

Abstract
Glucocorticoids have been used for decades in the treatment of brain tumor patients and belong to the most powerful class of agents in reducing tumor-associated edema and minimizing side effects and the risk of encephalopathy in patients undergoing radiation therapy. Unfortunately, corticosteroids are associated with numerous and well-characterized adverse effects, constituting a major challenge in patients requiring long-term application of corticosteroids. Novel anti-angiogenic agents, such as bevacizumab (Avastin®), which have been increasingly used in cancer patients, are associated with significant steroid-sparing effects, allowing neuro-oncologists to reduce the overall use of corticosteroids in patients with progressive malignant brain tumors. Recent experimental studies have revealed novel insights into the mechanisms and effects of corticosteroids in cancer patients, including modulation of tumor biology, angiogenesis and steroid-associated neurotoxicity. This article summarizes current concepts of using corticosteroids in brain cancer patients and highlights potential pitfalls in their effects on both tumor and neural progenitor cells.
Keywords: anti-angiogenesis, brain cancer, cancer stem cells, corticosteroids, dexamethasone, giloma, neural stem cells, neurotoxicity
Traditional concepts of steroid use in brain cancer patients
Corticosteroids have been widely used in cancer therapy and are particularly beneficial in brain cancer patients with significant peritumoral edema and associated neurological deficits [1,2]. Despite their longstanding use and tremendous impact in clinical oncology over several decades, little is known about the mechanisms by which corticosteroids exert their biological and clinical effects.
Steroids were introduced into the care of brain cancer patients nearly 50 years ago based on their powerful effects on tumor-induced edema. Ingraham pioneered the use of cortisone to treat postoperative cerebral edema in neurosurgical patients in 1952 [3], and Kofman first used prednisone for peritumoral edema from brain metastases in 1957 [4]. More than 40 years ago, it was demonstrated that dexamethasone effectively alleviated cerebral edema due to brain tumors [1], which eventually revolutionized care of brain tumor patients. Interestingly, Galicich’s work was intended to target tumor cell growth based on experimental evidence that large doses of dexamethasone were able to inhibit the growth of experimental brain tumors [1]. Dexamethasone, which was first synthesized in 1958, to date has remained the most favorable drug for brain cancer patients, and offered a unique benefit in the design of these early trials owing to its low index of sodium and water retention compared with other corticosteroids available at that time [5].
Subsequently, prednisone, prednisolone, dexamethasone and methylprednisolone have demonstrated antineoplastic effects against hematologic malignancies [6]. Glucocorticoids have also proven to be beneficial in controlling tumor-associated pain, limiting nausea and vomiting, and improving appetite in cancer patients [7]. Importantly, steroids have been commonly used in patients with carcinomatous meningitis and CNS lymphoma [8].
Dexamethasone has become the drug of choice in neuro-oncology, in part owing to its long half-life, low mineralocorticoid activity, and a relatively low tendency to induce psychosis [9]. Interestingly, and despite its common use, there have only been a few prospective clinical trials to determine the optimal dose of dexamethasone in brain cancer patients [10–12]. The efficacy of steroids in reducing the tumor-associated edema has been well demonstrated [13–15]. Approximately 70% of patients with cerebral metastases symptomatically improve after starting steroids. Administration of steroids 1–2 days prior to an elective surgical procedure has the potential to reduce edema formation and to improve clinical condition by the time of the craniotomy [5]. Although clinical response (as measured by a reduction in the number of intracranial pressure plateau waves) can occur within 24 h, the pressure may not be consistently lower until 2–4 days after initial dosing [13,16].
Typically, large doses of 10–20 mg of dexamethasone are given intravenously at initial presentation of patients with acute neurological symptoms secondary to a brain tumor or spinal cord lesion. The daily dose may be increased up to 100 mg per day, if necessary, although maintenance dosing typically consists of oral or intravenous dexamethasone at a daily dosage of 4–24 mg in divided doses [9,17,18].
There is evidence that lower doses may be equally effective, especially in patients with less severe edema [10]. If beneficial, neurological improvement can be expected within 48 h with this regimen. For asymptomatic patients with radiographic evidence of peritumoral edema, corticosteroids are usually not necessary Although twice-daily dosing is sufficient in cancer patients, it is current inpatient neurosurgical practice to use an every 4–6-h intravenous dosing schedule.
Main molecular mechanisms of corticosteroids
Numerous effects of corticosteroids have been described and can be classified into genomic and nongenomic effects (Figure 1). Genomic effects include transactivation, transrepression and post-transcriptional regulation. Nongenomic effects include activation of signaling cascades and constitute rather rapid downstream effects. Unbound glucocorticoids easily diffuse through the plasma membrane and bind to their associated cytoplasmic receptor. This binding allows for release of heat-shock protein 90 kDa, which exposes two nuclear localization signals and facilitates the movement of the glucocorticoid–receptor complex into the nucleus [19]. In the nucleus, glucocorticoids bind directly to specific DNA sequences called glucocorticoid response elements (GREs) that regulate the transcription of nuclear DNA. Downstream, this affects the production of mRNA and subsequent protein synthesis. Synthetic steroids have a higher affinity (>2–11-fold) for this response element than cortisol [20]. At transcriptional levels, glucocorticoids suppress synthesis of several cytokines and chemokines, such as granulocyte–macrophage colony-stimulating factor; IL-1β, -4, -5, -8 and -10; as well as eotaxin and lipocortin 1, which are involved in the regulation of the inflammatory reaction [21].
Figure 1. Glucocorticoid signaling pathways.

The figure details the genomic and nongenomic pathways of action of a glucocorticoid. Specifically, the pathways that can influence the downstream signaling pathways and determine tumor cell responses have been highlighted. Genomic effects include transactivation, transrepression and post-transcriptional regulation. Nongenomic effects include activation of signaling cascades and constitute rather rapid downstream effects. Unbound glucocorticoids easily diffuse through the plasma membrane and bind to their associated cytoplasmic receptor. This binding allows the release of hsp90, which exposes two nuclear localization signals and facilitates the movement of the glucocorticoid–receptor complex into the nucleus. In the nucleus, the glucocorticoid acts via a hormonal response element that regulates the transcription of nuclear DNA. Downstream, this affects the production of mRNA and resultant protein synthesis. Synthetic steroids have a higher affinity (two- to 11-fold greater) for this response element than cortisol. GCs bind and inactivate NF-κB (1) and prevents free NF-κB (2) from activating inflammatory pathways.
