation, similar to that produced by expression of activated Babo. However, the one available Actβ loss-of-function mutation does not produce substantial developmental delay, and the majority of larvae (>90%) instead undergo slightly precocious puparation (A. Ghosh and M.B.O., unpublished) consistent with potential upregulation of insulin signaling, as suggested above. Likewise, mutations in daw, a second Activin-like ligand that is produced in the PG (M.B.O., unpublished), do not elicit major developmental delay when fed a yeast-enriched diet. However, when strong daw alleles are combined with the one available Actß mutation, then only 25-35% of the larvae are able to initiate pupariation on rich food (Zhu et al., 2008) (M.B.O., unpublished). This observation suggests a functional redundancy between these ligands for regulating developmental timing, similar to their previously noted redundant roles in regulating neuroblast proliferation in the larval brain (Zhu et al., 2008). The residual pupation ability of the daw-Actß double mutants may be accounted for by the compensation mechanism described above or further functional redundancy provided by the two other Activin-like ligands, Myo and Mav. At present, no mutations are available in these genes.
Direct or indirect regulation of steroid production by Activin signaling?
Although our observations clearly show that loss of E biosynthetic enzyme expression underlies the dSmad2 developmental arrest phenotype, we cannot say with certainty that this downturn is due solely to the loss of insulin and PTTH signaling or whether dSmad2 might also participate directly in regulating biosynthetic enzyme gene expression. In addition, whether dSmad2 binds directly to target sequences within Torso and InR regulatory sequences also remains uncertain as no dSmad2 responsive elements have been identified for any gene in Drosophila. It is interesting to note, however, that one other molecular process, SUMOlyation, has recently been implicated in regulating E biosynthetic enzyme expression and localization in the PG (Talamillo et al., 2008). Knockdown of smt3, the sole SUMO-encoding gene in Drosophila, in the PG results in third instar larval arrest phenotype that is rescuable by feeding the larvae 20E. In addition, these larvae show low levels of Dib protein accumulation in the PG (Talamillo et al., 2008). These phenotypes are strikingly similar to those seen in dSmad2 RNAi larvae. Interestingly, previous studies have demonstrated that Medea can be SUMOlyated in vitro, providing a potential link between SUMOlyation and Activin signaling (Miles et al., 2008). However, we have been unable to detect any SUMOylated forms of Medea or dSmad2 under various signaling conditions in vivo or in S2 cells. Additional studies will be required to determine whether SUMOlyation and Activin signaling are linked in a common pathway or whether they act through independent means to control steroid production in the PG.
A conserved role for TGFβ/Activin signaling in regulating developmental transitions
Despite the uncertainty in determining whether one or more ligands act as a PG competence factor, the observation that TGFβ/Activin signaling regulates metamorphosis may highlight an ancient and conserved role for these factors in regulating developmental transitions in many organisms. For example, the nematode worm C. elegans employs the TGFβ pathway to make a nutritionally dependent decision on whether to continue the normal development program into a mature adult or to enter a developmentally arrested stage known as dauer. The activation of the TGFβ pathway promotes normal development whereas its inactivation results in dauer formation (for a review, see Fielenbach and Antebi, 2008). Interestingly, a key target of the TGFβ pathway is daf-9, which encodes a cytochrome P450 protein. DAF-9 is involved in the synthesis of dafachronic acid, an ecdysone-like steroid hormone that prevents the developmental arrest as a dauer (Gerisch et al., 2001; Jia et al., 2002). Likewise, in mammals, Activin signaling probably affects pubertal timing by controlling sex steroid synthesis. Activin signaling can stimulate the production of the sex steroid hormone estradiol by enhancing the activity of CYP 450 aromatase (Hutchinson et al., 1987; Miro et al., 1991; Miro and Hillier, 1992). These observations from C. elegans and mammalian studies, together with our finding that Drosophila Activin signaling also regulates cytochrome P450 expression, although probably by indirect means, strongly indicate that TGFβ/Activin signaling is a common means by which developmental transitions are regulated across species.
