

Abstract
Contigs have been assembled, and over 2800 clones selected for sequencing for human chromosomes 9, 10 and 13. Using the FPC (FingerPrinted Contig) software, the contigs are assembled with markers and complete digest fingerprints, and the contigs are ordered and localised by a global framework. Publicly available resources have been used, such as, the 1998 International Gene Map for the framework and the GSC Human BAC fingerprint database for the majority of the fingerprints. Additional markers and fingerprints are generated in-house to supplement this data. To support the scale up of building maps, FPC V4.7 has been extended to use markers with the fingerprints for assembly of contigs, new clones and markers can be automatically added to existing contigs, and poorly assembled contigs are marked accordingly. To test the automatic assembly, a simulated complete digest of 110 Mb of concatenated human sequence was used to create datasets with varying coverage, length of clones, and types of error. When no error was introduced and a tolerance of 7 was used in assembly, the largest contig with no false positive overlaps has 9534 clones with 37 out-of-order clones, that is, the starting coordinates of adjacent clones are in the wrong order. This paper describes the new features in FPC, the scenario for building the maps of chromosomes 9, 10 and 13, and the results from the simulation.
FPC (FingerPrinted Contigs) assembles clones into contigs by using either the end-labeled double digest method (Coulson et al. 1986; Gregory et al. 1997) or the complete digest method (Olson et al. 1986; Marra et al. 1999). Both methods produce a characteristic set of bands for each clone. To determine if two clones overlap, the number of shared bands is counted where two bands are considered “shared” if they have the same value within a tolerance. The probability that the N shared bands is a coincidence is computed, and if this score is below a user supplied cutoff, the clones are considered to overlap. If two clones have a coincidence score below the cutoff but do not overlap, it is a false-positive (F+) overlap. If two clones have a coincidence score above the cutoff but do overlap, it is a false-negative (F−) overlap. It is very important to set the cutoff to minimize the number of F+ and F− overlaps.
Over a decade ago, the first contigs built by restriction fragment fingerprints were published. Coulson et al. (1986) used the end-labeled double digest method with cosmid clones for mapping Caenorhabditis elegans. Olson et al. (1986) used the complete digest method with lambda clones for mapping yeast. Fingerprinting was used for mapping regions of human chromosomes; e.g., Carrona et al. (1989) used the double digest method with cosmid clones for mapping chromosome 19, and Stallings et al. (1990) used the complete digest method for mapping chromosome 16. Restriction fingerprinting was labor intensive and resulted in many gaps. To reduce this problem and anchor contigs, investigators used markers in conjunction with fingerprints in which a marker can be a sequence tag site (STS), polymerase chain reaction (PCR), hybridization, etc. Cosmid maps ordered by Alu-PCR have been constructed for 40% of human chromosome 21 (Soeda et al. 1995), and cosmid maps anchored to yeast artificial chromosomes (YAC) by STSs were constructed for 72.5% of the Y chromosome (Taylor et al. 1996).
Due to perceived difficulties in building physical maps, Venter et al. (1996) claimed it would be more efficient not to build physical maps. Their arguments were based on problems using the YAC to cosmid map building methodology. The strategy they proposed instead uses bacterial artificial clones (BACs) for sequencing, sequence tag connectors (STCs) to find overlapping clones, and fingerprints to ensure the integrity of each clone before sequencing. Because the fingerprints are necessary, the next step of assembling them into an initial set of contigs by using FPC is easy, and problems can be found and repaired using the interactive features. By using the expressed sequence tags (ESTs) from the 1998 International Gene Map (Deloukas et al. 1998) to select clones, the contigs will be located with almost no extra effort. The gaps can be closed by walking or by incorporating simulated digested sequence into the map.
In short, restriction fingerprinting once again is considered a reasonable way to order clones due to improved clone libraries and software. BAC libraries provide longer inserts that require fewer clones to cover a region and close gaps versus the shorter inserts with cosmids and do not have the instabilities of the longer YAC clones. Recent sequence ready maps have been constructed with BAC, PACs (P1 Artificial Chromosomes), markers, and global frameworks. Niederfuhr et al. (1998) constructed a sequence ready map of PACs for human chromosome 11p13 by using chromosome walking independently verified by fingerprint analysis. Cao et al. (1999) built a sequence ready map of chromosome 16p13.1-p11.2 by using BACs and previously mapped STSs. Zhu et al. (1999) built a sequence ready map of chromosome 7 of the rice blast fungus Magnaporthe grisea by using BAC contigs assembled by hybridization and integrated with fingerprinted BAC contigs Hoskins et al. (2000) integrated STS content, restriction fingerprinting, and polytene chromosome in situ hybridization to produce a Drosophila melanogaster map for 81% of the genome. Klein et al. (2000) used amplified fragment length polymorphism (AFLP)-based markers integrated with fingerprints to map sorghum. To provide confirmation of overlap and information to merge contigs, the Sanger Centre traditionally has used markers with fingerprints (e.g., see Mungall et al. 1997; Soderlund et al. 1998). An alternative approach by Ding et al. (1999) uses three separate sets of fingerprints to increase the sensitivity of overlap calculation.
In the spring of 1999, the Genome Sequencing Center (GSC) in St. Louis started mass-fingerprinting BACs from the RPCI-11 male library constructed at Roswell Park Cancer Institute (Buffalo NY) (see http://genome.wustl.edu/gsc and http://www.chori.org/bacpac, respectively). A FPC database, called the humanmap, of the fingerprinted clones periodically has been made available via ftp (file transfer protocol). We extract chromosome-specific clones from this database, where the clones have been assigned to a chromosome by screening the RPCI-11 library with ESTs from the 1998 International Gene Map (98GeneMap). The clones and ESTs are loaded into chromosome-specific FPC databases, along with other markers and clones fingerprinted in-house. The markers and fingerprints are used to assemble sequence ready contigs, which are ordered and localized on the chromosome by the ESTs, and clones are selected and sent for sequencing.
Using FPC for assembling the chromosome 9, 10, and 13 maps has confirmed that it works well based on agreement with marker and external data, and from comparing hand assembled contigs with automatic assembled contigs. Regardless, the phrase “works well” is vague, and it is difficult to be more precise without having the sequence to verify. Now that some sequence is available, an initial set of experiments has been performed using a simulated digest on human sequence. The experiments vary the coverage, length of clone, and amount of error. The results show the effect on F+ overlaps, F− overlaps, the number of contigs, and the number of bad contigs. The second part of the simulation assembles a set of simulated digest sequence with a FPC database of real fingerprints.
The first section of Results describes the new features in FPC that support the incremental building of maps composed of fingerprints and markers. The second section discusses the building of the physical maps for chromosomes 9, 10, and 13. The third section provides a characterization of the fingerprints for these three chromosomes. The fourth section presents results from a complete digest simulation.
RESULTS
FPC V4.7
As originated from ContigC (Sulston et al. 1988), two clones are considered to overlap if the following score is below a user supplied cutoff:
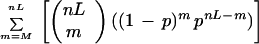 |
1 |
M is the number of shared bands, nL and nH are the lowest and highest number of bands in the two clones, respectively, t is the tolerance, gellen is approximately the number of possible values, b = 2t/gellen, and p = (1 − b)nH,. This equation is used by the routine that automatically builds contigs and also by various functions that allow the user to further evaluate clones interactively.
A FPC “complete build” bins clones into transitively overlapping sets where each clone in a set has an overlap with at least one other clone in the set and no clone has an overlap with any clone outside the set. The clones in a bin are given an appropriate ordering by building a CB (Consensus Band) map and the CB map is instantiated as a contig. Hence, a complete build guarantees that each contig is a transitively overlapping set of clones based on a given cutoff. The length of a clone in a contig is equal to the number of its bands, and the overlap between the coordinates of the two clones is approximately the number of shared bands. If clone CA has exactly or approximately the same bands as clone CB, CA can be “buried” in CB, and CB will be called the “parent.” Clones that do not have an overlap with any other clone are not placed in a contig and are called “singletons.” Markers that hybridize to a clone and displayed in the contig with the clone as illustrated in Figure 1. A clone can only be in one contig, but a marker can be attached to clones in multiple contigs. An externally ordered subset of the markers can be input into FPC as the “framework.” Contigs containing these markers can be listed by framework order in the project window.