BIM: Bcl-2-interacting mediator of cell death; cGR: Cytoplasmic glucocorticoid receptor; COX2: Cyclooxygenase 2; Dexa: Dexamethasone; GC: Glucocorticoid; GR: Glucocorticoid receptor; GRE: Glucocorticoid response element; hsp90: 90 kDa heat-shock protein; mGR: Membrane glucocorticoid receptor; PI-PLC: Phosphatidylinositol-specific phospholipase C.
Glucocorticoids also interact with other transcription factors such as NF-κB, activating protein 1, p53, CRE-binding protein, signal transducer and the STAT family of activators of transcription (STAT3, STAT5) and others, indirectly influencing the activity of the latter on their own target genes [22]. As described in detail later, among such transcription factors, NF-κB activates many of the inflammatory pathways through its regulation of the production of proinflammatory cytokines and chemokines. Inhibition of NF-κB results in an anti-inflammatory effect [19,23].
Vasogenic edema & corticosteroids
Corticosteroids are usually indicated in any patient with a malignant brain tumor and symptomatic peritumoral edema [24]. Dexamethasone is most commonly used by neuro-oncologists owing to its comparably minimal mineralocorticoid activity, possible lower risk of infection and occurrence of cognitive impairment.
The vasogenic edema surrounding brain tumors contributes significantly to the morbidity experienced by patients. Edema results from the flow of fluid into the extracellular space of the brain parenchyma through an incompetent blood–brain barrier (BBB) [25]. In high-grade gliomas and brain metastases, the BBB is typically disrupted, allowing passage of fluid into the extracellular space. The increased permeability of the BBB is primarily owing to opening of the interendothelial tight junctions, but also due to increased endothelial pinocytosis and endothelial fenestrations [26]. Defective endothelial tight junctions result from deficiency of normal astrocytes (which produce factors required for the formation of a normal BBB) and the tumor cell-associated production of additional factors, such as VEGF [27] and scatter factor/hepatocyte growth factor [28], which increase the permeability of tumor vessels [29]. Moreover, lack of pericyte coverage and other structural defects of the tumor BBB contribute to its increased permeability [30].
It has been suggested that corticosteroids produce their anti-edema effect by reducing the permeability of tumor capillaries (Figure 2) [26,31,32]. As indicated previously, corticosteroids diffuse through the plasma membrane and bind to the cytoplasmic receptor, allowing the steroid–receptor complex to move to the nucleus where it directly affects transcription of genes and also interacts with other transcription factors, mediating nontranscriptional regulation of other signaling cascades (Figure 1) [19]. Corticosteroids decrease endothelial permeability by upregulation of the tight junction (TJ) component occluding in endothelial cells [33] and partly by causing dephosphorylation of occludin and another TJ component, ZO1 [26]. Some reports indicate that corticosteroid-induced repression of NF-κB causes reduction of edema via inhibition of cytokine-induced barrier breakdown and expression of cell adhesion molecules, which mediate T-cell–BBB interactions and excessive leukocyte recruitment across the BBB [34]. Other nontranscriptional regulation mechanisms of action of glucocorticoids involve rearrangement and tight binding of VE-cadherin to the cytoskeleton, leading to decreased permeability of capillaries [35].
Figure 2. MRI effects of decadron.
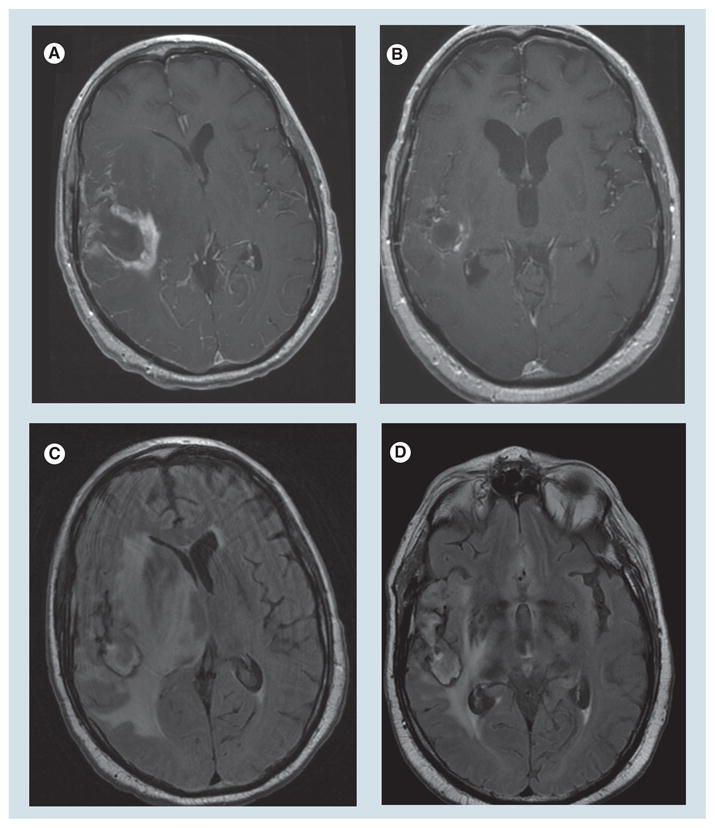
Axial post-gadolinium T1 images (A,B) and axial Fluid attenuated inversion recovery images (C,D) showing poststeroid (B,D) reduction in enhancement, mass effect and edema compared with presteroids (A,C).
Side effects of corticosteroids
Steroids are associated with a large number of potentially serious side effects [36]. Severity of adverse effects usually correlates with the total daily dose and duration of steroid application. As most complications resolve after cessation of steroid use, some side effects may persist, such as osteoporosis and cataract formation. Using the lowest possible doses may reduce the occurrence of these complications. Systemic side effects include a cushingoid appearance, truncal obesity, hirsutism, acne, impaired wound healing, striae, easy bruising and capillary fragility, immunosuppression, hypertension, glucose intolerance, electrolyte disturbance, fluid retention, peripheral edema, increased appetite, gastrointestinal bleeding, osteoporosis, avascular necrosis, growth retardation, cataracts, glaucoma and visual blurring. Typical neurologic complications of corticosteroids are summarized in Box 1. Among the common side effects of steroids, gastrointestinal complications, steroid myopathy, Pneumocystis jerovecii pneumonitis (PJP) and osteoporosis are of particular concern in brain tumor patients and will be discussed in more detail later.
Box 1. Neurological complications of corticosteroids.
