

Abstract
There is growing evidence that horizontal gene transfer is a potent evolutionary force in prokaryotes, although exactly how potent is not known. We have developed a statistical procedure for predicting whether genes of a complete genome have been acquired by horizontal gene transfer. It is based on the analysis of G+C contents, codon usage, amino acid usage, and gene position. When we applied this procedure to 17 bacterial complete genomes and seven archaeal ones, we found that the percentage of horizontally transferred genes varied from 1.5% to 14.5%. Archaea and nonpathogenic bacteria had the highest percentages and pathogenic bacteria, except for Mycoplasma genitalium, had the lowest. As reported in the literature, we found that informational genes were less likely to be transferred than operational genes. Most of the horizontally transferred genes were only present in one or two lineages. Some of these transferred genes include genes that form part of prophages, pathogenecity islands, transposases, integrases, recombinases, genes present only in one of the two Helicobacter pylori strains, and regions of genes functionally related. All of these findings support the important role of horizontal gene transfer in the molecular evolution of microorganisms and speciation.
Immediate public access to the data of the complete genome sequences opens up a new biological age. Koonin and Galperin (1997) have considered the birth of a new science: Genome-based biology. In addition to the implications for medicine, knowing the microbial complete genome sequence also provides a wealth of information for tracing evolutionary networks (Doolittle 1998). Thus, the majority of genes from complete genomes of archaea most resemble counterparts among eubacteria and not eukaryotes (Doolittle 1998). Of course, this calls into question the rooted “universal tree of life” determined from comparative analyses of the nucleotide sequences of genes encoding ribosomal RNAs and several proteins (Pennisi 1998; Doolittle 1999a,b). At the same time, the genetic relationship between archaea and bacteria strongly supports horizontal gene transfer (HGT) as an important factor in speciation and the molecular evolution of microorganisms. Whereas mutation usually causes only a very small genetic change in a cell, genetic transference usually involves much larger changes that may allow the organism to carry out new functions and can result in adaptation to a changing environment (Lawrence 1999). Lawrence and Ochman (1998) estimated at 18% the overall impact of HGT on the further evolution of the Escherichia coli genome, and Nelson and coworkers (Nelson et al. 1999) estimated that 24% of the genes of the bacteria hyperthermophile Thermotoga maritima are more similar to archaeal genes.
The genomic DNA of different organisms has a particular mean G+C content. In eubacteria this content varies from 25% to 75% and is related to phylogeny (Osawa et al. 1992). Although there is considerable heterogeneity in codon usage among genes in a genome (Li 1997), Grantham et al. (1980) proposed the genome hypothesis, which states that genes in a given genome use the same coding strategy for choices among synonymous codons. That is, the bias in codon usage is species specific. Both parameters (G+C content and codon usage) have been used to determine the acquisition of genomic portions by HGT (Kaplan and Fine 1998; Garcia-Vallvé et al. 1999, 2000). In this article, we have combined a set of statistical approaches to determine which genes significantly deviate from the mean G+C and/or from the average codon usage and to so identify recently transferred genes in 17 bacterial complete genomes and seven archaeal ones. We have excluded small genes and genes with anomalous amino acid compositions in order to obtain a prediction that lies outside the twilight zone of HGT prediction. Moreover, with access to the full data sets online, researchers can explore this twilight zone themselves when encountering anomalies in protein sequence family trees.
RESULTS
We have identified the amount of genes originated by HGT in 24 complete genomes (Table 1). The percentage of horizontally transferred genes in the 24 genomes varies from 1.56% to 14.47%. Broadly speaking, archaea and nonpathogenic bacteria show higher percentages than pathogenic bacteria, except for Mycoplasma genitalium. This pathogenic bacterium shows, with Bacillus subtilis, the highest percentage. The horizontally transferred genes may be either isolated or in blocks that we call alien genomic strips (see http://www.fut.es/∼debb/HGT/ for a complete location in every genome). The G+C content of these alien genomic strips may be higher or lower than the mean G+C content of their own genome. We have named these strips as regions with a high or low G+C content, respectively (Table 1). Table 2 shows the classification of the proposed horizontally transferred genes into functional categories. It is important to note both that in some organisms, especially those with the greatest number of horizontally transferred genes, the informational genes (i.e., those involved in information storage and processing) are less frequently transferred than other functional groups and that the majority of the proposed horizontally transferred genes are not present in any of the previously defined clusters of orthologous groups (Tatusov et al. 2000), that is, they are only present in one or two of the lineages compared.