Figure 1.

Contig with markers. The markers are ordered by placing each over the deepest stack of positive clones. The markers along the bottom are from the framework map. An * after a clone name indicates a parent clone. The clones highlighted in blue are the tiling path. The clone highlighted in cyan was selected with the mouse, which caused its markers and remarks to be highlighted in green and pink, respectively. The map units are the number of bands.
New Features
Recently, three salient changes were made to FPC to reduce the amount of human intervention. They are briefly described in this section. A review of the algorithms and additional details are provided in the Methods section.
Q Clones
The routine that orders clones is called the CB algorithm; an example of the output is shown in Figure 2. If there is a severe problem aligning a clone to the CB map, it is marked as a Q (questionable) clone. This information is saved in the FPC file and displayed in the project window. If there are many Q clones in the contig, the ordering almost certainly will be wrong. The CB algorithm can be executed on the contig by using a more stringent cutoff in order to assemble the contig into multiple good CB maps which can be ordered and/or split into new contigs.
Figure 2.

Consensus band map. In the top row, an = (not shown) or a ˜ indicates a potential exact or approximate buried clone, respectively. Qs indicates a Q (questionable) clone. The next three rows are the clone name. The last row is the number of extra bands that could not be placed. Below these four columns is a set of +, × , and 0. The + signs indicate a band within the tolerance of the consensus band to the right. The × indicate a band within twice the tolerance of the consensus band. The 0 indicate no band within the tolerance.
CpM (Cutoff plus Marker)
FPC provides the option of defining a set of rules on what constitutes a valid overlap, which are entered into the CpM table. The rules we use with complete digest BAC clones are as follows: two clones will be considered to overlap if they (1) have less than a 1e-10 score, (2) share at least one marker and score less than 1e-08, (3) share at least two markers and score less than 1e-07, or (4) share at least three markers and score less than 1e-06. When using the CpM table, the complete build guarantees that each contig is a transitively overlapping set of clones based on the CpM rules. The CpM table can significantly reduce the number of contigs. For example, the complete build of chromosome 13 with 13,944 clones resulted in 1443 contigs. When 2866 markers were included in the build, it resulted in 1298 contigs, a saving of 145 interactive merges. In both cases, it took approximately 30 seconds for a complete build on a Dec Alpha D4.0 500 Mhz with 128 MB RAM and 410 MB swap.
IBC (Incremental Build Contigs)
We are daily adding clones and markers to the FPC databases. The IBC routine automatically adds new clones to contigs and merges contigs based on the cutoff and CpM table, and then the clones in each modified contig are reordered by executing the CB algorithm. Contigs that have two or more clones picked for sequencing are given a status of NoCB; they are not automatically reordered because the user may have interactively moved clones to reflect the exact overlap between sequence ready clones and therefore, prefers the choice of executing the CB algorithm or arranging the merged contigs interactively. The IBC provides a summary of the modifications performed on each contig in the project window.
Merge and Split
FPC maintains two sets of CpM rules: The static set is used for the initial complete build and all subsequent IBCs. The variable set is changed interactively to find and repair F+ and F− overlaps, i.e., contigs to be split or merged, respectively. To reduce the amount of human intervention, it is necessary that the cutoff and CpM table be set to minimize the number of “merges and splits” that are needed. The best values to use can be determined by trying different values on an initial data set, as shown by the simulations.
The validity of merging contigs and ordering CB maps depends on the following two constraints: (1) the two clones are on the ends of two different contigs or CB maps, and (2) they qualify as an overlap based on reduced stringency rules. Human intervention is necessary in these cases because there are often ambiguities to be resolved.
Merge
To find contigs for potential manual merges, the Ends → Ends routine on the Main Analysis window will list all clones that obey both of the constraints. The Ctg → Ends routine on the Contig Analysis window will provide the same list for the displayed contig only.
Split (Removing Qs)
If a contig has Q clones, the CB algorithm can be run on the contig by using a more stringent set of rules that breaks it into multiple CB maps and removes most (or all) of the Q clones. The CB maps are ordered using the OKALL routine based on the two constraints. If the CB maps do not join into one contig, it is often the case that the original cutoff caused F+ overlaps. Disconnected contigs will be moved to new contigs.
Input
For generating the values corresponding to the bands on the gel image, we use the Image program (Sulston et al. 1989; http://www.sanger.ac.uk/Software/Image). Three sets of files are produced for each gel. (1) The band files contain the migration rates, which correspond to the position of the bands on the gel image. (2) The size files contain the estimated fragment sizes in base pairs, which are extrapolated from the rates. The larger the migration rate, the smaller the size and the sizes are not linearly related to the rates (see Methods). The size files are not relevant for the end-labeled double digest method. (3) The gel files contain the gel image.
Three FPC subdirectories contain these three sets of files. Either the size files or the band files can be used as the fingerprint input to FPC. Chromosomes 9, 10, and 13 use the band files for the fingerprint. The reasons are both historical and because the migration rates align to the gels that can be viewed in FPC. The problem with using the rates is that it is desirable to know the true sizes when picking clones for sequencing. Therefore, the sizes for a clone can be viewed via the size calculator that can be accessed from the gel image window of FPC (see Fig. 3) and was developed by Ken McDonald at the GSC (Marra et al. 1999).
Figure 3.

Gel image and size calculator.
The Building of Physical Maps for Chromosome 9, 10, and 13
As part of the international effort to determine the complete sequence of the human genome, our strategy has been to build maps by using large insert bacterial clones and then to sequence a minimally overlapping set of clones by shotgun sequencing. For chromosomes 9, 10, and 13, BAC clones from the RPCI-11 library are screened using a high density of STS-based markers. These “seed” markers are obtained from the 1998 International Gene Map, GDB (Genome DataBase) and random genomic markers that are generated at the Sanger Centre. The majority of BAC clones from the RPCI-11 library have been fingerprinted at the GSC in St. Louis, and these are supplemented with additional clones fingerprinted at the Sanger Centre by using the same protocol so that the fingerprints are compatible (Marra et al. 1997; Humphray et al. 2000). In order to extend the contigs by walking, STSs are designed using the available GSS (Genome Sequencing Survey; Mahairas et al. 1999) from selected clones at the ends of contigs. These STSs then are used to identify joins between existing contigs or to identify additional BAC clones from the library. The clones and markers are entered into a chromosome-specific AceDB (Durbin and Thierry-Mieg 1994).
Identifying chromosome-specific markers and clones is an ongoing process as we continually update the chromosome-specific FPC with new information. This is done as follows:
Weekly, a new Human FPC database is downloaded from the GSC. Nightly, clones are extracted from chromosome-specific AceDBs, and the get_GSC script is run to build chromosome-specific FPC input files for the clones that are in both AceDB and the human FPC database.
Nightly, the markers and clone hybridization results are extracted from AceDB and used to update FPC. The FPC framework map is updated by a file containing the 98GeneMap for a chromosome.
A few times a week, the project leader will do the following: (1) Update.cor is executed to add new fingerprints to the FPC database, which are obtained as described in step 1 and from fingerprints generated in-house. (2) The IBC routine is executed to update and merge contigs based on the new clones and markers. The altered contigs are interactively confirmed or rejected. Contigs with a status of NoCB are generally reordered using the CB algorithm. Contigs with many Q clones are assembled at a more stringent overlap and split into multiple contigs where appropriate. (3) The end clones are compared to determine contigs to merge. The bands from the end clones are viewed in the CB (Build or Selected) window and/or the fingerprint window to ensure that the bands can be arranged in a consistent order. Potential joins generally are confirmed by external data and/or the framework map. (4) Sequence ready clones are selected. A clone sent for sequencing must have all its bands confirmed by overlapping clones within the gel image window, where F+ and F− bands can be detected.