Common
Myopathy
Visual blurring
Tremor
Behavioral changes
Insomnia
Reduced taste and smell
Cerebral atrophy
Uncommon
Psychosis
Hallucinations
Hiccups
Dementia
Seizures
Dependency
Epidural lipomatosis
Neuropathy
Gastrointestinal bleeding
Most patients receiving corticosteroids are routinely treated with medications (usually histamine H2 antagonists) to reduce the risk of gastric ulcer and hemorrhage. An association between steroid usage and peptic ulceration has not been clearly shown. No prospective trials have been carried out, but in retrospective analyses of clinical trials evaluating corticosteroid usage in a number of diseases, no significant association between steroid usage and gastrointestinal bleeding could be identified [37–39]. In practice, however, it is prudent to use H2 antagonists in patients treated with steroids for a prolonged duration, especially in the immediate postoperative period and in those patients receiving unusually high doses of corticosteroids. In most patients, twice-daily corticosteroid dosing with meals may reduce the risk of possible stomach irritation and spare patients the added side effects and expense of H2 antagonists. Other rare problems associated with corticosteroid use include pancreatitis, small bowel perforation and fatty liver disease.
Myopathy
Steroid myopathy is common and troublesome at times, impacting significantly on the quality of life of cancer patients. It is the most common side effect of dexamethasone in patients with primary brain tumors and occurs in as many as 10% of those patients [40,41]. The majority of patients develop weakness between the ninth and twelfth week of treatment. Individual susceptibility varies dramatically. Some patients develop significant weakness after taking a low dose of steroids for only a few weeks, while other patients receive large doses for months to years and never develop symptoms. Those who do not develop myopathy also seem to be patients with a lower risk of developing cushingoid features and fluid retention.
It is thought that fluorinated glucocorticoids, such as dexamethasone, are more likely to produce muscle weakness and atrophy than nonfluorinated glucocorticoids, such as hydrocortisone or prednisone [42–45]. While there may be improvement in steroid myopathy with substitution of nonfluorinated glucocorticoids, such as prednisone for dexamethasone, prednisone may not be as effective in controlling brain edema. The precise pathophysiology of steroid myopathy remains unknown. It is likely that steroids exert their effects through inhibition of protein synthesis (partly through inhibition of peptide initiation), increased protein catabolism and possible induction of glutamine synthetase activity [46,47]. Steroid myopathy may improve if the drug can be withdrawn or the dose reduced. Recovery can take a few months after discontinuation of the steroid. Improvement in patients treated at reduced doses can take even longer. In animal models, muscle activity may reduce steroid-induced muscle wasting, suggesting the possibility that an exercise or a physical therapy program may help reduce the severity of myopathy in patients [48].
Infections
The use of moderate to high doses of glucocorticoids can result in clinically significant suppression of the immune system and vulnerability to opportunistic infections. P. jirovecii (previously known as Pneumocystis carinii) is an archiascomycetous fungus capable of causing life-threatening PJP in immunocompromised patients [49,50]. PJP is relatively rare in patients with solid tumors [51] but there is increasing evidence that patients with brain tumors receiving corticosteroids may be at an increased risk of PJP [52–54].
Osteoporosis
Patients receiving chronic corticosteroids are at increased risk of developing osteoporosis. In the past, this has not been considered to be a significant problem owing to the limited life expectancy of most patients with malignant brain tumors. However, as the survival rates improve, an increasing number of patients are developing complications of osteoporosis such as fractures on the lumbar spine and hip. The mechanism of bone loss is multifactorial, but the most important effects are due to the direct actions of glucocorticoids in skeletal cells and include reduction in calcium absorption, with secondary hyperparathyroidism and decreased gonadal hormones. Molecular mechanisms such as reduction in IGH-1 and prostaglandin E2, both of which stimulate bone growth, are also implicated. Patients receiving chronic corticosteroid therapy should be given calcium supplements (1500 mg/day) with vitamin D (800 international units daily) or an activated form of vitamin D (e.g., alfacalcidiol at 1 μg/day or calcitriol at 0.5 μg/day) [55]. In addition, bisphosphonates such as etidronate, alendronate, risedronate and zoledonate [55,56] should be considered. For patients who develop severe pain from compression fractures, kyphoplasty may play an important treatment consideration.
Mood disturbance
Mild neuropsychiatric effects of steroids such as anxiety, insomnia and irritability are probably the most common and pervasive; however, the more dramatic euphoric and psychotic presentations are the more memorable and significant [57]. Patients with a history of psychiatric problems are at greatest risk. Seizures are unusual features of steroid effects, but can be seen at high dose and are usually limited to patients with a history of a seizure disorder. Discrimination of these steroid complications from manifestations of gliomas, cerebral irradiation or changing intracranial pressure can be difficult in clinical practice.
Management of steroid-induced neuropsychiatric side effects involves discontinuing or tapering the implicated agent as soon as is practical. Neuroleptics or lithium may be considered in consultation with a psychiatrist. Tricyclic antidepressants should be avoided as they may confound the problem.
Effects of corticosteroids on the immune system
Dexamethasone, like any typical glucocorticoid, is capable of causing immunosuppression, thereby inhibiting immune and inflammatory responses. Experimental studies have shown a pro-apoptotic effect of dexamethasone on T lymphocytes [58], suggesting that glucocorticoids may direct T-cell positive and negative selection in the thymus, limit activation-induced cell death during the contraction phase of an adaptive immune response and induce generalized thymocyte apoptosis after polyclonal T-cell activation [59]. Dexamethasone is also capable of inducing a shift in an immune response towards a Th2 humoral response from a Th1 cellular response by influencing the levels of cytokines produced by the lymphocytes [60]. Moreover, dexamethasone causes a reduction of splenic and lymph node B-cell numbers and attenuation of early B-cell progenitor proliferation. Glucocorticoids also enhance the activity of macrophages and induce tolerogenic dendritic cells, exerting a potent anti-inflammatory effect [61].
Effects of corticosteroids on tumor cells, cancer stem cells & neural progenitor cells
There is experimental and clinical evidence that corticosteroids have direct effects on tumor cell proliferation and apoptosis. Data from in vitro experiments suggest a variable, time-dependent inhibitory effect of dexamethasone on the proliferation of glioma cells [62]. Stimulatory effects of dexamethasone on glioma cells lines have also been observed at low cell densities and steroid concentration under experimental conditions [63]. However, the molecular mechanisms underlying the effects of corticosteroids on tumor cell proliferation are still poorly understood. Preliminary in vitro data indicate that dexamethasone can either inhibit or stimulate the proliferation of different tumor cell lines, depending on the genetics of the cell line [Rao K, Pastorino S, Kesari S, Unpublished Data].