PTTH and insulin signaling impinge on steroidogenic regulation in distinct ways
Previously, we have found that complete loss of PTTH signaling delayed metamorphosis but did not completely block it (McBrayer et al., 2007). This observation led us to speculate that a second metamorphic signal, perhaps supplied by insulin, eventually facilitated pupation in the PTTH negative larvae. Our finding here that simultaneous knockdown of both pathways by eliminating dSmad2 in the PG leads to third instar developmental arrest and that resupplying activity in either pathway promotes metamorphosis is consistent with this idea. Even more intriguing is our finding that the two metamorphic signals provided by PTTH and insulin appear to regulate the steroid biosynthetic capacity of the gland in two distinct ways: PTTH at the level of biosynthetic enzyme mRNA accumulation and insulin at the biosynthetic enzyme protein level (Fig. 6). The observation that loss of PTTH signal reception in dSmad2 PG knockdown larvae reduces the steady-state mRNA levels of the E biosynthetic enzymes is consistent with our earlier PTTH-neuron ablation results with one exception. In our present studies, we only observed downregulation of dib, spok and nvd in response to reduction in Torso expression, whereas in PTTH-neuron ablated larva, transcription of phm and sad are also reduced (McBrayer et al., 2007). One possible explanation for this discrepancy is that the five biosynthetic enzymes exhibit different sensitivities to the strength of the PTTH signal. In PTTH-neuron ablation experiments, all signaling is lost, whereas in the dSmad2 knockdown, it is likely that some Torso expression remains and provides enough signal to activate phm and sad. Consistent with this view, we find that in the rescued larvae, where we resupply PTTH signaling in the PG using activated Ras, the mRNA levels of the phm and sad genes are dramatically upregulated compared with the other three E biosynthetic enzymes (Fig. 4F), indicating they are very sensitive to the PTTH signal.
Fig. 6.

Model for TGFβ/Activin regulation of metamorphosis. The onset of metamorphosis is regulated by the activities of multiple signaling pathways within the PG, including PTTH, insulin and TGFβ/Activin. PTTH signaling plays a primary role in upregulation of the E biosynthetic gene transcription, while insulin signaling regulates their translation and/or stability. Activin signaling ensures proper expression of Torso and InR in the PG. Without Activin signaling, the PG is unable to respond to either PTTH or to insulin signal, leading to a failure to express sufficient levels of the E biosynthetic enzymes that trigger metamorphosis.
The effects of insulin signaling on protein levels is intriguing and has not been examined previously, although there are two reports to indicate that insulin signaling also affects transcription of at least two biosynthetic enzymes, dib and phm (Colombani et al., 2005; Walkiewicz and Stern, 2009). However, the reported effects were modest (between 30% to 2 fold) and it was not clear whether the determinations were normalized for differences in body size and/or ring gland size, which are crucial when considering the small reported differences. In our measurements, we used ring gland-brain complexes that were of similar size and staging, and did not detect any differences in transcription of biosynthetic enzymes between the dSmad2 knockdown animals and those in which InR expression was restored to the PG. Instead, we observe significant differences in biosynthetic enzyme protein levels. The known role of insulin in modulating translational capacity of cells is consistent with the idea that these changes in biosynthetic enzyme levels are the result of translational differences. Whether this effect is through Tor, which has been shown to be an important mediator of developmental timing in the PG (Layalle et al., 2008), and its modulation of S6 kinase, remains to be determined. In addition, we cannot exclude effects on protein stability, and further studies examining protein turnover will also be required to address this issue fully. Overall, this two-tiered regulatory control of biosynthetic enzyme expression may better enable the larva to fine-tune its ecdysone level depending on conditions, or perhaps may serve as a coincidence detector to ensure that both developmental and nutritional conditions are appropriate before triggering a terminal developmental program such as metamorphosis.