Table 1.
Species, Disease Caused, Genome Size, and Number of Open Reading Frames of the Bacterial and Archaeal Genomes Used
| Species | Disease caused | Genome size (bp) | Number of open reading frames | HGT | Percentage HGT | GC+ | GC− |
|---|---|---|---|---|---|---|---|
| Proteobacteria | |||||||
| Escherichia coli | · · · | 4,639,221 | 4289 | 381 | 9.62 | 68 | 258 |
| Haemophilus influenzae | Pneumonia | 1,830,138 | 1709 | 96 | 6.19 | 31 | 33 |
| Helicobacter pylori 26695 | Ulcer | 1,667,867 | 1553 | 89 | 6.41 | 0 | 68 |
| Helicobacter pylori J99 | Ulcer | 1,643,831 | 1491 | 80 | 5.81 | 5 | 59 |
| Rickettsia prowazekii | Typhus | 1,111,523 | 834 | 28 | 3.62 | 10 | 8 |
| Gram-positive bacteria | |||||||
| Bacillus subtilis | · · · | 4,214,814 | 4100 | 537 | 14.47 | 85 | 402 |
| Mycoplasma genitalium | Urethritis | 580,074 | 480 | 67 | 14.47 | 46 | 19 |
| Mycoplasma pneumoniae | Pneumonia | 816,394 | 677 | 39 | 5.93 | 0 | 32 |
| Mycobacterium tuberculosis | Tuberculosis | 4,411,529 | 3918 | 187 | 5.01 | 55 | 53 |
| Spirochaete | |||||||
| Borrelia burgdorferi | Lyme disease | 910,724 | 850 | 12 | 1.56 | 0 | 5 |
| Treponema pallidum | Syphilis | 1,138,011 | 1031 | 77 | 8.32 | 30 | 38 |
| Chlamydiae | |||||||
| Chlamydia trachomatis | Trachoma, epidymitis | 1,042,519 | 894 | 36 | 4.32 | 8 | 13 |
| Chlamydia pneumoniae | Pneumonia, bronchitis | 1,230,230 | 1052 | 55 | 5.70 | 28 | 16 |
| Aquifex aeolicus | · · · | 1,551,335 | 1522 | 72 | 4.84 | 6 | 43 |
| Deinococcus radiodurans | · · · | 2,648,638 | 2580 | 95 | 3.92 | 6 | 38 |
| Synechocystis PCC6803 | · · · | 3,573,470 | 3169 | 219 | 7.50 | 12 | 151 |
| Thermotoga maritima | · · · | 1,860,725 | 1846 | 198 | 11.63 | 69 | 100 |
| Ureaplasma urealyticum | Adverse pregnancy outcome, neonatal disease, and superative arthritis | 751,719 | 610 | 32 | 5.70 | 8 | 9 |
| Archaea | |||||||
| Aeropyrum pernix | · · · | 1,669,695 | 2694 | 370 | 14.01 | 108 | 203 |
| Archaeoglobus fulgidus | · · · | 2,178,400 | 2407 | 179 | 8.44 | 16 | 116 |
| Methanobacterium thermoautotrophicum | · · · | 1,751,377 | 1869 | 179 | 10.73 | 30 | 111 |
| Methanococcus jannaschii | · · · | 1,664,970 | 1715 | 77 | 5.00 | 18 | 38 |
| Pyrococcus abyssi | · · · | 1,765,118 | 1765 | 124 | 7.35 | 22 | 63 |
| Pyrococcus horikoshii | · · · | 1,738,505 | 2064 | 154 | 7.68 | 68 | 42 |
Note: HGT is the total number of proposed transferred genes, GC+ and GC− are proposed transferred genes that belong to regions with a high or low G+C content, respectively. The percentage HGT was calculated by dividing the number of proposed transferred genes by the sum of nonproposed ones, excluding genes smaller than 300 bp, and proposed transferred genes.