Dexamethasone and other glucocorticoid hormones have shown to decrease proliferation of embryonic neural stem cells with associated long-term effects on brain development [64]. Molecular studies are currently being carried out to examine whether dexamethasone has any direct influence on cancer stem cell function and behavior. Dexamethasone, specifically through the glucocorticoid receptor, has an inhibitory effect on proliferation, but not differentiation of neural progenitor cells in the process of neurogenesis [65]. Dexamethasone can also inhibit astroglial differentiation from neural precursor cells [65,66]. This effect is supported by both in vitro and in vivo data and is considered to be the cause of cognitive dysfunction [67]. High circulating levels of glucocorticoid hormones adversely affect cognition by directly acting on hippocampal function. Chronic exposure to elevated glucocorticoid levels results in Cushing’s syndrome, and is associated with deficits in cognitive function and in emotion. The hippocampus plays a crucial role in the behavioral manifestations of this syndrome and is particularly prone to dysfunction, due to the highly tissue specific distribution of glucocorticoid receptors [68].
Drug interactions
Dexamethasone and phenytoin are frequently given together as a standard therapy for patients with newly detected primary or secondary brain tumors, and phenytoin is sometimes administered prophylactically, especially perioperatively. Phenytoin increases the metabolic clearance rate of cortisol and dexamethasone and reduces the plasma half-life of dexamethasone by up to 50% by inducing liver enzymes such as CYP3A4, which metabolizes dexamethasone into the hydroxyl metabolite [69–72]. It has been postulated that this may be a mechanism for a protective effect of phenytoin in reducing the risk of development of steroid myopathy [73]. Carbamazepine and phenobarbital may also induce the hepatic metabolism of dexamethasone [74]. Patients with brain tumors on anticonvulsants may therefore need higher doses of dexamethasone to control brain edema. However, particular care should be taken in dose escalation of steroids. For instance, under coadministration of dexamethasone and phenytoin, the levels of the latter are also altered. Contrasting data show both increased or decreased levels of phenytoin in the presence of dexamethasone [73–75]. Although the exact mechanisms are still unclear, these phenomena have important clinical consequences, as prediction of phenytoin levels in an individual patient taking dexamethasone becomes difficult. Therefore, careful monitoring of phenytoin levels is highly recommended, as well as tapering of both dexamethasone and phenytoin after successful control of the tumor-associated brain edema [76].
It has recently been demonstrated that dexamethasone induces alterations in transporter levels [77]. Specifically, dexamethasone differentially alters the expression of efflux proteins depending on the type of tissue or origin of the cells. An important implication is that glucocorticoid administration may lead to altered pharmacokinetics and restricted penetration of concomitant chemotherapeutic agents and may diminish the effectiveness of drugs that are required to penetrate the BBB.
Dexamethasone is also currently being investigated for its role in modulating the action of other chemotherapies. Dexamethasone has been established to protect cells from the apoptotic action of temozolomide in established glioma cell lines in vitro [75]. Other experiments currently underway aim to observe how dexamethasone antagonizes or synergizes with the action of important chemotherapies like rapamycin, Tarceva® and Gleevec®, and apoptotic drugs like staurosporine. Early experimental results suggest protection of glioma cells from apoptosis by exposure to dexamethasone. Pretreatment and subsequent cotreatment strategies for experiments have indicated an additive effect of dexamethasone in combination with growth factor signaling inhibitors [Rao K, Pastorino S, Kesari S, Unpublished Data].
Novel anti-edema agents
The large number of complications associated with corticosteroid therapy has led to the search for alternative therapies for peritumoral edema. Corticotropin-releasing factor (CRF) [76,77] reduces peritumoral edema by a direct effect on blood vessels through CRF 1 and 2 receptors, independent of the release of adrenal steroids, and has been effective in animal models [76]. Phase I/II trials of this agent suggested that it is relatively well tolerated [77,78]. Several Phase III trials are currently in progress examining the efficacy of this drug in the treatment of acute and chronic peritumoral edema. Recently, preliminary studies suggest that cyclooxygenase-2 inhibitors might be effective in treating cerebral edema [79,80]. Clinical studies using cyclooxygenase-2 inhibitors for peritumoral edema are planned, although the cardiac complications of this class of drugs have delayed these studies. Since VEGF plays an important role in the pathogenesis of peritumoral edema, it is possible that inhibitors of VEGF, such as VEGF antibodies (e.g., bevacizumab [Avastin®]) or inhibitors of VEGF receptors, such as cediranib (Recentin™), sorafenib (Nexavar®) and sunitinib (Sutent®) may be helpful in reducing peritumoral edema (Table 1) [81]. It is hoped that some of these agents will prove to be more effective and less toxic alternatives to corticosteroids [82].
Table 1.
Novel angiogenesis inhibitors associated with steroid-sparing effects.
| Drugs | Target |
|---|---|
| Anti-VEGF ligands | |
| Bevacizumab (Avastin®) | VEGF-A |
| Aflibercept (VEGF-Trap) | VEGF-A/B, PLGF |
| Receptor tyrosine kinase inhibitors | |
| Cediranib, AZD2171 (Recentin™) | VEGF-R, PDGF-R, c-Kit |
| Dasatinib (Sprycel®) | PDGF-R, Src, Bcr-Abl |
| Pazopanib, GW786034 (Votrient®) | VEGF-R, c-Kit |
| Sorafenib (Nexavar®) | VEGF-R, PDGF-R, c-Kit, Raf |
| Sunitinib (Sutent®) | VEGF-R, PDGF-R, c-Kit, FLT-3 |
| Vandetanib, ZD6474 (Zactima) | VEGF-R, EGF-R, RET |
| Vatalanib, PTK787/ZK222584 | VEGF-R, PDGF-R, c-Kit |
| Tandutinib (MLN 518) | PDGF-R, c-Kit, FLT-3 |
| XL184 | VEGF-R, c-Met |
FLT-3: Fms-tyrosine kinase; PDGF-R: PDGF receptor; VEGF-R: VEGF receptor.
Corticosteroid effect on neuroimaging findings
The efficacy of steroids in reducing the edema associated with tumors is well confirmed [13–15]. More than 70% of patients with cerebral metastases symptomatically improve after starting steroids. In general, symptoms reflecting generalized neurologic dysfunction or brain edema improve more consistently than do focal symptoms such as hemiparesis. Neuroimaging studies may show a dramatic improvement in enhancement, peritumoral edema, and mass effect (Figure 2), although they may lag behind clinical improvement and not show a decrease in edema for at least 1 week [16]. Corticosteroids significantly impact the ability for contrast dye to cross the BBB in neuroimaging studies. As a result, the dose and duration of glucocorticoids must be carefully taken into account when they are interpreted [83,84]. If primary CNS lymphoma is a possible diagnostic consideration, dexamethasone should be withheld unless cerebral edema and mass effect are clinically significant. The reason for this is the prompt antineoplastic effect of dexamethasone. In some cases, the response can be so dramatic as to impact on the ability to biopsy the tumor. In only a few days, the enhancing tumor can improve to such a degree that it may not appear on the localizing scan carried out prior to biopsy.