Supplementary Material
Acknowledgements
We thank MaryJane Shimell, Aidan Peterson and Naoki Yamanaka for comments on the manuscript. We thank Scott Gesualdi and Theo Haerry for helpful discussions and communication of results prior to publication. Y.Y.G. was supported by training grant T32 HD007480 and by RO1 GM095746 to M.B.O. M.B.O. was an Investigator with the Howard Hughes Medical Institute during part of this work. Deposited in PMC for release after 6 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.063412/-/DC1
References
- Andres A. J., Thummel C. S. (1992). Hormones, puffs and flies: the molecular control of metamorphosis by ecdysone. Trends Genet. 8, 132-138 [DOI] [PubMed] [Google Scholar]
- Baehrecke E. H. (1996). Ecdysone signaling cascade and regulation of Drosophila metamorphosis. Arch. Insect Biochem. Physiol. 33, 231-244 [DOI] [PubMed] [Google Scholar]
- Britton J. S., Lockwood W. K., Li L., Cohen S. M., Edgar B. A. (2002). Drosophila's insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev. Cell 2, 239-249 [DOI] [PubMed] [Google Scholar]
- Broadus J., McCabe J. R., Endrizzi B., Thummel C. S., Woodard C. T. (1999). The Drosophila beta FTZ-F1 orphan nuclear receptor providescompetence for stage-specific responses to the steroid hormone ecdysone. Mol. Cell 3, 143-149 [DOI] [PubMed] [Google Scholar]
- Brummel T., Abdollah S., Haerry T. E., Shimell M. J., Merriam J., Raftery L., Wrana J. L., O'Connor M. B. (1999). The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev. 13, 98-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell P. E., Walkiewicz M., Stern M. (2005). Ras activity in the Drosophila prothoracic gland regulates body size and developmental rate via ecdysone release. Curr. Biol. 15, 1785-1795 [DOI] [PubMed] [Google Scholar]
- Chang Y. Y., Neufeld T. P. (2009). An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 20, 2004-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez V. M., Marqués G., Delbecque J. P., Kobayashi K., Hollingsworth M., Burr J., Natzle J. E., O'Connor M. B. (2000). The Drosophila disembodied gene controls late embryonic morphogenesis and codes for a cytochrome P450 enzyme that regulates embryonic ecdysone levels. Development 127, 4115-4126 [DOI] [PubMed] [Google Scholar]
- Childs S. R., Wrana J. L., Arora K., Attisano L., O'Connor M. B., Massague J. (1993). Identification of a Drosophila activin receptor. Proc. Natl. Acad. Sci. USA 90, 9475-9479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancio M. J., Watson R. D., Bollenbacher W. E. (1986). Competency of Manduca sexta prothoracic glands to synthesize ecdysone during development. Mol. Cell. Endocrinol. 44, 171-178 [DOI] [PubMed] [Google Scholar]
- Colombani J., Bianchini L., Layalle S., Pondeville E., Dauphin-Villemant C., Antoniewski C., Carre C., Noselli S., Leopold P. (2005). Antagonistic actions of ecdysone and insulins determine final size in Drosophila. Science 310, 667-670 [DOI] [PubMed] [Google Scholar]
- Das P., Maduzia L. L., Wang H., Finelli A. L., Cho S. H., Smith M. M., Padgett R. W. (1998). The Drosophila gene medea demonstrates the requirement for different classes of smads in dpp signaling. Development 125, 1519-1528 [DOI] [PubMed] [Google Scholar]
- Fehon R. G., Dawson I. A., Artavanis-Tsakonas S. (1994). A Drosophila homologue of membrane-skeleton protein 4.1 is associated with septate junctions and is encoded by the coracle gene. Development 120, 545-557 [DOI] [PubMed] [Google Scholar]
- Fielenbach N., Antebi A. (2008). C. elegans dauer formation and the molecular basis of plasticity. Genes Dev. 22, 2149-2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerisch B., Weitzel C., Kober-Eisermann C., Rottiers V., Antebi A. (2001). A hormonal signaling pathway influencing C. elegans metabolism, reproductive development, and life span. Dev. Cell 1, 841-851 [DOI] [PubMed] [Google Scholar]
- Gilbert L. I. (2004). Halloween genes encode P450 enzymes that mediate steroid hormone biosynthesis in Drosophila melanogaster. Mol. Cell. Endocrinol. 215, 1-10 [DOI] [PubMed] [Google Scholar]