Nightly, the contigs are loaded into AceDB, the new clones picked for sequencing are sent to the Sanger Centre Oracle tracking database, and the tracking database sends the status of sequence ready clones to FPC for update.
As a result of the above steps, the FPC databases always have the most current set of markers, clones and sequence ready clone status.
By midsummer of 2000, the physical map of chromosome 9 had 903 clones picked for sequencing from 25 anchored contigs; there are 2591 seed markers for one per 47 kb and 719 frameworks (691 placements) for one per 173 kb; the estimated total length is 145 Mb of which 20 Mb is heterochromatin; only the euchromatic portion was seeded so the length used is 125 Mb. The physical map of chromosome 10 has 1162 clones picked for sequencing from 23 anchored contigs; there are 3165 seed markers for one per 45 kb and 1287 frameworks (1212 placement) for one per 111 kb; the estimated total length is 144Mb. The physical map of chromosome 13 has 808 clones picked for sequencing from 20 anchored contigs; there are 2011 seed markers for one per 49 kb and 667 frameworks (633 placement) for one per 146 kb; the estimated total length is 114 Mb of which 16 Mb is heterochromatin; only the euchromatic portion was seeded so the length used is 98 Mb.
Characterization of the Fingerprints for Chromosomes 9, 10, and 13
The size files for chromosomes 9, 10, and 13 were used to create FPC databases. Summing the size fragments for each clone, the average size is 159 kb with an average deviation of 17 kb. The average size calculated by Peter de Jong's laboratory is 164 kb (see http://www.chori.org/bacpac/11framehmale.htm). The difference in size calculation is due to the fact that fragments <600 bp and very large fragments generally are not detected, and fragments >32,627 bp are stored by FPC as 32,627 bp because of a size limitation (the rates are generally used which have values less than 4300). The average number of bands is 36 bands per clone, and the average fragment size for the three chromosomes is 4082 bp. The average number of exact duplicate bands is 5.4, and the average number within a 7 tolerance is 5.6 per clone (see Methods).
The vector has three fragments of estimated sizes (6511, 510, and 449 bp) and migration rates (1387, 3695, and 3766 bp), respectively. FPC can remove vector bands, which was done for chromosomes 9, 10, and 13. EcoRI is used for the partial digest to obtain the clones, and HindIII is used for the complete digest so each clone will have two end fragments. One is attached to 1020 bp of vector sequence, and the other is attached to 332 bp of vector, so the second end may not be detected by image analysis.
To estimate the deviation (i.e., uncertainty, tolerance) in migration rate and the number of F+ and F− bands, duplicate fingerprints were compared where the Sanger Centre produced one gel and GSC produced the other. Only pairs of gels that have below a 1e-10 score were used to eliminate bad or misnamed gels; this resulted in 4203 pairs of fingerprints. To estimate the tolerance, we compared duplicate fingerprints by using a tolerance of 7, the percentage of bands having a difference of 0 to 7 is (16, 20, 18, 15, 12, 9, 6, 4) respectively. To estimate the rate of F+ and F− overlaps, the number of extra bands was computed, i.e., bands that do not match any band from the corresponding gel. This resulted in 17% of the bands that could be either a F+ band, a F− band, an end band, or the difference in their rates is >7, e.g., if one band differs from the real value by +4 and the corresponding band from the other gel differs by −4, their difference in value is 8, which will result in two extra bands. Note that these estimates of tolerance and extra bands is an upper limit for chromosomes 9, 10, and 13 because it is the combined error of two sets of bands from two different labs. As more complete sequence becomes available, we will be able to compare simulated digested sequence with the clones to get the tolerance variation and F+/F− rate for individual clones.
In a characterization of the mouse BAC/PAC library (Osoegawa et al. 2000), it was estimated that 1% are chimeric or have rearrangements. We assume a similar number is estimated for the human library.
Simulation
Two data sets were created from sequences obtained from http://www.ncbi.nlm.nih.gov/genome/seq. The first set is six sequences from chromosomes 6, 7, 8, 12, 21 and 22 with sizes ranging from 1.5 to 11 Mb and a total length of 23 Mb. The second set is from the concatenation of 40 sequences resulting in one 110-Mb sequence.
A Perl program was written that digests each sequence with a given set of parameters. The clones can be created with an exact or random overlap. For “exact overlap,” the clones start every x number of bases to give the desired overlap. For “random overlap,” (1) a pool of clones is created by finding all EcoRI cut sites and generating all possible clones within the allowable range, e.g., between 145 and 185 kb; (2) the number of clones, N, needed for a specific coverage is determined, and (3) N clones are randomly picked from the pool of clones. The clones are named with their chromosome number followed by their location within the ordered clones (e.g., Zac22_3 is a clone from the sequence 22 and should assemble third from the end). A routine has been added to FPC that tests for the correctness of the contigs by using the information in the name and the real coordinates from the sequence (the real coordinates were entered as clone remarks). The clones are digested into fragments with HindIII.
Tables 1–6 show results based on different input parameters. The results for each table are a subset of the following measurements. (1) The number of contigs, F−, F+, and chimeric: F− is the number of adjacent clones that are not statistically overlapping. Occasionally, another clone pair will bridge the F− overlap; therefore, it does not cause a break. The number of bridging clones will be indicated by an “-n” after the number, e.g., 13-2 will result in 11 breaks in the contigs. F+ is the number of pairs of clones that are statistically overlapping but do not in reality overlap. Chimeric is the number of contigs that have clones from different chromosomes or regions of a sequence. If there is at least one F+ between clones from different regions, there will be at least one chimeric contig. These measures are a result of the cutoff. (2) The number of Q clones: a large number of Qs are generally a result of the cutoff since F+'s may result in no possible linear order. Occasionally, Qs can result from poor data and the CB algorithm, which is a fast approximation. (3) Order is a triple of numbers: the first number is the out-of-order clones, i.e., when clone CA should start immediately before clone CB, but CB starts before CA. The second number is the nonburied out-of-order clones. This measure is relevant because the tiling path of sequence clones generally is picked from nonburied clones. The last number is the count of adjacent clones that do not have overlapping coordinates in a contig. The order triplet is a result of positioning the clones by the CB algorithm and also increases in regions with F+ overlaps. (4) The number of gaps: because picking clones is random, there may be gaps. This random picking does not model hot spots or cold spots. The number of gaps is the result of one random picking and is not the average of N trials.
Table 1.
Rates and Sizes with Perfect Overlap
| Type | Tol | Cutoff | Ctgs | F− | F+ | Chia | Qsb | Orderc |
|---|---|---|---|---|---|---|---|---|
| Rates | 0 | 1e-08 | 5 | 0 | 1 | 1 | 71 (1) | 32, 29, 0 |
| 0 | 1e-09 | 6 | 0 | 0 | 0 | 0 | 0, 0, 0 | |
| 7 | 1e-06 | 6 | 0 | 0 | 0 | 0 | 2, 0, 0 | |
| Sizes | 0.00 | 1e-09 | 6 | 0 | 0 | 0 | 0 | 0, 0, 0 |
| 0.07 | 1e-06 | 6 | 0 | 0 | 0 | 0 | 1, 0, 0 |
(Tol) tolerance; (Ctgs) contigs; (F−) false-negative; (F+) false-positive; (Qs) questionable.
The data set has perfect data, 80% overlap, and 689 clones with lengths 160 kb.
Chimeric contigs, i.e., sequences from different chromosomes.
In parentheses is the number of contigs with Q clones.