Table 2.
Functional Distribution of Genes Proposed as Being Acquired by Horizontal Gene Transfer
| Organism | HGT | Info. (%) | Cell (%) | Meta. (%) | Poor (%) | − (%) |
|---|---|---|---|---|---|---|
| Archaeoglobus fulgidus | 179 | 6 (2.2) | 22 (8.1) | 17 (2.7) | 31 (6.0) | 103 (14.5) |
| Aquifex aeolicus | 72 | 7 (3.2) | 12 (3.8) | 15 (3.6) | 20 (6.4) | 18 (6.9) |
| Borrelia burgdorferi | 12 | 2 (1.1) | 3 (1.7) | 1 (0.8) | 3 (2.2) | 3 (1.3) |
| Bacillus subtilis | 537 | 54 (11.5) | 34 (5.8) | 76 (8.0) | 42 (7.3) | 331 (21.8) |
| Chlamydia pneumoniae | 55 | 6 (3.4) | 4 (3.0) | 10 (4.9) | 6 (5.3) | 29 (6.8) |
| Chlamydia trachomatis | 36 | 5 (2.9) | 5 (3.8) | 9 (4.8) | 0 (0.0) | 17 (5.7) |
| Escherichia coli | 381 | 23 (4.8) | 41 (6.6) | 42 (3.8) | 27 (5.4) | 248 (15.7) |
| Haemophilus influenzae | 96 | 3 (1.1) | 19 (7.0) | 10 (2.2) | 3 (1.4) | 61 (12.0) |
| Helicobacter pylori 26695 | 89 | 11 (5.0) | 3 (1.1) | 5 (1.6) | 3 (1.6) | 67 (11.6) |
| Helicobacter pylori J99 | 80 | 7 (3.2) | 6 (2.3) | 5 (1.6) | 4 (2.2) | 58 (11.5) |
| Mycoplasma genitalium | 67 | 26 (19.1) | 10 (16.7) | 11 (12.6) | 6 (9.1) | 14 (10.7) |
| Methanococcus jannaschii | 77 | 8 (3.6) | 11 (7.5) | 6 (1.6) | 7 (1.8) | 45 (7.8) |
| Mycoplasma pneumoniae | 39 | 8 (5.1) | 0 (0.0) | 3 (2.7) | 2 (2.7) | 26 (9.6) |
| Methanobacterium thermoautotrophicum | 179 | 4 (1.7) | 19 (8.8) | 26 (5.6) | 44 (11.1) | 86 (15.5) |
| Mycobacterium tuberculosis | 187 | 6 (1.6) | 20 (4.9) | 35 (4.0) | 17 (3.2) | 109 (6.2) |
| Pyrococcus horikoshii | 154 | 10 (4.2) | 11 (6.1) | 8 (2.1) | 23 (4.9) | 102 (12.7) |
| Rickettsia prowazekii | 28 | 8 (4.4) | 3 (1.9) | 12 (6.6) | 4 (3.6) | 1 (0.5) |
| Synechocystis PCC6803 | 219 | 14 (5.3) | 31 (5.2) | 15 (2.6) | 22 (5.0) | 137 (10.6) |
| Thermotoga maritima | 198 | 13 (5.3) | 18 (6.5) | 55 (10.7) | 47 (12.0) | 65 (15.6) |
| Treponema pallidum | 77 | 8 (4.5) | 17 (8.7) | 2 (1.4) | 17 (10.6) | 33 (9.2) |
The functional classification available in the COG database (Tatusov et al. 2000) was used. The table shows the number and the group percentage of the genes proposed as being acquired by HGT. The functional groups used were: Info, Information storage and processing; Cell, cellular processes; Meta, metabolism; Poor, poorly characterized; −, not present in any cluster of orthologous group.