- Gilbert L. I., Song Q., Rybczynski R. (1997). Control of ecdysteroidogenesis: activation and inhibition of prothoracic gland activity. Invert. Neurosci. 3, 205-216 [DOI] [PubMed] [Google Scholar]
- Gilbert L. I., Rybczynski R., Warren J. T. (2002). Control and biochemical nature of the ecdysteroidogenic pathway. Annu. Rev. Entomol. 47, 883-916 [DOI] [PubMed] [Google Scholar]
- Goberdhan D. C., Paricio N., Goodman E. C., Mlodzik M., Wilson C. (1999). Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. 13, 3244-3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y., Miyamoto K., Abe Y., Nakamura T., Sugino H., Eto Y., Shibai H., Igarashi M. (1988). Induction of follicle stimulating hormone receptor by erythroid differentiation factor on rat granulosa cell. Biochem. Biophys. Res. Commun. 156, 668-674 [DOI] [PubMed] [Google Scholar]
- Hutchinson L. A., Findlay J. K., de Vos F. L., Robertson D. M. (1987). Effects of bovine inhibin, transforming growth factor-beta and bovine activin-A on granulosa cell differentiation. Biochem. Biophys. Res. Commun. 146, 1405-1412 [DOI] [PubMed] [Google Scholar]
- Ikeya T., Galic M., Belawat P., Nairz K., Hafen E. (2002). Nutrient-dependent expression of insulin-like peptides from neuroendocrine cells in the CNS contributes to growth regulation in Drosophila. Curr. Biol. 12, 1293-1300 [DOI] [PubMed] [Google Scholar]
- Ishizaki H., Suzuki A. (1994). The brain secretory peptides that control moulting and metamorphosis of the silkmoth, Bombyx mori. Int. J. Dev. Biol. 38, 301-310 [PubMed] [Google Scholar]
- Jia K., Albert P. S., Riddle D. L. (2002). DAF-9, a cytochrome P450 regulating C. elegans larval development and adult longevity. Development 129, 221-231 [DOI] [PubMed] [Google Scholar]
- Jones D., Jones G. (2007). Farnesoid secretions of dipteran ring glands: What we do know and what we can know. Insect Biochem. Mol. Biol. 37, 771-798 [DOI] [PubMed] [Google Scholar]
- Kaplowitz P. B., Slora E. J., Wasserman R. C., Pedlow S. E., Herman-Giddens M. E. (2001). Earlier onset of puberty in girls: relation to increased body mass index and race. Pediatrics 108, 347-353 [DOI] [PubMed] [Google Scholar]
- Kawakami A., Kataoka H., Oka T., Mizoguchi A., Kimura-Kawakami M., Adachi T., Iwami M., Nagasawa H., Suzuki A., Ishizaki H. (1990). Molecular cloning of the Bombyx mori prothoracicotropic hormone. Science 247, 1333-1335 [DOI] [PubMed] [Google Scholar]
- Knight P. G., Glister C. (2001). Potential local regulatory functions of inhibins, activins and follistatin in the ovary. Reproduction 121, 503-512 [DOI] [PubMed] [Google Scholar]
- Larsson J., Karlsson S. (2005). The role of smad signaling in hematopoiesis. Oncogene 24, 5676-5692 [DOI] [PubMed] [Google Scholar]
- Layalle S., Arquier N., Léopold P. (2008). The TOR pathway couples nutrition and developmental timing in Drosophila. Dev. Cell 15, 568-577 [DOI] [PubMed] [Google Scholar]
- Li W. X. (2005). Functions and mechanisms of receptor tyrosine kinase torso signaling: lessons from drosophila embryonic terminal development. Dev. Dyn. 232, 656-672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal E., Vandersmissen H. P., Badisco L., Van de Velde S., Verlinden H., Iga M., Van Wielendaele P., Huybrechts R., Simonet G., Smagghe G., et al. (2010). Control of ecdysteroidogenesis in prothoracic glands of insects: a review. Peptides 31, 506-519 [DOI] [PubMed] [Google Scholar]
- Mauras N., Rogol A. D., Haymond M. W., Veldhuis J. D. (1996). Sex steroids, growth hormone, insulin-like growth factor-1: neuroendocrine and metabolic regulation in puberty. Horm. Res. 45, 74-80 [DOI] [PubMed] [Google Scholar]