The triplet represents the out-of-order coordinates for adjacent clones, out-of-order coordinates for adjacent nonburied clones, and adjacent clones that do not have overlapping coordinates.
Table 6.
Different Random Sets
| Set | Ctgs | F− | F+ | Qs | Order | Order/no Qsa |
|---|---|---|---|---|---|---|
| 1 | 42 + 1 | 102 − 59 | 59 | 112 (13) | (2770, 3400, 9) | (186, 220, 0) |
| 2 | 43 + 2 | 95 − 50 | 108 | 48 (12) | (2861, 3414, 1) | (395, 475, 0) |
| 3 | 38 + 1 | 88 − 49 | 91 | 145 (14) | (3024, 3634, 25) | (414, 458, 1) |
| 4 | 44 + 4 | 92 − 44 | 123 | 243 (16) | (2871, 3416, 10) | (391, 458, 1) |
| 4b | 27 + 0 | 35 − 9 | 236 | 8 (1) | (111, 16, 0) | (84, 11, 0) |
| 4c | 6 + 0 | 7 − 2 | 312 | 22 (1) | (508, 422, 1) | (4, 1, 0) |
(Ctgs) contigs; (F−) false-negative; (F+) false-positive; (Qs) questionable.
Each of the first four sets is a different random selection of 16,692 clones from the same pool, a 25× coverage with clone lengths of 130–190 kb, and one marker per 45 kb, and there is one chimeric contig. The error includes end fragments, 4% F+, 4% F, and the tolerance distribution of (16, 20, 18, 15, 12, 9, 6, 4). All the data sets were assembled with a 7 tolerance, 1e-10 cutoff with cutoff plus marker table (1 1e-08, 2, 1e-07, 3 1e-06). On the average, 17% of the clones are buried. The data sets assembled without markers resulted in 143, 155, 137, and 138 contigs, respectively.
The second order triplet is for the contigs with no Q clones.
The data set is the same as the previous one but without error. The set with error has 28 F+ clones with an average of 7 F+ overlaps per clone, whereas the set without error has 32 F+ clones with an average of 13 F+ overlaps per clone.
The data set is the same as the previous one that has no error but is assembled with a tolerance of 0 and a cutoff of 1e-12. The largest contig with no F+'s has 4738 clones and two out-of-order pairs.
Table 1 shows the results of using exact overlapped error-free data with a 5× coverage of the six sequence data set. Using a tolerance of 0 and a cutoff of 1e-08, there is one F+ overlap, i.e., clones Zac21_61 and Zac22_250 have a cutoff of 2e-09. This F+ causes the clones from sequence 21 and sequence 22 to be binned in the same contig. The CB algorithm perfectly orders the sequence 22 clones up to Zac22_250, but when it incorporates Zac21_61, there is no way to arrange its bands in a consistent manner, and so it is marked as a Q clone. It subsequently brings in all the other sequence 21 clones, and because there is no third dimension, there is no way to order the clones, and they end up in a stack. There are 71 clones in sequence 21, and there are 71 Q clones. The algorithm recovers and gives a perfect ordering to the rest of the sequence 22 clones, as is shown in Figure 4.
Figure 4.

Simulation results with stack of Q clones. A F+ overlap occurs between Zac22_250 and Zac21_61 causing all the sequence 21 clones to end up in a stack, as there is no linear order for both the sequences 22 and 21 clones in the same space. The clones highlighted all have a statistically good overlap with Zac22_250.
When using a tolerance of 0 and a cutoff of 1e-09, the clones assemble into six perfect contigs. With a tolerance of 0, the only problem that can occur is if two different bands of the same size are physically near each other and are computed to be the same band. This did not occur as the out-of-order values are all zero. With a tolerance of 7, there will be many more bands incorrectly called the same due to being within the tolerance. This is illustrated in the third entry that uses a tolerance of 7 and has two out-of-order clones. Because the tolerance is used in the equation above, it influences the cutoff; when using a tolerance of 7 and a cutoff of 1e-06, the clones assemble into six almost perfect contigs. The rest of the tables use a tolerance of 7.
The simulation produces either migration rates or size values. The first three entries use migration rates. The last two entries are the same set of clones except the sizes are used instead of the migration rate. With sizes, a variable tolerance is used (in reality, the tolerance is dependent on the size of the fragment; a table of tolerance values can be specified in FPC).
Table 2 shows results of using markers versus not using them. The data set is a 15× coverage of the six sequences; there are no gaps, and there is a marker approximately every 20 kb. It also lists the number of splits and merges needed to reduce it to six contigs. The first entry uses a 1e-06 cutoff and results in nine contigs, two of which have Q clones. The following gives a detailed account of reducing the nine contigs to six contigs. (1) For the first contig with Q clones, a cutoff of 1e-07 assembled the contig into two CB maps with no Q clones. Using a 1e-05 cutoff with the OKALL, the CB maps could not be ordered so were split into two contigs. The second contig with Q clones also splits into two contigs; as a result, there are 11 contigs. (2) The Ends → Ends routine was used to find contigs to merge. When run with a 1e-05, two pairs of contigs were listed as potential merges, and both sets of contigs were merged. Another two contigs were identified for merging by using a 1e-04 cutoff. The four merges resulted in seven contigs. (3) To find the last two contigs to merge, a cutoff of 1e-03 was used with the Ends → Ends function. It listed 12 clones from the end of one contig that statistically overlapped with clones at the end of another contig. The clone pairs shared between 14 and 18 bands each, and the bands assembled almost perfectly. One contig was from sequence 7, the other from sequence 21; this would have been an incorrect merge. To list the correct contig pair to merge, a cutoff of 3e-03 was needed which also listed many incorrect contig pairs. Note that the cutoffs used for merging in this example are not sufficiently stringent and would probably only be used in reality with supporting data such as markers.
Table 2.
Markers and Interactive Operations
| Cutoff | Markersa | Ctgsb | F−c | F+ | Chi | Qs | Order | Split | Merge |
|---|---|---|---|---|---|---|---|---|---|
| 1e-06 | No | 9 + 1 | 7 − 1 | 3 | 2 | 146 (2) | 45, 17, 0 | 2 | 5 |
| 1e-06 | Yes | 5 + 0 | 2 − 1 | 3 | 2 | 283 (2) | 116, 74, 0 | 2 | 1 |
| 1e-07 | No | 16 + 1 | 13 − 2 | 0 | 0 | 0 (0) | 16, 1, 0 | 0 | 10 |
| 1e-07 | Yes | 7 + 0 | 2 − 1 | 0 | 0 | 0 (0) | 19, 3, 0 | 0 | 1 |
(Ctgs) contigs; (F−) false-negative; (F+) false-positive; (Qs) questionable.
The data set has perfect data, 15× random coverage, 2098 clones with lengths between 145 and 185 kb, and a marker every 20 kb and uses a tolerance of 7.
The CpM table uses the (marker, cutoff) pairs of (1 1e–05) (2 1e–04) (3 1e–03).
The +n is the number of singletons.
The −n indicates the number of bridging clones, i.e., clones that cover F− overlaps and prevent breaks.
Table 3 shows the effect of varying the coverage of the 110-Mb sequence by using clones of 145–185 kb in length. As would be expected, as the coverage increases, the number of F− overlaps decreases, and hence, the number of contigs decreases. As markers are added to the assembly, the number of F−'s further decreases. When considering F+ overlaps, we also can consider the number of clones and regions that have F+ overlaps. For example, in the 10× coverage with a 1e-08 cutoff, there are five clones in one region that overlap with seven clones in another region. There is a clone in a third region that overlaps a clone in the second region, so there are 13 overlapping clones in three regions for total of 29 F+ overlaps. For the 20×, 30×, 40×, and 50× coverages, there are 33, 54, 72, and 93 clones with F+ overlaps, respectively; the clones are from the same nine regions in all cases. When a 1e-10 is used, each coverage has two regions that overlap, and they are the same two regions in all cases.