A correspondence analysis of codon usage for genes of B. subtilis (Kunst et al. 1997; Moszer et al. 1999) shows that they can be divided into three different classes: class 1, in which the majority of the genes are located; class 2, in which the highly expressed genes are present, characterized by a coincidence between codon usage and the most abundant tRNAs; and class 3, which contains a large proportion of genes with an unknown function and genes forming part of prophages. A reliable HGT prediction must include genes of class 3, but not of class 2. This is the case with our prediction for the genes of B. subtilis, where 365 genes predicted as being acquired by HGT belong to class 3, 169 belong to class 1, and only 3 belong to class 2. A similar situation occurs for genes of E. coli. Some other genes that are expected to be acquired by HGT and are also included in our predictions are: 88 alien genes defined by Karlin et al. (1998) distributed in seven clusters in the B. subtilis genome, some of the genes that form part of described prophages or pathogenicity islands (such as the cag pathogenicity island of H. pylori; Karlin et al. 1998), genes associated with virulence (such as mviN gene from Treponema pallidum), transposases, integrases and recombinases, DNA transfer proteins (such as tfoX gene from H. influenzae), genes present only in one of the two H. pylory strains (such as the jhp0937–jhp0953 region of H. pylory J99), genes that belong to the same alien genomic strip and are functionally related (such as the B. subtilis nasB operon required for nitrate and nitrite assimilation [Ogawa et al. 1995] and the E. coli nik operon for specific transport of nickel [Navarro et al. 1993]) and genes described previously as being acquired by HGT (such as phosphofructokinase 1 and 2 from C. trachomatis [Stephens et al. 1998] and erythroid ankyrin from Synechocystis sp [Ponting et al. 1999]).
Figure 1 shows a correspondence analysis of relative synonymous codon usage for four of the 24 genomes analyzed. Analysis involves plotting genes into two axes according to the most important sources of codon usage variation. Greater distances from the origin and between points correspond to greater differences in codon usage. Although each genome has a different type of pattern in these plots, genes proposed as being acquired by HGT can be split into two groups according to their G+C content. When one of these groups is large enough, it can be differentiated from the majority of genes of its organism. This is the case of the transferred genes with a low G+C content of E. coli, B. subtilis, Thermotoga maritima, and Methanobacterium thermoautotrophicum.
Figure 1.

Correspondence analysis of relative synonymous usage for genes of (A) Escherichia coli, (B) Bacillus subtilis, (C) Thermotoga maritima, and (D) Methanobacterium thermoautotrophicum. Triangles correspond to genes proposed as being acquired by horizontal gene transfer and included in regions with a low G+C content. Circles correspond to genes proposed as being acquired by HGT and included in regions with a high G+C content. Dots correspond to genes that are not proposed as being acquired by HGT. For the sake of clarity, genes proposed as being acquired by HGT that do not belong to a region that is high or low in the G+C content are not shown.
DISCUSSION
In any prediction there are, obviously, false positives (genes that appear transferred but are not) and false negatives (transferred genes we have missed). Our HGT list should be considered as a first approximation. Identifying evolution by HGT involves detailed study of the protein level, including orthologous analysis and confirmation of an inappropriate position in a phylogenetic tree (Smith et al. 1992; Syvanen 1994). For example, we determined previously that the xynA gene from B. subtilis has been transferred, probably from an actinomyces bacterium (Garcia-Vallvé et al. 1999).