- McBrayer Z., Ono H., Shimell M., Parvy J. P., Beckstead R. B., Warren J. T., Thummel C. S., Dauphin-Villemant C., Gilbert L. I., O'Connor M. B. (2007). Prothoracicotropic hormone regulates developmental timing and body size in Drosophila. Dev. Cell 13, 857-871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles W. O., Jaffray E., Campbell S. G., Takeda S., Bayston L. J., Basu S. P., Li M., Raftery L. A., Ashe M. P., Hay R. T., et al. (2008). Medea SUMOylation restricts the signaling range of the dpp morphogen in the drosophila embryo. Genes Dev. 22, 2578-2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miro F., Hillier S. G. (1992). Relative effects of activin and inhibin on steroid hormone synthesis in primate granulosa cells. J. Clin. Endocrinol. Metab. 75, 1556-1561 [DOI] [PubMed] [Google Scholar]
- Miro F., Smyth C. D., Hillier S. G. (1991). Development-related effects of recombinant activin on steroid synthesis in rat granulosa cells. Endocrinology 129, 3388-3394 [DOI] [PubMed] [Google Scholar]
- Mirth C. K., Riddiford L. M. (2007). Size assessment and growth control: how adult size is determined in insects. BioEssays 29, 344-355 [DOI] [PubMed] [Google Scholar]
- Mirth C., Truman J. W., Riddiford L. M. (2005). The role of the prothoracic gland in determining critical weight for metamorphosis in Drosophila melanogaster. Curr. Biol. 15, 1796-1807 [DOI] [PubMed] [Google Scholar]
- Mondal D., Pradhan L., LaRussa V. F. (2004). Signal transduction pathways involved in the lineage-differentiation of NSCs: can the knowledge gained from blood be used in the brain? Cancer Invest. 22, 925-943 [DOI] [PubMed] [Google Scholar]
- Motola D. L., Cummins C. L., Rottiers V., Sharma K. K., Li T., Li Y., Suino-Powell K., Xu H. E., Auchus R. J., Antebi A., et al. (2006). Identification of ligands for DAF-12 that govern dauer formation and reproduction in C. elegans. Cell 124, 1209-1223 [DOI] [PubMed] [Google Scholar]
- Nguyen M., Parker L., Arora K. (2000). Identification of maverick, a novel member of the TGF-beta superfamily in Drosophila. Mech. Dev. 95, 201-206 [DOI] [PubMed] [Google Scholar]
- Niwa R., Namiki T., Ito K., Shimada-Niwa Y., Kiuchi M., Kawaoka S., Kayukawa T., Banno Y., Fujimoto Y., Shigenobu S., et al. (2010). Non-molting glossy/shroud encodes a short-chain dehydrogenase/reductase that functions in the ‘black box’ of the ecdysteroid biosynthesis pathway. Development 137, 1991-1999 [DOI] [PubMed] [Google Scholar]
- Ono H., Rewitz K. F., Shinoda T., Itoyama K., Petryk A., Rybczynski R., Jarcho M., Warren J. T., Marques G., Shimell M. J., et al. (2006). Spook and spookier code for stage-specific components of the ecdysone biosynthetic pathway in diptera. Dev. Biol. 298, 555-570 [DOI] [PubMed] [Google Scholar]
- Parvy J. P., Blais C., Bernard F., Warren J. T., Petryk A., Gilbert L. I., O'Connor M. B., Dauphin-Villemant C. (2005). A role for betaFTZ-F1 in regulating ecdysteroid titers during post-embryonic development in Drosophila melanogaster. Dev. Biol. 282, 84-94 [DOI] [PubMed] [Google Scholar]
- Perrimon N., Lu X., Hou X. S., Hsu J. C., Melnick M. B., Chou T. B., Perkins L. A. (1995). Dissection of the torso signal transduction pathway in Drosophila. Mol. Reprod. Dev. 42, 515-522 [DOI] [PubMed] [Google Scholar]
- Petryk A., Warren J. T., Marques G., Jarcho M. P., Gilbert L. I., Kahler J., Parvy J. P., Li Y., Dauphin-Villemant C., O'Connor M. B. (2003). Shade is the Drosophila P450 enzyme that mediates the hydroxylation of ecdysone to the steroid insect molting hormone 20-hydroxyecdysone. Proc. Natl. Acad. Sci. USA 100, 13773-13778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O., Marr M. T., Ruhf M. L., Tjian R. (2003). Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 17, 2006-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rewitz K. F., Yamanaka N., Gilbert L. I., O'Connor M. B. (2009). The insect neuropeptide PTTH activates receptor tyrosine kinase torso to initiate metamorphosis. Science 326, 1403-1405 [DOI] [PubMed] [Google Scholar]
- Rulifson E. J., Kim S. K., Nusse R. (2002). Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 296, 1118-1120 [DOI] [PubMed] [Google Scholar]