Table 3.
Varying Coverage
| Cov | Gap | Clones | Bury (%)a | Largest (order)b | F+ clonesc |
|---|---|---|---|---|---|
| 10× | 18 | 6676 | 62 | 506 (4, 2, 0) | 13 (3), 11 (2) |
| 20× | 3 | 13353 | 74 | 3529 (17, 2, 0) | 33 (9), 18 (2) |
| 30× | 0 | 20030 | 79 | 5306 (15, 2, 0) | 54 (9), 32 (2) |
| 40× | 0 | 26707 | 83 | 7110 (15, 2, 0) | 72 (9), 44 (2) |
| 50× | 0 | 33384 | 85 | 9534 (37, 4, 0) | 93 (9), 57 (2) |
| Cov | 1e-08 | 1e-10 | 1e-10, CpMd | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ctgs | F− | F+ | Qs | Ctgs | F− | F+ | Qs | Ctgs | F− | Qs | |
| 10× | 135 | 144 | 29 | 30 (1) | 193 | 216 | 28 | 2 (1) | 92 | 80 | 4 (1) |
| 20× | 48 | 60 | 76 | 190 (3)e | 95 | 106 | 65 | 2 (1) | 24 | 25 | 18 (1) |
| 30× | 22 | 28 | 262 | 1229 (4) | 51 | 59 | 228 | 45 (1) | 9 | 11 | 81 (1) |
| 40× | 15 | 19 | 524 | 674 (3) | 34 | 38 | 465 | 62 (1) | 5 | 7 | 365 (1) |
| 50× | 12 | 15 | 941 | 1086 (3) | 27 | 28 | 778 | 204 (1) | 4 | 3 | 724 (1) |
(Cov) coverage; (CpM) cutoff plus marker; (Ctgs) contigs; (F−) false-negative; (F+) false-positive; (Qs) questionable.
The data set has perfect data, 145–185 kb clone lengths, a marker every 20 kb, and a tolerance of 7.
Percentage buried in which a buried clone has 90% or more shared bands with the parent clone.
The largest contig with no Q clones and its order triple.
The number of clones that have an F+ overlap for the 1e-08 and 1e-10 cutoff, respectively. In parentheses is the number of regions that have F+ overlaps. The number of F+ overlaps is the same for 1e-10 with or without markers.
The CpM rules are (1 1e-08) (2 1e-07) (3 1e-06).
There are four chimeric contigs, but the fourth does not have Q clones. The three F+'s occur at the end of two contigs and bridge the contigs with little difficulty, i.e., F+'s at the ends of contigs causing an incorrect merge may result in a number of extra bands and gaps that looks the same as error in the data.
For the 20× coverage and 1e-08 cutoff, there is a contig with F+'s but no Q clones. The F+'s occur at the ends of two contigs causing them to incorrectly merge into one contig, and the bands in between could be arranged with an acceptable amount of extra bands and gaps. The CB algorithm only labels a clone as a Q clone if 50% of its bands will not order consistently; this low number is to accommodate the usual amount of error. That is, perfect data with F+'s at the ends of regions have a similar appearance as data with error.
Table 4 shows the results of different clone lengths on a 20× coverage of the 110-Mb sequence. The data set of 40 kb clones has significantly more gaps and F− overlaps resulting in many contigs and singletons. From the 5048 singletons in the 1e-08 assembly, 4522 have less than five bands, which will not score as an overlap with any clone at this cutoff. Of the 12,184 singletons in the 1e-10 assembly, 11,537 have less than seven bands and will not score as an overlap with any clone. There are many occurrences of a clone being a singleton, yet its absence does not cause two contigs, i.e., the dual F− overlaps are bridged by two other clones; this is unusual in longer clones, where a clone may have a F− with one adjacent clone but not both. The number of contigs reduces as the length of the clones increases since longer clones require less overlap; e.g., with a 1e-10 cutoff, two clones with ten bands each need 90% shared bands to qualify as an overlap, whereas two clones having 30 bands each need 63% shared bands to qualify as an overlap. The data set with clones of length 100 ± 60 kb (i.e., 40–160 kb) approximately simulates mixing cosmids and BACs. There are many more F− overlaps because a clone with ten bands never scores below a 1e-08 when compared with a clone with 40 bands, and hence, never appears to overlap. In all the simulations based on length, the out-of-order triplet remains low, with a slight increase when the variation in sizes increases (data not shown).
Table 4.
Varying Length
| Len +/− | (kb) | Gap | Clones | Na | 1e-08 | 1e-10 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ctgs | F− | F+ | Ctgs | F− | F+ | |||||
| 40 | 10 | 455 | 55084 | 10 | 3k + 5kb | 8.5k − 630 | 207 | 3.5k + 12k | 16k − 1k | 98 |
| 100 | 20 | 11 | 22033 | 26 | 212 + 27 | 295 − 62 | 145 | 358 + 71 | 531 − 122 | 54 |
| 100 | 60 | 62 | 22033 | 26 | 217 + 244 | 1.5k − 1k | 292 | 380 + 668 | 3k − 1.5k | 130 |
| 165 | 20 | 3 | 13353 | 43 | 48 + 3 | 60 − 8 | 76 | 95 + 4 | 106 − 10 | 65 |
| 165 | 50 | 4 | 13353 | 43 | 38 + 1 | 72 − 36 | 94 | 72 + 4 | 144 − 71 | 58 |
| 165 | 80 | 4 | 13353 | 43 | 33 + 5 | 171 − 133 | 92 | 55 + 13 | 319 − 251 | 28 |
| 200 | 50 | 0 | 11016 | 52 | 26 + 0 | 37 − 13 | 77 | 46 + 0 | 71 − 27 | 56 |
(Ctgs) contigs; (F−) false-negative; (F+) false-positive.
The data set has perfect data, 20× coverage, and uses a 7 tolerance.
Average number of bands per clone.
A Nk is approximately N times 1000 clones.
Table 5 varies the error and uncertainty in the fragments for a 15× coverage of the 110-Mb sequence by using clones of 145–185 kb in length. The error conditions used are as follows. (1) End fragments >600 bp were not removed. (2) To simulate the inclusion of vector bands, we used a distribution of 85% for 1387, 33% for 3695, and 33% for 3766. (3) An F+ rate of 4%. (4) An F− rate of 4%. (5) To simulate uncertainty in the rate measurement, we used a distribution of (16, 20, 18, 15, 12, 9, 6, 4) was used for tolerances of 0 through 7, respectively. The table shows that none of the types of error by themselves is significant. The worst out-of-order triples are with the addition of ends (548, 93, 0) and tolerance (249, 224, 0), whereas the data set with all the types of error has a triplet of (1520, 1792, 0). With all the error, the scores for the CB maps decreases, where the score is the total number of consecutive bands minus the number of extra bands and gaps (i.e., o's). The score is displayed next to the number of Qs in the CB map (see Fig. 2). The scores for these CB maps fall as low as 0.86, and the number of extra bands per clone typically ranges between four and ten; consequently, the number of out-of-order pairs increases significantly. The average difference between starting coordinates for the out-of-order clones is 1 for the data sets without error and 2 for data set with multiple types of error. Recall that in the previous section a test was run on multiple gels to estimate the tolerance and number of extra bands; this test was run on the clones with the same end points from the data set with error and gave similar tolerances and the same percentage of extra bands.
Table 5.