False positives may include pseudogenes or segments of fossilized DNA whose ability to mutate has no restriction and genes with a G+C content or codon usage bias caused by forces other than HGT. Other forces responsible for the heterogeneity in codon usage among genes in a genome are compositional asymmetries between genes lying on the leading versus lagging strand (Rocha et al. 1999; Lafay et al. 1999), selection for translational efficiency, mutation biases, and random drift (Li 1997). The heterogeneity in codon usage among genes in a genome is shown in Figure 1. Genes from E. coli and B. subtilis (Fig. 1A,B) are clustered into three distinct groups or classes (Medigue et al. 1991; Moszer 1998). For both genomes, and for Methanococcus janaschii and Haemophilus influenzae (McInerney 1997), mutational bias and translational selection are the most important sources of variation. It is important to distinguish the set of highly expressed genes of these organisms from the horizontally transferred genes. Both differ from the mean codon usage values. However, the highly expressed genes do not deviate in G+C content and are not included in our HGT list. Genes from Borrelia burgdorferi and Treponema pallidum are clustered into two groups according to the strand (leading or lagging) to which they belong (McInerney 1998; Lafay et al. 1999). Genes from Thermotoga maritima (Fig. 1C) and Pyrococcus abyssi are clustered into three groups according to which of the two cysteine codons (TGT and TGC) are used more frequently. Finally, for most of the other complete genomes (Fig. 1D shows that of M. thermoautotrophicum), there is a nonspecific pattern, with none of the above characteristics. The fact that some horizontally transferred genes are indistinguishable from the majority of genes in the correspondence analysis means that new algorithms need to be developed to compare the codon usage between genes or between a gene and a group of genes. We have therefore used the Mahalanobis distance coupled with a Montecarlo method (see the Methods section) irrespective of the most important factors of codon usage variation. This has allowed us to establish the limits for excluding extraneous genes from codon usage.
According to Lawrence and Ochman (1998), a statistical procedure cannot detect as horizontally transferred genes those genes whose parameters closely resemble those of the receiving organism or those genes that have adjusted to the base composition and codon usage of the resident genome, called the amelioration process (Lawrence and Ochman 1997). These genes would be false negatives in our prediction. This may be the case of some genes from Chlamydia trachomatis and Rickettsia prowazekii. Using analysis of sequences and protein phylogenetic trees, Koonin and coworkers (Wolf et al. 1999; Stephens et al. 1998) identified some genes that were acquired by HGT in both organisms. Except for C. trachomatis pyrophosphate-dependent phosphofructokinases genes (ct205 and ct207), none of these genes are extraneous in the G+C content or codon usage and are not included in our HGT list. Our results should, therefore, be seen as a conservative prediction of horizontally transferred genes.
Evidence for the importance of HGT in the molecular evolution of microorganisms is increasing (Jain et al. 1999; Martin 1999). Lawrence and Ochman (1998) found that all the phenotypic characteristics that distinguish E. coli and Salmonella enterica are encoded by horizontally transferred genes. Our finding that the majority of the horizontally transferred genes are not present in any of the previously defined clusters of orthologous groups (Tatusov et al. 2000) clearly suggests the important role of HGT in speciation—the process of formation of new species. This view is underlined by the comparison of two different strains of H. pylori. We have predicted as horizontally transferred genes some of the regions that are only present in one of the two strains. Finally, genes of our defined alien genomic strips that are functionally related reflect the acquisition of novel metabolic capabilities in a single transfer event (Lawrence 1999).
The predominant evolutionary process in parasitic bacteria is genome reduction (Koonin et al. 1997). This is reflected in the small size of their genomes and agrees with our finding that pathogenic bacteria have lower percentages of horizontally transferred genes. Mycoplasma genitalium is the exception. The high percentage of horizontally transferred genes for this pathogenic bacterium may be due to its small genome size of only 580,074 pb. Another cause of this difference between pathogenic and nonpathogenic bacteria could be habitat. Species with a broad range habitat, such as E. coli and B. subtilis, have more opportunities to interchange genes. Despite the lower percentages, HGT has played a significant role in the emergence of pathogenic bacteria (Fuchs 1998). The finding that genes from pathogenicity islands have been acquired by HGT (such as the cag pathogenicity island of H. pylori) supports this hypothesis.
Many archaea inhabit extreme environments with similar conditions to those in which life originated. Although members of the archaea may be seen as evolutionary relics of Earth's earliest life forms, none of the organisms living today are primitive. All extant life forms are modern organisms well adapted to their ecological niches. Many authors have found many horizontally transferred genes in archaea (Koonin et al. 1997; Aravind et al. 1998). Koonin et al. (1997) have found large fractions of genes of apparent bacterial or eukaryotic origin in archaea genomes, which suggests a chimeric origin for the archaea. Our percentages of horizontally transferred genes in bacteria and archaea are similar. This shows that HGT is a wide-ranging phenomenon.