Expert commentary
Glucocorticoids are powerful agents that have been widely used for decades by neuro-oncologists in the management of brain tumor patients. Despite their long history in medicine, the exact mechanisms by which glucocorticoids exert their anti-inflammatory and anti-edema actions are not completely understood. Moreover, glucocorticoids may directly influence the properties and behavior of brain tumor cells and normal neural progenitors, carrying the risk of interfering with the success of cancer treatment. Glucocorticoids are associated with numerous and potentially serious side effects, especially in patients treated over long time periods. Several novel molecular targeted agents, such as bevacizumab (Avastin), designed to inhibit pathways involved in angiogenesis, have shown to reduce brain tumor-associated cerebral edema, allowing the reduction of glucocorticoids use in brain tumor patients.
Five-year view
There is a surprising scarcity of high-level data to support many of the common practices of clinicians treating brain tumor patients with steroids, including overall indication, dosage and duration of therapies. The ubiquitous effects of these agents, as well as their mechanisms of action, raise the question of interaction of steroids with the plethora of targeted therapies that are being developed for the treatment of the brain tumors. This article highlights the need to investigate these interactions in order to optimize therapies and minimize complications. In the future, controlled clinical trials addressing these questions will be needed.
Key issues.
Widely used in the treatment of cancer, corticosteroids are beneficial mainly owing to their anti-edema effect.
They provide relief from symptoms related to intracranial pressure and edema owing to the presence of primary or secondary tumors.
Despite being the drug of choice, little is known about their mechanism of action.
The action of corticosteroids involve genomic, as well as nongenomic, mechanisms. Bound to their cytoplasmatic receptor, they enter the nucleus and directly affect DNA transcription. Alternatively, glucocorticoids can interact with few transcription factors and modulate signaling pathways in various cell types, influencing inflammation and immune responses, as well as cell permeability, cell proliferation and survival.
Corticosteroids produce their anti-edema effect by reducing the permeability of tumor capillaries. Corticosteroids affect both endothelial and pericyte function by regulation of tight junction components, such as occludin and ZO1 as well as the rearrangement of VE-cadherin binding to the cytoskeleton. Anti-edema effect of glucocorticoids are also mediated by NF-κB, which regulates T-cell–blood–brain barrier interactions and leukocyte recruitment across the blood–brain barrier
Corticosteroids are potent anti-inflammatory and immunosuppressive agents: their effect is exerted by either direct modulation of proliferation, survival and apoptosis, and/or by regulating expression of cytokines.
Corticosteroids have direct effects on tumor cell proliferation and apoptosis. Dexamethasone and other glucocorticoid hormones have been shown to decrease proliferation of embryonic neural stem cells with associated long-term effects on brain development. Through the glucocorticoid receptors, dexamethasone has an inhibitory effect on proliferation, but not differentiation on neural progenitor cells during neurogenesis. Corticosteriods can also inhibit astroglial cells.
The use of corticosteroids is associated with potentially serious side effects such as gastrointestinal bleeding, myopathy, osteoporosis infections and neuropsychiatric effects.
Dosing is critical, and evidence demonstrates that lower dosing may be equally effective, especially in patients with less severe edema. Clinical trials should be performed to determine the optimal dose in brain tumor patients.
Given the pleiotropic effects on multiple signaling pathways and their direct action on cell proliferation, survival and apoptosis, it is reasonable to think that glucocorticoids may affect the efficacy of anticancer treatments. Current investigations suggest that dexamethasone can sensitize gliomas to tyrosine kinase inhibitors and that the extent of the sensitization is dependent on the genetics of the tumor.
Corticotropin-releasing factor, cyclooxygenase-2 inhibitors and VEGF receptor inhibitors are now being investigated as alternative therapies for their anti-edema properties. It is hoped that some of these agents will prove to be more effective and less toxic alternatives to corticosteroids.
Footnotes
For reprint orders, please contact reprints@expert-reviews.com
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
• • of considerable interest
- 1.Galicich JH, French LA, Melby JC. Use of dexamethasone in treatment of cerebral edema associated with brain tumors. Lancet. 1961;81:46–53. [PubMed] [Google Scholar]
- 2•.Ryken TC, McDermott M, Robinson PD, et al. The role of steroids in the management of brain metastases: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2010;96(1):103–114. doi: 10.1007/s11060-009-0057-4. Thoroughly discusses dose effectiveness of steroid therapy in the treatment of neurological symptoms of metastatic brain tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3•.Ingraham FD, Matson DD, Mc Laurin RL. Cortisone and ACTH as an adjunct to the surgery of craniopharyngiomas. N Engl J Med. 1952;246(15):568–571. doi: 10.1056/NEJM195204102461502. Initial study indicating usefulness of steroid in controlling and preventing postsurgical complications in brain tumors. [DOI] [PubMed] [Google Scholar]
- 4.Kofman S, Garvin JS, Nagamani D, Taylor SG., 3rd Treatment of cerebral metastases from breast carcinoma with prednisolone. J Am Med Assoc. 1957;163(16):1473–1476. doi: 10.1001/jama.1957.02970510039008. [DOI] [PubMed] [Google Scholar]