Varying Error
| Error | Ctgs | Bury (%) | F− | F+ | Qs | Order |
|---|---|---|---|---|---|---|
| None | 145 + 9 | 69 | 156 − 9 | 36 | 4 (1) | (66, 12, 0) |
| Ends | 156 + 10 | 63 | 182 − 20 | 36 | 4 (1) | (548, 93, 0) |
| Vectora | 133 + 9 | 67 | 153 − 17 | 38 | 24 (1) | (212, 76, 0) |
| 4% F+ | 158 + 10 | 67 | 176 − 14 | 36 | 0 (0)b | (258, 59, 0) |
| 4% F− | 153 + 15 | 68 | 191 − 30 | 36 | 0 (0)b | (521, 78, 0) |
| Tolc | 174 + 17 | 51 | 257 − 73 | 30 | 0 (0)b | (249, 224, 0) |
| Alld | 276 + 43 | 9 | 547 − 229 | 21 | 13 (5) | (1520, 1792, 0) |
(Ctgs) contigs; (F−) false-negative; (F+) false-positive; (Qs) questionable; (Tol) tolerance.
The data set has a 15× coverage, 10,015 clones, seven gaps, 145–185 kb lengths, one chimeric contig, and uses 7 tolerance and a 1e-10 cutoff.
Adds a band of 1387, 3695, and 3766 to 85%, 33%, and 33% of the clones, respectively.
The F+'s occurred at the ends of two contigs, so the incorrectly merged contig assembled without causing a stack, i.e., Q clones.
Adds or subtracts a tolerance of 0 through 7 for (16, 20, 18, 15, 12, 9, 6, 4) of the bands, respectively.
Uses all of the previous error.
The previous tables are based on one possible set of clones picked from a pool. For Table 5, a second set of clones was picked from the pool of 299k clones (data not shown); the data sets have a F+ number >100 instead of ∼30 due to a large number of clones being randomly selected from the F+ regions, but the number of contigs is similar and the relations between the data sets are the same, e.g., all the error resulted in more than three times the number of out-of-order pairs.
Table 6 shows the results from four different random selections of 16,692 clones from a pool of 449k clones. It has a 25× coverage of the 110-Mb sequence; the clones have lengths of 130–190 kb, one marker per 45 kb, and contains all the error shown in Table 5 except for the vector bands. The number of F+'s varies from 59 to 123, and the number of contigs varies from 38 to 44. The average number of contigs is 41 with only one two clone contig. This is lower than we observe in reality, e.g., a complete build of the 25× coverage of clones from the chromosome 9 FPC database (not all the clones are from this chromosome, only ∼11,857 are from anchored contigs) results in 1430 contigs with 720 two clone contigs. The simulated data sets do not contain chimeric clones or bad fingerprints, the clone lengths are within a fixed range, there are no uncloneable regions or incorrect hybridization, etc. This simulation is representative of only ∼3% of the genome and probably does not include difficult regions. Therefore, this simulation is not representative of all situations, but it does provide some rough guidelines on how FPC will perform given data of varying parameters and shows the benefit of reducing error in the data. For a general equation, see Lander and Waterman (1988).
Humanmap plus Simulated Digested Sequence
The size files for a 7× coverage of the humanmap, i.e., 134,895 clones, were assembled using a 1e-10 cutoff, which resulted in 18,458 contigs. The simulated clones from Table 1, i.e., the 5× coverage using 80% overlap of the six sequences described in the previous section, were added to the database. Referring to the simulated clones as ZACs, the results from executing the IBC are as follows. (1) Nineteen contigs have ZAC clones, and the ZAC clones caused 106 original contigs to be merged into the 19 contigs. That is, many overlapping BACs do not qualify as an overlap because they do not have enough shared bands. One or more ZAC clones overlap BACs from the ends of two different original contigs causing them to join, albeit by a small amount. (2) Within the 19 contigs, there are 15 gaps between the original BAC contigs. The 15 gaps have sizes 4, 10, 10, 10, 14, 18, 34, 38, 42, 42, 46, 49, 57, 63, 74 where the gap sizes are the number of bands between BAC contigs. Figure 5 shows one of the gaps. (3) The ZAC clones are in a perfect order even though they are assembled with the less perfect BAC clones. None of the contigs has ZAC clones from two different sequences.
Figure 5.

Contig from humanmap and sequence. The ZAC clones are highlighted in blue. The two clones highlighted in cyan are the nearest clones from two BAC contigs, as is indicated in the clone text windows, where the oldctg field is the clone's contig previous to the last IBC.
DISCUSSION
The ordering of clones based on restriction fragment data is a computationally intractable problem in the sense that the only way to guarantee the correct solution would be to try all possible solutions which would take an unacceptable amount of time. As with so many computational genomic problems, the solution is both statistical and approximate and requires a threshold. For example, sequence comparison, sequence assembly and gene identification all require a cutoff to minimize the F+ and F− results. Fingerprint assembly requires a cutoff to reduce the F+ and F− overlaps. The optimal cutoff will vary based on the fingerprint method, data acquisition, image analysis package, number of bands, and the amount of error and must be optimized for a given set of experimental parameters. Some tolerance/cutoff variations are 7/1e-14 (Klein et al. 2000), 5/1e-06 (Ding et al. 1999), 7/1e-09 (Zhu et al. 1999), and 8/1e-09 (Marra et al. 1997), and we use a tolerance 7 cutoff 1e-10 for complete digest BAC data for human chromosomes 9, 10, and 13.
FPC orders clones by building a CB map and aligning the clones to the map. Each band in the CB map is the consensus of two or more bands, so all regions must have at least a 2× coverage. Any region that has a 1× coverage will be condensed as the region will not be represented in the CB map, and the bands from the 1× region will be listed as extra bands. The CB map is the same as a partially ordered restriction map when using complete digest data. If the data has error in it, the map probably will have error in it. Occasionally, the map will have error even when the data are perfect; this occurs when two bands are called the same but are actually different. The CB algorithm does not try to correct these errors because it was designed to give a fast approximate ordering for data that have varying amounts of error. Given a set of clones with no error and no F+ overlaps, the simulations show that FPC performs perfectly except for an occasional out-of-order pair, i.e., the first coordinates of two adjacent clones are in the wrong order. The largest such contig assembled with a tolerance of 7 has 9534 clones and 37 out-of-order pairs. The largest contig assembled with a tolerance of 0 has 4738 clones and two out-of-order pairs. In both cases, the starting coordinates of out-of-order pairs differ by one.
The simulations show that a large number of Q clones are a strong indication of one or more F+ overlaps. If two different parts of the genome are mapped to the same space, there is no linear order and often results in a stack of clones. A second cause of Q clones is error; as it increases, the number of Q clones increases. In reality, it is hard to determine the cause of the Q clones. The mix of problems makes this difficult to sort out interactively, and if there are multiple F+ overlaps, it is computationally an intractable problem to determine what overlaps to remove. The approach used here is to create a set of good CB maps (few Q clones) and then order the CB maps. The absence of Q clones does not guarantee that there are no F+ overlaps due to the fact that if they occur at the ends of regions, the clones may assemble with no more gaps and extra bands than would occur from data with error.
The simulation of the clones with lengths of 40 kb compared with 165 kb supports the fact that assembling cosmid clones results in too many contigs and gaps, whereas using this method with BACs is very reasonable. As would be expected, adding more clones or markers decreases the number of contigs, and reducing the error improves the quality of the map.
In FPC, the clones are partially ordered, and as a result the markers are partially ordered. The location of a marker is above the deepest stack of clones for which it is positive. The ability to have both markers and clones in FPC provides an easy way to use them in conjunction for assembly, which reduces the number of manual merges needed and aids making decisions for the remaining manual merges. A third type of data, the global framework markers, allow contigs to be anchored and ordered. Our FPC databases contain all three types of data plus the latest status of the sequence ready clones. Nightly, FPC exchanges information with chromosome-specific AceDBs and the tracking database. Therefore, FPC always provides an updated snapshot of the sequence ready contigs for chromosomes 9, 10, and 13.