Finally, our results are consistent with earlier claims of the important role of HGT in the evolution of microorganims (Lawrence and Ochman 1998; Lawrence 1999). Moreover, the functional classification of genes acquired by HGT agrees with the complexity hypothesis of Jain et al. (1999), who found that operational genes (those involved in housekeeping) are more successfully transferred than informational genes (those involved in transcription, translation, and related processes).
METHODS
Sequence files of the 24 complete genomes were retrieved from the NCBI (ftp://ncbi.nlm.nih.gov/genbank/genomes/bacteria/). For each genome we calculated the mean values and standard deviations (σ) of codon usage, relative synonymous codon usage (RSCU values), total and positional G+C contents (G+C[T], G+C[1], G +C[2] and G+C[3]), and amino acid compositions. We excluded genes shorter than 300 base pairs, which can have extraneous values of G+C content, codon usage, or amino acid composition.
We considered genes as extraneous in terms of the G+C content if their G+C(T) content deviated by >1.5σ from the mean value of their genome or if deviations of G+C(1) and G+C(3) were of the same sign and at least one was >1.5σ. We also ran an 11-gene window through each genome. Five or more extraneous genes in a given window indicated the presence of an alien genomic strip. Finally, we filtered these strips to disregard short isolated segments and to include genes that we did not consider extraneous but that had a deviation of their G+C content of the same sign as the deviation of the strip to which they belong.
We used the Mahalanobis distance as a measure of the distance between the codon usage of a gene (X) and the mean of an organism. This distance takes into account the coupling effect among different codon frequencies, and we adapted it from the prediction of protein structural classes (Chou and Zhang 1995). In our method, each gene corresponds to a vector or to a point in the 61-D space whose coordinates are the relative frequency of use of the 61 codons. The stop codons are not included, and each organism corresponds to a vector or a point whose coordinates are the mean values.
The Mahalanobis distance uses the 61 × 61 covariance matrix (S), whose elements si,j are given by
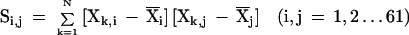 |
where N is the number of genes of an organism, and 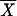 i are the mean values for each codon. The Mahalanobis distance can be calculated as follows:
i are the mean values for each codon. The Mahalanobis distance can be calculated as follows:
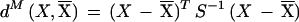 |
where X and 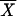 are vectors of 61 dimensions that contain the relative frequency of each codon for a gene and the mean values for an organism, respectively, the superscript T is the transposition operator, and S−1 is the inverse matrix of S. A higher value of this distance represents more differences in codon usage.
are vectors of 61 dimensions that contain the relative frequency of each codon for a gene and the mean values for an organism, respectively, the superscript T is the transposition operator, and S−1 is the inverse matrix of S. A higher value of this distance represents more differences in codon usage.
We calculated the Mahalanobis distance from each gene to the mean value of its own organism. These distances did not follow a normal distribution, so we could not apply the criteria regarding deviations >1.5σ from the mean value to identify extraneous genes from codon usage. Instead we used a Montecarlo procedure (Guillespie 1977). This entailed generating a random sample of 10,000 sequences from the means and standard deviations of the codon usage of each genome. The Mahalanobis distances of these sets of random sequences had a normal distribution, and so, we could calculate a mean value and a standard deviation. We considered as extraneous genes those that had a Mahalanobis distance of >2σ from the mean value.
What large deviations from the mean values of amino acid composition represent is very ambiguous. They may be caused either by functional constraints or by the result of the extraneous codon usage or G+C content of a horizontally transferred gene. We therefore chose the restricting criterion: We excluded from our set of genes predicted as being acquired by HGT those isolated genes whose derived protein has deviations of >3σ in at least one amino acid content. Only genes included in some of the alien genomic strips could present such deviation.
Genes were said to originate from HGT if they were extraneous from G+C content and codon usage, if they were longer than 300 bp and did not deviate from amino acid composition, or if they were included in our defined alien genomic strips.
The genes proposed as being originated from HGT were represented by correspondence analysis (Hill 1974). Protein-coding sequences are considered as points in a 59-dimensional space (the stop codons and codons for methionine and tryptophan are not included), and each dimension corresponds to the relative frequency of use of each codon, measured with the relative synonymous codon usage (RSCU) values. Using the ×2 distance between each pair of genes, we can project the cloud of points into a two-dimensional space with a minimum loss of information and maximum scattering.