- 5.Bell BA, Smith MA, Kean DM, et al. Brain water measured by magnetic resonance imaging. Correlation with direct estimation and changes after mannitol and dexamethasone. Lancet. 1987;1(8524):66–69. doi: 10.1016/s0140-6736(87)91908-8. [DOI] [PubMed] [Google Scholar]
- 6.Inaba H, Pui CH. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 2010;11(11):1096–1106. doi: 10.1016/S1470-2045(10)70114-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markman M, Sheidler V, Ettinger DS, Quaskey SA, Mellits ED. Antiemetic efficacy of dexamethasone. Randomized, double-blind, crossover study with prochlorperazine in patients receiving cancer chemotherapy. N Engl J Med. 1984;311(9):549–552. doi: 10.1056/NEJM198408303110901. [DOI] [PubMed] [Google Scholar]
- 8.Todd FD, 2nd, Miller CA, Yates AJ, Mervis LJ. Steroid-induced remission in primary malignant lymphoma of the central nervous system. Surg Neurol. 1986;26(1):79–84. doi: 10.1016/0090-3019(86)90068-6. [DOI] [PubMed] [Google Scholar]
- 9.Ryken TC, McDermott M, Robinson PD, et al. The role of steroids in the management of brain metastases: a systematic review and evidence-based clinical practice guideline. J Neurooncol. 2010;96(1):103–114. doi: 10.1007/s11060-009-0057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vecht CJ, Hovestadt A, Verbiest HB, van Vliet JJ, van Putten WL. Dose–effect relationship of dexamethasone on Karnofsky performance in metastatic brain tumors: a randomized study of doses of 4, 8, and 16 mg per day. Neurology. 1994;44(4):675–680. doi: 10.1212/wnl.44.4.675. [DOI] [PubMed] [Google Scholar]
- 11.Graham PH, Capp A, Delaney G, et al. A pilot randomised comparison of dexamethasone 96 mg vs 16 mg per day for malignant spinal-cord compression treated by radiotherapy: TROG 01.05 Superdex study. Clin Oncol (R Coll Radiol) 2006;18(1):70–76. doi: 10.1016/j.clon.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 12.Marantidou A, Levy C, Duquesne A, et al. Steroid requirements during radiotherapy for malignant gliomas. J Neurooncol. 2010;100(1):89–94. doi: 10.1007/s11060-010-0142-8. [DOI] [PubMed] [Google Scholar]
- 13.Miller JD, Leech P. Effects of mannitol and steroid therapy on intracranial volume–pressure relationships in patients. J Neurosurg. 1975;42(3):274–281. doi: 10.3171/jns.1975.42.3.0274. [DOI] [PubMed] [Google Scholar]
- 14.Miller JD, Sakalas R, Ward JD, et al. Methylprednisolone treatment in patients with brain tumors. Neurosurgery. 1977;1(2):114–117. doi: 10.1227/00006123-197709000-00005. [DOI] [PubMed] [Google Scholar]
- 15.Yeung WT, Lee TY, Del Maestro RF, Kozak R, Bennett J, Brown T. Effect of steroids on iopamidol blood–brain transfer constant and plasma volume in brain tumors measured with x-ray computed tomography. J Neurooncol. 1994;18(1):53–60. doi: 10.1007/BF01324604. [DOI] [PubMed] [Google Scholar]
- 16.Gutin PH. Corticosteroid therapy in patients with cerebral tumors: benefits, mechanisms, problems, practicalities. Semin Oncol. 1975;2(1):49–56. [PubMed] [Google Scholar]
- 17.Sturdza A, Millar BA, Bana N, et al. The use and toxicity of steroids in the management of patients with brain metastases. Support Care Cancer. 2008;16(9):1041–1048. doi: 10.1007/s00520-007-0395-8. [DOI] [PubMed] [Google Scholar]
- 18•.Roth P, Wick W, Weller M. Steroids in neurooncology: actions, indications, side-effects. Curr Opin Neurol. 2010;23(6):597–602. doi: 10.1097/WCO.0b013e32833e5a5d. Focuses on the use of glucocorticoids to manage tumor-associated edema and emphasizes dosing, side effects and the need for more systematic studies. [DOI] [PubMed] [Google Scholar]
- 19•.Barnes PJ. Molecular mechanisms and cellular effects of glucocorticosteroids. Immunol Allergy Clin North Am. 2005;25(3):451–468. doi: 10.1016/j.iac.2005.05.003. Extensively addresses the effects of glucocorticoids on the immune system, investigating the mechanism of action. [DOI] [PubMed] [Google Scholar]
- 20.Karssen AM, de Kloet ER. Synthetic glucocorticoids. In: Fink G, editor. Encyclopedia of Stress. Academic Press; CA, USA: 2000. pp. 566–570. [Google Scholar]
- 21.Cosio BG, Torrego A, Adcock IM. Molecular mechanisms of glucocorticoids. Arch Bronconeumol. 2005;41(1):34–41. doi: 10.1016/s1579-2129(06)60392-3. [DOI] [PubMed] [Google Scholar]
- 22•.Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75(1):1–12. doi: 10.1016/j.steroids.2009.09.002. Discusses the recent findings regarding the molecular mechanisms of action of glucocorticoids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schaaf MJ, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83(1–5):37–48. doi: 10.1016/s0960-0760(02)00263-7. [DOI] [PubMed] [Google Scholar]
- 24.Batchelor T, DeAngelis LM. Medical management of cerebral metastases. Neurosurg Clin N Am. 1996;7(3):435–446. [PubMed] [Google Scholar]
- 25.Wen PY, Schiff D, Kesari S, Drappatz J, Gigas DC, Doherty L. Medical management of patients with brain tumors. J Neurooncol. 2006;80(3):313–332. doi: 10.1007/s11060-006-9193-2. [DOI] [PubMed] [Google Scholar]
- 26.Papadopoulos MC, Saadoun S, Binder DK, Manley GT, Krishna S, Verkman AS. Molecular mechanisms of brain tumor edema. Neuroscience. 2004;129(4):1011–1020. doi: 10.1016/j.neuroscience.2004.05.044. [DOI] [PubMed] [Google Scholar]
- 27••.Machein MR, Plate KH. VEGF in brain tumors. J Neurooncol. 2000;50(1–2):109–120. doi: 10.1023/a:1006416003964. Focuses on the evidence that angiogenesis in brain tumors is mediated by VEGF. The involvement of VEGF in the genesis of peritumoral edema is discussed. [DOI] [PubMed] [Google Scholar]
- 28.Lamszus K, Laterra J, Westphal M, Rosen EM. Scatter factor/hepatocyte growth factor (SF/HGF) content and function in human gliomas. Int J Dev Neurosci. 1999;17(5–6):517–530. doi: 10.1016/s0736-5748(99)00008-8. [DOI] [PubMed] [Google Scholar]
- 29.Cloughesy TF, Black KL. Peritumoral edema. In: Berger MS, Wilson CB, editors. The Gliomas. WB Saunders; PA, USA: 1999. pp. 107–114. [Google Scholar]