METHODS
RHMAPPER and the Z Extensions
For chromosomes 9, 10, and 13, we use the order of ESTs established for the Genebridge4 panel of the 1998 International Gene Map (Deloukas et al. 1998), as displayed at http://www.sanger.ac.uk/RHserver. The ordering of the ESTs for the Genebridge4 panel was computed as follows. The International Consortium scored ∼30000 ESTs by radiation hybrid mapping using the Genebridge4 panel. The data were entered into the RHdb database (http://www.ebi.ac.uk/RHdb), which we downloaded and assembled. The genetic order is used as the framework with some additional markers to fill in large gaps. The RHMAPPER software (Slonim et al. 1997) was used to bin additional markers within the framework; these are referred to as placement markers. The Z extensions (Soderlund et al. 1998) were used to compute the totally linked markers, bin only the canonical marker for each group, and automate the mass binning of thousands of markers.
Rates to Sizes
A gel has marker lanes of known fragments in between the lanes of digested clones. Image uses this information to determine the migration rates and convert the rates to sizes as follows (S. Leonard, pers. comm.). A set of standard migration rates and corresponding fragment sizes are known for the bands that appear in each marker lane. A mapping then is established for each marker lane between the standard values and actual position on the gel. For nonmarker lanes, the mapping is calculated by linear interpolation of the mapping for the neighboring marker lanes. The migration rate and fragment size for any band then may be calculated by interpolating the known values at the band position by using a Bi-cubic spline. For fragment size, it is the log of the fragment size that actually is interpolated.
This maps a small set of rates into a much larger set of sizes, which explains the high number of duplicates (J. Mullikin, pers. comm.). The maximum number of rate values is dependent on the length of the gel, which for the set of clones described in this document is 1000. The 1000 values are scaled to 4000 (early versions of FPC displayed all gels using length 4000 kb) and mapped into ∼32 kb sizes. The value used for the gellen (see equation above) is the default 3300.
FPC V4.7
CB Algorithm
The CB algorithm for ordering clones is basically the same as described in Soderlund et al. (1997). It builds a CB map as shown in Figure 2. To summarize the algorithm:
All clones in the input set are compared, and the coincidence score is computed. If two clones have a score below the user-supplied cutoff, the clones are treated as overlapping clones. In the following discussion, clones that have a good overlap score are called “friends.”
CB maps are built so that each map is a set of transitively overlapping clones. As clones are integrated into the map, an approximate partially ordered CB map simultaneously is computed. When a clone is added to the map, it is aligned to the CB map within the region of its friends already in the map. If a good alignment cannot be found, it is marked as a Q (questionable) clone and assigned coordinates within the region (regardless of the fact that it does not align). This means that the resulting contig generally will have all friends overlap regardless of the quality of data. If clones are determined incorrectly to be friends, they will incorrectly overlap.
The original algorithm had a “shuffle” step to reorder bands as the initial ordering can be faulty. The current algorithm tries N different orderings and uses the best. This is faster, uses less memory, and gives better results. By default, N is ten.
The current algorithm uses the CpM table if the user requests. Two clones are friends if they share x markers and their cutoff is below the cutoff corresponding to the entry in the table for x markers.
The input set of clones can be all the singletons in the database (Build Contig on the Main Analysis window) or for a contig (Calc on the Contig Analysis window).
Incremental Build Contigs (IBC)
If a set of contigs has been built and then more clones have been added to the FPC database, the IBC algorithm will add clones to contigs and merge contigs retaining the transitively overlapping clone property of the database. That is, if there has been no user editing of the contigs to split or merge them, then the results of the IBC is exactly the same as destroying all contigs and performing a complete build on all clones in the data set. The benefits of the IBC versus executing a complete build are:
It is faster on a large data set with a relatively small number of new clones. To add 1733 new clones to a database of 11,096 clones took 6 minutes with the IBC, whereas it took 20 minutes for the complete build. These times are CPU on a Dec Alpha V4.0D 500 Mhz with 128 Mb RAM and 410 Mb swap.
It displays the results of the additions and merges.
Contig numbers do not change unless a merge occurred.
If a user has manually split or merged contigs, these changes will be retained. The one exception is if a contig was split, and then a new clone rejoins the two contigs.
The disadvantage of the IBC is that it does not take into account new gels for existing clones (see the User's Guide for more detail).
If markers are incrementally added to the FPC database, then it is advantageous to have the IBC routine consider all clones that have new markers to see if any of these causes a join. If markers are added via the file operation “Replace markers” or “Merge markers” (see FPC User's Guide), each marker added to a clone has its state set to “new.” If the CpM table is on and the IBC is run, clones with new markers are compared against all other clones and appropriate joins and additions are made.
Framework
An ordered set of markers can be entered into FPC via the “Replace framework” (see FPC User's Guide). The markers are shown as anchors along the bottom of each contig and can be listed in the project window along with the contigs they hit. The markers can be from radiation hybrid mapping, or ordered by a program such as SAM (System for Assembling Markers; Soderlund and Dunham 1995), or any other means for ordering markers.
Get_GSC
This script takes as input a set of clones, and maintains an index file of clones already extracted from the human FPC database files. It outputs band files and size files for all clones that are in both the input file and the humanmap file and are not in the index file. The file fpc/fpp/trace.c has the location of the humanmap gel directory hardwired into it so that if the gel is not in the local Gel directory, it looks in the shared directory. The humanmap tar file was obtained from http://genome.wustl.edu/gsc/human. It contains the band, size, and gel files for all fingerprinted clones and an assembled FPC file.
In addition, we use the humanmap files for aiding in the mapping of chromosomes 1, 6, 9, 10, 13, 20, and X. We use it to build “walking FPCs” which takes as input a file of chromosome-specific clones and builds the FPC input files for all the clones in each contig that contain a clone from the input file; these databases are used to find clones for walking. We add simulated sequence to the Human_sizes (built with sizes instead of rates) to determine what contigs are hit. And we add our PACs and marker data to the humanmap to find additional merges to select clones for sequencing.
Availability
The FPC software and manual is available at http://www.cs.clemson.edu/∼cari/fpc.html. Information on the maps can be found at http://www.sanger.ac.uk/HGP/ChrN in which N is chromosome 9, 10, or 13. Contact Sean Humphray (sjh@sanger.ac.uk) about chromosome 9, Lisa French (lon@sanger.ac.uk) about chromosome 10, and Andrew Dunham (ad1@sanger.ac.uk) about chromosome 13.
Acknowledgments
We thank David Bentley and all of the physical mapping group for ideas contributing to the design of FPC and building of the chromosome maps. We thank Richard Durbin, LaDeana Hillier, Jim Mullikin, and Gernot Presting for comments on the manuscript. Carol Scott wrote the script to extract data from AceDB and enter data into AceDB and set up the interactions between the databases as cronjobs. LaDeana Hillier and Ken McDonald from the Genome Sequencing Center in St. Louis wrote the original simulation program. The addition of simulated digest data to the humanmap was part of an experiment performed with David Bentley and Carol Scott. Simon Gregory, Gareth Howell, and Andy Mungall have contributed ideas to the use of the humanmap. The Sanger Centre is supported by the Wellcome Trust.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL cari@cs.clemson.edu; FAX (864) 656-4293.
Article published online before print: Genome Res., 10.1101/gr.137500.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.137500.