Files containing statistical calculations for each organism and gene and lists of the horizontally transferred genes are available on our HGT-DB web server (http://www.fut.es/∼debb/HGT/).
Acknowledgments
S.G-V. has been the recipient of a fellowship (FI/96–7.030) from the Catalan Governmental Agency CIRIT (Generalitat de Catalunya). We thank Kevin Costello (of the Language Service of the Rovira I Virgili University) for his help with writing the manuscript and Dr. I. Moszer for supplying us with a list of B. subtilis genes separated into different classes. This research has not been awarded grants by any research-supporting institution.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL romeu@quimica.urv.es; FAX 34-977-55-81-88.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.130000.
REFERENCES
- Aravind L, Tatusov RL, Wolf YI, Walker DR, Koonin EV. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet. 1998;14:442–444. doi: 10.1016/s0168-9525(98)01553-4. [DOI] [PubMed] [Google Scholar]
- Chou KC, Zhang CT. Prediction of protein structural classes. Crit Rev Biochem Mol Biol. 1995;30:275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- Doolittle RF. Microbial genomes opened up. Nature. 1998;392:339–342. doi: 10.1038/32789. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. Lateral genomics. Trends Biochem Sci. 1999a;24:M5–M8. [PubMed] [Google Scholar]
- ————— Phylogenetic classification and the universal tree. Science. 1999b;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Fuchs TM. Molecular mechanisms of bacterial pathogenicity. Naturwissenschaften. 1998;85:99–108. doi: 10.1007/s001140050463. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallvé S, Palau J, Romeu A. Horizontal gene transfer in glycosyl hydrolases inferred from codon usage in Escherichia coli and Bacillus subtilis. Mol Biol Evol. 1999;9:1125–1134. doi: 10.1093/oxfordjournals.molbev.a026203. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallvé S, Romeu A, Palau J. Horizontal gene transfer of glycosyl hydrolases of the rumen fungi. Mol Biol Evol. 2000;17:352–361. doi: 10.1093/oxfordjournals.molbev.a026315. [DOI] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Mercier R, Pavé A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980;8:r49–r62. doi: 10.1093/nar/8.1.197-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillespie DT. Estocastic simulation of coupled chemical reaction. J Phys Chem. 1977;81:2340–2352. [Google Scholar]
- Hill MO. Correspondence analysis: A neglected multivariate method. Appl Stat. 1974;23:340–353. [Google Scholar]
- Jain R, Rivera MC, Lake JA. Horizontal gene transfer among genomes: The complexity hypothesis. Proc Natl Acad Sci. 1999;96:3801–3806. doi: 10.1073/pnas.96.7.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan JB, Fine DH. Codon usage in Actinobacillus actinomycetemcomitans. FEMS Microbiol Lett. 1998;163:31–36. doi: 10.1111/j.1574-6968.1998.tb13022.x. [DOI] [PubMed] [Google Scholar]
- Karlin S, Campbell AM, Mrazek J. Comparative DNA analysis across diverse genomes. Annu Rev Genet. 1998;32:185–225. doi: 10.1146/annurev.genet.32.1.185. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Galperin MY. Prokaryotic genomes: The emerging paradigm of genome-based microbiology. Curr Opin Genet Dev. 1997;7:757–763. doi: 10.1016/s0959-437x(97)80037-8. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Mushegian AR, Galperin MY, Walker DR. Comparison of archaeal and bacterial genomes: Computer analysis of protein sequence predicts novel functions and suggests a chimeric origin for the archaea. Mol Microbiol. 1997;25:619–637. doi: 10.1046/j.1365-2958.1997.4821861.x. [DOI] [PubMed] [Google Scholar]
- Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, BessiÈres P, Bolotin A, Borchert S, et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Lafay B, Lloyd AT, McLean MJ, Devine KM, Sharp PM, Wolfe KH. Proteome composition and codon usage in spirochaetes: Species-specific and DNA strand–specific mutational biases. Nucleic Acids Res. 1999;27:1642–1649. doi: 10.1093/nar/27.7.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG. Gene transfer, speciation, and the evolution of bacterial geneomes. Curr Opin Microbiol. 1999;2:519–523. doi: 10.1016/s1369-5274(99)00010-7. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Ochman H. Amelioration of bacterial genomes: Rates of change and exchange. J Mol Evol. 1997;44:383–397. doi: 10.1007/pl00006158. [DOI] [PubMed] [Google Scholar]
- ————— Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W-H. Molecular evolution. Sunderland, MA: Sinauer; 1997. [Google Scholar]
- Martin W. Mosaic bacterial chromosomes: A challenge en route to a tree of genomes. BioEssays. 1999;21:99–104. doi: 10.1002/(SICI)1521-1878(199902)21:2<99::AID-BIES3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- McInerney JO. Prokaryotic genome evolution as assessed by multivariate analysis of codon usage patterns. Microb Comp Genomics. 1997;2:1–10. [Google Scholar]
- ————— Replicational and transcriptional selection on codon usage in Borrelia burgdorferi. Proc Natl Acad Sci. 1998;95:10698–10703. doi: 10.1073/pnas.95.18.10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medigue C, Rouxel T, Vigier P, Henault A, Danchin A. Evidence for horizontal gene transfer in Escrerichia coli speciation. J Mol Biol. 1991;222:851–856. doi: 10.1016/0022-2836(91)90575-q. [DOI] [PubMed] [Google Scholar]
- Moszer I. The complete genome of Bacillus subtilis: From sequence annotation to data management and analysis. FEBS Lett. 1998;430:28–36. doi: 10.1016/s0014-5793(98)00620-6. [DOI] [PubMed] [Google Scholar]
- Moszer I, Rocha EPC, Danchin A. Codon usage and lateral gene transfer in Bacillus subtilis. Curr Opin Microbiol. 1999;2:524–528. doi: 10.1016/s1369-5274(99)00011-9. [DOI] [PubMed] [Google Scholar]
- Navarro C, Wu LF, Mandrand-Berthelot MA. The nik operon of Escherichia coli encodes a periplasmic binding–protein-dependent transport system for nickel. Mol Microbiol. 1993;9:1181–1191. doi: 10.1111/j.1365-2958.1993.tb01247.x. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, et al. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Ogawa K, Akagawa E, Yamane K, Sun ZW, Lacelle M, Zuber P, Nakano MM. The nasB operon and nasA gene are required for nitrate/nitrite assimilation in Bacillus subtilis. J Bacteriol. 1995;177:1409–1413. doi: 10.1128/jb.177.5.1409-1413.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa S, Jukes TH, Watanabe K, Muto A. Recent evidence for evolution of the genetic code. Microbiol Rev. 1992;56:229–264. doi: 10.1128/mr.56.1.229-264.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Aravind L, Schultz J, Bork P, Koonin EV. Eukaryotic signalling domain homologues in archaea and bacteria: Ancient ancestry and horizontal gene transfer. J Mol Biol. 1999;289:729–745. doi: 10.1006/jmbi.1999.2827. [DOI] [PubMed] [Google Scholar]
- Pennisi E. Genome data shake tree of life. Science. 1998;280:672–674. doi: 10.1126/science.280.5364.672. [DOI] [PubMed] [Google Scholar]
- Rocha EPC, Danchin A, Viari A. Universal replication biases in bacteria. Mol Microbiol. 1999;32:11–16. doi: 10.1046/j.1365-2958.1999.01334.x. [DOI] [PubMed] [Google Scholar]
- Smith MW, Feng D-F, Doolittle RF. Evolution by acquisition: The case for horizontal gene transfers. Trends Biochem Sci. 1992;17:489–493. doi: 10.1016/0968-0004(92)90335-7. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- Syvanen M. Horizontal gene transfer: Evidence and possible consequences. Annu Rev Genet. 1994;28:237–261. doi: 10.1146/annurev.ge.28.120194.001321. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Aravind L, Koonin EV. Trends Genet. 1999;15:173–175. doi: 10.1016/s0168-9525(99)01704-7. [DOI] [PubMed] [Google Scholar]