- Sauman I., Reppert S. M. (1996). Molecular characterization of prothoracicotropic hormone (PTTH) from the giant silkmoth antheraea pernyi: developmental appearance of PTTH-expressing cells and relationship to circadian clock cells in central brain. Dev. Biol. 178, 418-429 [DOI] [PubMed] [Google Scholar]
- Shi Y. B., Wong J., Puzianowska-Kuznicka M., Stolow M. A. (1996). Tadpole competence and tissue-specific temporal regulation of amphibian metamorphosis: roles of thyroid hormone and its receptors. BioEssays 18, 391-399 [DOI] [PubMed] [Google Scholar]
- Sisk C. L., Foster D. L. (2004). The neural basis of puberty and adolescence. Nat. Neurosci. 7, 1040-1047 [DOI] [PubMed] [Google Scholar]
- Smith W. A., Gilbert L. I. (1989). Early events in peptide-stimulated ecdysteroid secretion by the prothoracic glands of manduca sexta. J. Exp. Zool. 252, 264-270 [DOI] [PubMed] [Google Scholar]
- Talamillo A., Sanchez J., Cantera R., Perez C., Martin D., Caminero E., Barrio R. (2008). Smt3 is required for Drosophila melanogaster metamorphosis. Development 135, 1659-1668 [DOI] [PubMed] [Google Scholar]
- ten Dijke P., Korchynskyi O., Valdimarsdottir G., Goumans M. J. (2003). Controlling cell fate by bone morphogenetic protein receptors. Mol. Cell. Endocrinol. 211, 105-113 [DOI] [PubMed] [Google Scholar]
- Ting C. Y., Herman T., Yonekura S., Gao S., Wang J., Serpe M., O'Connor M. B., Zipursky S. L., Lee C. H. (2007). Tiling of r7 axons in the Drosophila visual system is mediated both by transduction of an activin signal to the nucleus and by mutual repulsion. Neuron 56, 793-806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkiewicz M. A., Stern M. (2009). Increased insulin/insulin growth factor signaling advances the onset of metamorphosis in Drosophila. PLoS ONE 4, e5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J. T., Petryk A., Marques G., Jarcho M., Parvy J. P., Dauphin-Villemant C., O'Connor M. B., Gilbert L. I. (2002). Molecular and biochemical characterization of two P450 enzymes in the ecdysteroidogenic pathway of drosophila melanogaster. Proc. Natl. Acad. Sci. USA 99, 11043-11048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J. T., Petryk A., Marques G., Parvy J. P., Shinoda T., Itoyama K., Kobayashi J., Jarcho M., Li Y., O'Connor M. B., et al. (2004). Phantom encodes the 25-hydroxylase of drosophila melanogaster and bombyx mori: a P450 enzyme critical in ecdysone biosynthesis. Insect Biochem. Mol. Biol. 34, 991-1010 [DOI] [PubMed] [Google Scholar]
- Warren J. T., Yerushalmi Y., Shimell M. J., O'Connor M. B., Restifo L. L., Gilbert L. I. (2006). Discrete pulses of molting hormone, 20-hydroxyecdysone, during late larval development of Drosophila melanogaster: correlations with changes in gene activity. Dev. Dyn. 235, 315-326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wismar J., Habtemichael N., Warren J. T., Dai J. D., Gilbert L. I., Gateff E. (2000). The mutation without children(rgl) causes ecdysteroid deficiency in third-instar larvae of Drosophila melanogaster. Dev. Biol. 226, 1-17 [DOI] [PubMed] [Google Scholar]
- Wu Q., Brown M. R. (2006). Signaling and function of insulin-like peptides in insects. Annu. Rev. Entomol. 51, 1-24 [DOI] [PubMed] [Google Scholar]
- Xiao S., Robertson D. M., Findlay J. K. (1992). Effects of activin and follicle-stimulating hormone (FSH)-suppressing protein/follistatin on FSH receptors and differentiation of cultured rat granulosa cells. Endocrinology 131, 1009-1016 [DOI] [PubMed] [Google Scholar]
- Yoshiyama T., Namiki T., Mita K., Kataoka H., Niwa R. (2006). Neverland is an evolutionally conserved rieske-domain protein that is essential for ecdysone synthesis and insect growth. Development 133, 2565-2574 [DOI] [PubMed] [Google Scholar]
- Zhu C. C., Boone J. Q., Jensen P. A., Hanna S., Podemski L., Locke J., Doe C. Q., O'Connor M. B. (2008). Drosophila activin- and the activin-like product dawdle function redundantly to regulate proliferation in the larval brain. Development 135, 513-521 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}