- 30.Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood–brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1(3):223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- 31.Hedley-Whyte ET, Hsu DW. Effect of dexamethasone on blood-brain barrier in the normal mouse. Ann Neurol. 1986;19(4):373–377. doi: 10.1002/ana.410190411. [DOI] [PubMed] [Google Scholar]
- 32.Heiss JD, Papavassiliou E, Merrill MJ, et al. Mechanism of dexamethasone suppression of brain tumor-associated vascular permeability in rats. Involvement of the glucocorticoid receptor and vascular permeability factor. J Clin Invest. 1996;98(6):1400–1408. doi: 10.1172/JCI118927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forster C, Silwedel C, Golenhofen N, et al. Occludin as direct target for glucocorticoid-induced improvement of blood–brain barrier properties in a murine in vitro system. J Physiol. 2005;565(Pt 2):475–486. doi: 10.1113/jphysiol.2005.084038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pitzalis C, Pipitone N, Perretti M. Regulation of leukocyte–endothelial interactions by glucocorticoids. Ann NY Acad Sci. 2002;966:108–118. doi: 10.1111/j.1749-6632.2002.tb04208.x. [DOI] [PubMed] [Google Scholar]
- 35.Blecharz KG, Drenckhahn D, Forster CY. Glucocorticoids increase VE-cadherin expression and cause cytoskeletal rearrangements in murine brain endothelial cEND cells. J Cereb Blood Flow Metab. 2008;28(6):1139–1149. doi: 10.1038/jcbfm.2008.2. [DOI] [PubMed] [Google Scholar]
- 36.Koehler PJ. Use of corticosteroids in neuro-oncology. Anticancer Drugs. 1995;6(1):19–33. doi: 10.1097/00001813-199502000-00002. [DOI] [PubMed] [Google Scholar]
- 37.Conn HO, Blitzer BL. Nonassociation of adrenocorticosteroid therapy and peptic ulcer. N Engl J Med. 1976;294(9):473–479. doi: 10.1056/NEJM197602262940905. [DOI] [PubMed] [Google Scholar]
- 38.Carson JL, Strom BL, Schinnar R, Duff A, Sim E. The low risk of upper gastrointestinal bleeding in patients dispensed corticosteroids. Am J Med. 1991;91(3):223–228. doi: 10.1016/0002-9343(91)90119-i. [DOI] [PubMed] [Google Scholar]
- 39.Conn HO, Poynard T. Adrenocorticosteroid administration and peptic ulcer: a critical analysis. J Chronic Dis. 1985;38(6):457–468. doi: 10.1016/0021-9681(85)90028-1. [DOI] [PubMed] [Google Scholar]
- 40.Dropcho EJ, Soong SJ. Steroid-induced weakness in patients with primary brain tumors. Neurology. 1991;41(8):1235–1239. doi: 10.1212/wnl.41.8.1235. [DOI] [PubMed] [Google Scholar]
- 41.Vick NA. Steroid toxicity. J Neurooncol. 1988;6(2):199. doi: 10.1007/BF02327397. [DOI] [PubMed] [Google Scholar]
- 42.Askari A, Vignos PJ, Jr, Moskowitz RW. Steroid myopathy in connective tissue disease. Am J Med. 1976;61(4):485–492. doi: 10.1016/0002-9343(76)90327-2. [DOI] [PubMed] [Google Scholar]
- 43.Kelly FJ, McGrath JA, Goldspink DF, Cullen MJ. A morphological/biochemical study on the actions of corticosteroids on rat skeletal muscle. Muscle Nerve. 1986;9(1):1–10. doi: 10.1002/mus.880090102. [DOI] [PubMed] [Google Scholar]
- 44.Koski CL, Rifenberick DH, Max SR. Oxidative metabolism of skeletal muscle in steroid atrophy. Arch Neurol. 1974;31(6):407–410. doi: 10.1001/archneur.1974.00490420073008. [DOI] [PubMed] [Google Scholar]
- 45.Ruff RL, Weissmann J. Endocrine myopathies. Neurol Clin. 1988;6(3):575–592. [PubMed] [Google Scholar]
- 46.Kimura K, Kanda F, Okuda S, Chihara K. Insulin-like growth factor 1 inhibits glucocorticoid-induced glutamine synthetase activity in cultured L6 rat skeletal muscle cells. Neurosci Lett. 2001;302(2–3):154–156. doi: 10.1016/s0304-3940(01)01667-6. [DOI] [PubMed] [Google Scholar]
- 47•.Owczarek J, Jasinska M, Orszulak-Michalak D. Drug-induced myopathies. An overview of the possible mechanisms. Pharmacol Rep. 2005;57(1):23–34. Addresses different clinical indications for glucocorticoids and provides information on beneficial and undesired effects exerted by these drugs. [PubMed] [Google Scholar]
- 48.Layzer RB. In: Neuromuscular Manifestations of Systemic Disease. Plum F, Baringer JR, Gilman S, editors. FA Davis Company; PA, USA: 1985. [Google Scholar]
- 49.Thomas CF, Jr, Limper AH. Pneumocystis pneumonia. N Engl J Med. 2004;350(24):2487–2498. doi: 10.1056/NEJMra032588. [DOI] [PubMed] [Google Scholar]
- 50.Edman JC, Kovacs JA, Masur H, Santi DV, Elwood HJ, Sogin ML. Ribosomal RNA sequence shows Pneumocystis carinii to be a member of the fungi. Nature. 1988;334(6182):519–522. doi: 10.1038/334519a0. [DOI] [PubMed] [Google Scholar]
- 51.Henson JW, Jalaj JK, Walker RW, Stover DE, Fels AO. Pneumocystis carinii pneumonia in patients with primary brain tumors. Arch Neurol. 1991;48(4):406–409. doi: 10.1001/archneur.1991.00530160074017. [DOI] [PubMed] [Google Scholar]
- 52.Mathew BS, Grossman SA. Pneumocystis carinii pneumonia prophylaxis in HIV negative patients with primary CNS lymphoma. Cancer Treat Rev. 2003;29(2):105–119. doi: 10.1016/s0305-7372(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 53.Schiff D. Pneumocystis pneumonia in brain tumor patients: risk factors and clinical features. J Neurooncol. 1996;27(3):235–240. doi: 10.1007/BF00165480. [DOI] [PubMed] [Google Scholar]
- 54.Sepkowitz KA, Brown AE, Telzak EE, Gottlieb S, Armstrong D. Pneumocystis carinii pneumonia among patients without AIDS at a cancer hospital. JAMA. 1992;267(6):832–837. [PubMed] [Google Scholar]
- 55.Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res (Hoboken) 2010;62(11):1515–1526. doi: 10.1002/acr.20295. [DOI] [PubMed] [Google Scholar]
- 56.Lipton A. New therapeutic agents for the treatment of bone diseases. Expert Opin Biol Ther. 2005;5(6):817–832. doi: 10.1517/14712598.5.6.817. [DOI] [PubMed] [Google Scholar]
- 57.Truhan AP, Ahmed AR. Corticosteroids: a review with emphasis on complications of prolonged systemic therapy. Ann Allergy. 1989;62(5):375–391. [PubMed] [Google Scholar]
- 58.Cifone MG, Migliorati G, Parroni R, et al. Dexamethasone-induced thymocyte apoptosis: apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase C, acidic sphingomyelinase, and caspases. Blood. 1999;93(7):2282–2296. [PubMed] [Google Scholar]