REFERENCES
- Cao Y, Kang H, Xu X, Wang M, Dho S, Huh J, Lee B, Kalush F, Bocskai D, Ding Y, et al. A 12-MB complete coverage BAC contig map in human chromosome 16p13.1-p11.2. Genome Res. 1999;9:763–774. [PMC free article] [PubMed] [Google Scholar]
- Carrano A, Lamerdin J, Ashworth L, Watkins B, Branscomb E, Slezak T, Raff M, de Jong P, Keith D, McBride L, et al. A high-resolution, fluorescence-based, semiautomated method for DNA fingerprinting. Genomics. 1989;4:129–136. doi: 10.1016/0888-7543(89)90291-7. [DOI] [PubMed] [Google Scholar]
- Coulson A, Sulston J, Brenner S, Karn J. Towards a physical map of the genome of the nematode C. elegans. Proc Natl Acad Sci. 1986;83:7821–7825. doi: 10.1073/pnas.83.20.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deloukas P, Schuler GD, Gyapay G, Beasley EM, Soderlund C, Rodriguez-Tome P, Hui L, Matise TC, McKusick KB, Beckmann JS, et al. A physical map of 30,000 human genes. Science. 1998;282:744–746. doi: 10.1126/science.282.5389.744. [DOI] [PubMed] [Google Scholar]
- Ding Y, Johnson M, Colayco R, Chen Y, Melnyk J, Schmitt H, Shizuya H. Contig assembly of bacterial artificial chromosome clones through multiplexed fluorescent-labeled fingerprinting. Genomics. 1999;56:237–246. doi: 10.1006/geno.1998.5734. [DOI] [PubMed] [Google Scholar]
- Durbin R, Thierry-Mieg J. The AceDB Genome Database. In: Suhai S, editor. Computational Methods in Genome Research. New York: Plenum Press; 1994. pp. 7821–7825. [Google Scholar]
- Gregory S, Howell G, Bentley D. Genome mapping by fluorescent fingerprinting. Genome Res. 1997;7:1162–1168. doi: 10.1101/gr.7.12.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins R, Nelson C, Berman B, Laverty T, George R, Ciesiolka L, Naeemuddin M, Arenson A, Durbin J, David R, et al. A BAC-based physical map of the major autosomes of Drosophila melanogaster. Science. 2000;287:2271–2274. doi: 10.1126/science.287.5461.2271. [DOI] [PubMed] [Google Scholar]
- Humphray, S., Knaggs, S., and Ragoussis, J. Contiguation of bacterial clones. In Genomic Protoc. (ed. R. Elaswwarapu and M. Satrkey), Human Press Inc., Towawa, NJ. (In press).
- Klein P, Klein R, Cartinhour S, Ulanch P, Dong J, Obert J, Morishige D, Schleuter S, Childs K, Ale M, et al. A high-throughput AFLP-based method for constructing integrated genetic and physical maps: Progress towards a sorghum genome map. Genome Res. 2000;10:789–807. doi: 10.1101/gr.10.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E, Waterman M. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics. 1988;2:231–239. doi: 10.1016/0888-7543(88)90007-9. [DOI] [PubMed] [Google Scholar]
- Mahairas G, Wallace J, Smith K, Swartzell S, Holzman T, Keller A, Shaker R, Furlong J, Young J, Zhao S, et al. Sequence-tagged connectors: A sequence approach to mapping and scanning the human genome. Proc Natl Acad Sci. 1999;96:9739–9744. doi: 10.1073/pnas.96.17.9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M, Kucaba T, Dietrich N, Green E, Brownstein B, Wilson R, McDonald K, Hillier L, McPherson J, Waterston R. High throughput fingerprint analysis of large-insert clones. Genome Res. 1997;7:1072–1084. doi: 10.1101/gr.7.11.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M, Kucaba T, Sakhon M, Hillier L, Martienssen R, Chinwalla A, Crockett J, Fedele J, Grover H, Gund C, et al. zA map for sequence analysis of the Arabidopsis thaliana genome. Nat Genet. 1999;22:265–275. doi: 10.1038/10327. [DOI] [PubMed] [Google Scholar]
- Mungall A, Humphray S, Ranby S, Edwards C, Heathcott R, Clee C, Holloway E, Peck A, Harrision P, Green L, et al. From long range mapping to sequence ready contigs on human chromosome 6. DNA Seq– J Seq Mapp. 1997;8:151–154. doi: 10.3109/10425179709034066. [DOI] [PubMed] [Google Scholar]
- Niederfuhr A, Hummerich H, Gawin B, Boyle S, Little P, Gessler M. A sequence-ready 3-Mb PAC contig covering 16 breakpoints of the wilms turmor/anirida region of human chromosome 11p13. Genomics. 1998;53:155–163. doi: 10.1006/geno.1998.5486. [DOI] [PubMed] [Google Scholar]
- Olson M, Dutchik J, Graham M, Brodeur G, Helms C, Frank M, MacCollin M, Scheinman R, Frank T. Random-clone strategy for genomic restriction mapping in yeast. Proc Natl Acad Sci. 1986;83:7826–7830. doi: 10.1073/pnas.83.20.7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osoegawa K, Tateno M, Woon P, Frengen E, Mammoser A, Catanese J, Hayashizaki Y, de Jong P. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 2000;10:116–128. [PMC free article] [PubMed] [Google Scholar]
- Slonim D, Kruglyak L, Stein L, Lander E. Building human genome maps with radiation hybrids. J Comput Biol. 1997;4:487–504. doi: 10.1089/cmb.1997.4.487. [DOI] [PubMed] [Google Scholar]
- Soderlund C, Dunham I. SAM: A system for iteratively building marker maps. CABIOS. 1995;11:645–655. doi: 10.1093/bioinformatics/11.6.645. [DOI] [PubMed] [Google Scholar]
- Soderlund C, Gregory S, Dunhum I. Sequence ready clones. In: Bishop M, editor. Guide to Human Genome Computing. San Diego, CA: Academic Press; 1997. pp. 151–177. [Google Scholar]
- Soderlund C, Longden I, Mott R. FPC: A system for building contigs from restriction fingerprinted clones. CABIOS. 1997;13:523–535. doi: 10.1093/bioinformatics/13.5.523. [DOI] [PubMed] [Google Scholar]
- Soderlund C, Lau T, Deloukas P. Z extensions to the RHMAPPER package. Bioinformatics. 1998;14:538–539. doi: 10.1093/bioinformatics/14.6.538. [DOI] [PubMed] [Google Scholar]
- Soeda E, Hou D, Osoegawa K, Atsuchi Y, Yamagata T, Shimokawa T, Kishida H, Soeda E, Okano S, Chumakov I, et al. Cosmid assembly and anchoring to human chromosome 21. Genomics. 1995;25:73–84. doi: 10.1016/0888-7543(95)80111-x. [DOI] [PubMed] [Google Scholar]
- Stallings R, Torney D, Hildebrand C, Longmire J, Deaven L, Jett J, Doggett N, Moyzis R. Physical mapping of human chromosomes by repetitive sequence fingerprinting. Proc Natl Acad Sci. 1990;87:6218–6222. doi: 10.1073/pnas.87.16.6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J, Mallet F, Staden R, Durbin R, Horsnell T, Coulson A. Software for genome mapping by fingerprinting techniques. CABIOS. 1988;4:125–132. doi: 10.1093/bioinformatics/4.1.125. [DOI] [PubMed] [Google Scholar]
- Sulston J, Mallett F, Durbin R, Horsnell T. Image analysis of restriction enzyme fingerprints autoradiograms. CABIOS. 1989;5:101–132. doi: 10.1093/bioinformatics/5.2.101. [DOI] [PubMed] [Google Scholar]
- Taylor K, Hornigold N, Conway D, Williams D, Ulinowski Z, Agochiya M, Fattorini P, de Joug P, Little P, Wolfe J. Mapping the human Y chromosome by fingerprinting cosmid clones. Genome Res. 1996;6:235–248. doi: 10.1101/gr.6.4.235. [DOI] [PubMed] [Google Scholar]
- Venter JC, Smith HO, Hood L. A new strategy for genome sequencing. Nature. 1996;381:364–366. doi: 10.1038/381364a0. [DOI] [PubMed] [Google Scholar]
- Zhu H, Blackmon B, Sasinowski M, Dean R. Physical map and organization of chromosome 7 in the rice blast fungus, Magnapothe grisea. Genome Res. 1999;9:739–750. [PMC free article] [PubMed] [Google Scholar]