- 59.Herold MJ, McPherson KG, Reichardt HM. Glucocorticoids in T cell apoptosis and function. Cell Mol Life Sci. 2006;63(1):60–72. doi: 10.1007/s00018-005-5390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Franchimont D, Galon J, Gadina M, et al. Inhibition of Th1 immune response by glucocorticoids: dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. J Immunol. 2000;164(4):1768–1774. doi: 10.4049/jimmunol.164.4.1768. [DOI] [PubMed] [Google Scholar]
- 61.Baschant U, Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol. 2010;120(2–3):69–75. doi: 10.1016/j.jsbmb.2010.03.058. [DOI] [PubMed] [Google Scholar]
- 62.Kaup B, Schindler I, Knupfer H, Schlenzka A, Preiss R, Knupfer MM. Time-dependent inhibition of glioblastoma cell proliferation by dexamethasone. J Neurooncol. 2001;51(2):105–110. doi: 10.1023/a:1010684921099. [DOI] [PubMed] [Google Scholar]
- 63.McLean JS, Frame MC, Freshney RI, Vaughan PF, Mackie AE, Singer I. Phenotypic modification of human glioma and non-small cell lung carcinoma by glucocorticoids and other agents. Anticancer Res. 1986;6(5):1101–1106. [PubMed] [Google Scholar]
- 64.Sundberg M, Savola S, Hienola A, Korhonen L, Lindholm D. Glucocorticoid hormones decrease proliferation of embryonic neural stem cells through ubiquitin-mediated degradation of cyclin D1. J Neurosci. 2006;26(20):5402–5410. doi: 10.1523/JNEUROSCI.4906-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65••.Kim JB, Ju JY, Kim JH, et al. Dexamethasone inhibits proliferation of adult hippocampal neurogenesis in vivo and in vitro. Brain Res. 2004;1027(1–2):1–10. doi: 10.1016/j.brainres.2004.07.093. Study showing that steroids can directly affect neural stem cell proliferation. [DOI] [PubMed] [Google Scholar]
- 66•.Sabolek M, Herborg A, Schwarz J, Storch A. Dexamethasone blocks astroglial differentiation from neural precursor cells. Neuroreport. 2006;17(16):1719–1723. doi: 10.1097/01.wnr.0000236862.08834.50. Study showing that steroids affect differentiation of astrocytes. [DOI] [PubMed] [Google Scholar]
- 67.Yu IT, Lee SH, Lee YS, Son H. Differential effects of corticosterone and dexamethasone on hippocampal neurogenesis in vitro. Biochem Biophys Res Commun. 2004;317(2):484–490. doi: 10.1016/j.bbrc.2004.03.071. [DOI] [PubMed] [Google Scholar]
- 68.Forget H, Lacroix A, Somma M, Cohen H. Cognitive decline in patients with Cushing’s syndrome. J Int Neuropsychol Soc. 2000;6(1):20–29. doi: 10.1017/s1355617700611037. [DOI] [PubMed] [Google Scholar]
- 69.Chalk JB, Ridgeway K, Brophy T, Yelland JD, Eadie MJ. Phenytoin impairs the bioavailability of dexamethasone in neurological and neurosurgical patients. J Neurol Neurosurg Psychiatry. 1984;47(10):1087–1090. doi: 10.1136/jnnp.47.10.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Choi Y, Thrasher K, Werk EE, Jr, Sholiton LJ, Olinger C. Effect of diphenylhydantoin on cortisol kinetics in humans. J Pharmacol Exp Ther. 1971;176(1):27–34. [PubMed] [Google Scholar]
- 71.Haque N, Thrasher K, Werk EE, Jr, Knowles HC, Jr, Sholiton LJ. Studies on dexamethasone metabolism in man: effect of diphenylhydantoin. J Clin Endocrinol Metab. 1972;34(1):44–50. doi: 10.1210/jcem-34-1-44. [DOI] [PubMed] [Google Scholar]
- 72.Ruegg S. Dexamethasone/phenytoin interactions: neurooncological concerns. Swiss Med Wkly. 2002;132(29–30):425–426. doi: 10.4414/smw.2002.10085. [DOI] [PubMed] [Google Scholar]
- 73.Ruff RL. Endocrine myopathies. In: Banker BQ, Engle AG, editors. Myology: Basic and Clinical. McGraw-Hill; NY, USA: 1986. pp. 1871–1879. [Google Scholar]
- 74.Penry JK, Newmark ME. The use of antiepileptic drugs. Ann Intern Med. 1979;90(2):207–218. doi: 10.7326/0003-4819-90-2-207. [DOI] [PubMed] [Google Scholar]
- 75.Das A, Banik NL, Patel SJ, Ray SK. Dexamethasone protected human glioblastoma U87MG cells from temozolomide induced apoptosis by maintaining Bax:Bcl-2 ratio and preventing proteolytic activities. Mol Cancer. 2004;3(1):36. doi: 10.1186/1476-4598-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tjuvajev J, Uehara H, Desai R, et al. Corticotropin-releasing factor decreases vasogenic brain edema. Cancer Res. 1996;56(6):1352–1360. [PubMed] [Google Scholar]
- 77.Villalona-Calero MA, Eckardt J, Burris H, et al. A Phase I trial of human corticotropin-releasing factor (hCRF) in patients with peritumoral brain edema. Ann Oncol. 1998;9(1):71–77. doi: 10.1023/a:1008251426425. [DOI] [PubMed] [Google Scholar]
- 78.Hariharan S, Shapiro W, Chang S, et al. Phase II randomized dose-ranging trial of human corticotrophin releasing factor in symptomatic brain tumor patients. Neurology. 2000;54:A12. [Google Scholar]
- 79.Nathoo N, Barnett GH, Golubic M. The eicosanoid cascade: possible role in gliomas and meningiomas. J Clin Pathol. 2004;57(1):6–13. doi: 10.1136/jcp.57.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Portnow J, Suleman S, Grossman SA, Eller S, Carson K. A cyclooxygenase-2 (COX-2) inhibitor compared with dexamethasone in a survival study of rats with intracerebral 9L gliosarcomas. Neuro Oncol. 2002;4(1):22–25. doi: 10.1215/15228517-4-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81•.Gerstner ER, Duda DG, di Tomaso E, et al. VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat Rev Clin Oncol. 2009;6(4):229–236. doi: 10.1038/nrclinonc.2009.14. Addresses the usefulness of anti-VEGF agents as a suitable alternative to glucocorticoids to treat cancer-related conditions associated with increased vascular permeability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 83.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for Phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8(7):1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 84.van den Bent MJ, Vogelbaum MA, Wen PY, MacDonald DR, Chang SM. End point assessment in gliomas: novel treatments limit usefulness of classical MacDonald’s Criteria. J Clin Oncol. 2009;27(18):2905–2908. doi: 10.1200/JCO.2009.22.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]