

Abstract
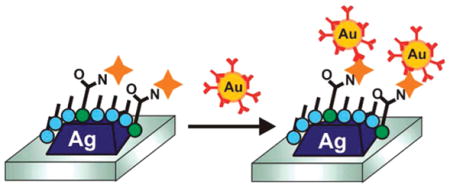
A method to amplify the wavelength shift observed from localized surface plasmon resonance (LSPR) bioassays is developed using gold nanoparticle-labeled antibodies. The technique, which involves detecting surface-bound analytes using gold nanoparticle conjugated antibodies, provides a way to enhance LSPR shifts for more sensitive detection of low-concentration analytes. Using the biotin and antibiotin binding pair as a model, we demonstrate up to a 400% amplification of the shift upon antibody binding to analyte. In addition, the antibody-nanoparticle conjugate improves the observed binding constant by 2 orders of magnitude, and the limit of detection by nearly 3 orders of magnitude. This amplification strategy provides a way to improve the sensitivity of plasmon-based bioassays, paving the way for single molecule-based detection and clinically relevant diagnostics.
INTRODUCTION
The need to probe biomolecule interactions in areas as diverse as proteomics, disease detection, and drug discovery is an important motivation for biosensor research. The development of therapeutics capable of treating early stage disease, as well as the discovery that ultralow quantities of biomolecules can lead to diseased states, necessitates that these biosensors operate with extreme sensitivity. The biological community has tackled this need for sensitive signal transduction modalities through the development of creative biomolecule labeling strategies, including colorimetric and luminescent techniques. The current gold standard for biomolecule detection is the ELISA assay, which detects antigen-mediated antibody dimer formation using enzyme-conjugated antibodies. Other methods, such as the biobarcode assay, utilize the same basic sandwich principle, but achieve lower detection levels by using a silver-staining amplification technique.1
Biosensors that take advantage of the plasmonic properties of noble metal films and nanostructures have emerged as an alternative to traditional signal transduction modalities. Surface plasmon resonance (SPR) sensors, which detect changes in the reflectance intensity or angle of thin gold films, have been commercialized for nearly two decades and are commonly used to probe the kinetics and strength of binding interactions. Localized SPR sensors, which employ noble metal nanoparticles, are increasingly used as an alternative to SPR sensors because the highly localized electromagnetic fields that occur at nanoparticle surfaces can enable improved detection of nanoscale biological analytes. LSPR sensors can sensitively monitor binding events in real time and have been used to detect a variety of processes,2 including self-assembled monolayer formation,3 protein-ligand and antibody-protein interactions,4–6 DNA hybridization,7 protein conformational changes,8 and gas detection.9 The spectral position of the nanoparticle extinction peak (λmax) is highly dependent on the local refractive index at the nanoparticle surface. Biomolecule binding to specific surface-bound ligands changes the local refractive index, causing changes in the λmax that are easily monitored using UV-visible spectroscopy. However, sensitivity losses arise when LSPR sensors utilize longer surface ligands and molecules, such as in the ELISA-style antibody sandwich binding, that place the detecting molecule far from the surface and outside the sensing volume.
Several methods exist for enhancing the sensitivity of plasmonic sensors. Careful design of spectroscopic instrumentation can reduce noise levels in λmax to as low as 10−3–10−4 nm, enabling the detection of spectral shifts of only hundredths of nanometers.8,10 Rational design of nanostructures can also lead to sensitivity improvements via narrowing of spectral linewidths and hence reduction of the standard deviation of λmax determinations. For example, single optically isolated nanoparticles can possess narrow spectral linewidths on the order of 0.1 eV.11 Even narrower linewidths, down to 0.032 eV, have been reported for gold nanohole arrays.12 In a resonance-enhanced assay, resonance coupling between a molecular absorption band in the analyte and the nanoparticle plasmon band leads to wavelength shifts that are amplified by a factor of 3.13 Yet another technique uses enzymatic amplification triggered by binding reactions to achieve DNA detection at femtomolar levels.14 This article outlines a technique in which plasmonically active nanoparticle labels are used to enhance the wavelength shift upon biomolecule binding through both resonant coupling of plasmons and an increase in refractive index change.
Nanoparticle labels have previously been used to enhance the response from SPR sensors.15–18 Gold nanoparticles ranging from 11 to 40 nm in diameter were conjugated either to a detection antibody or directly to the analyte. Limits of detection using gold labeled biomolecules were generally enhanced by about 3 orders of magnitude, with picomolar detection reported. A similar enhancement was demonstrated using gold nanoparticle labeled biotin molecules on a streptavidin-functionalized LSPR sensor.19 The observed shift upon binding of the biotinylated gold nanoparticles was enhanced 300% as compared to streptavidin binding alone. A second LSPR study found a 10-fold improvement in LOD, from 79 to 7 nM, in the detection of biotinylated BSA upon exposure to gold nanoparticle conjugated streptavidin. 20 Here, we conjugate gold nanoparticles to antibodies and demonstrate that these gold nanoparticle antibody conjugates provide LSPR enhancements similar to those observed previously using gold nanoparticle conjugates of biotin and streptavidin. Specifically, the LSPR response upon antibody binding to a biotin-functionalized surface is enhanced up to 400%. The labeling of antibodies, rather than biotin/streptavidin, provides a signal enhancement scheme that more closely approximates the antigen-antibody assays traditionally used for biosensing. In addition, we report a binding curve for the biotin-antibiotin interaction which reveals both enhanced wavelength shifts and a stronger binding interaction upon the addition of a gold nanoparticle label.
EXPERIMENTAL METHODS
Nanoparticle Fabrication
Nanosphere lithography (NSL) was used to create monodisperse, surface-confined Ag nanoprism arrays. Glass coverslips (Fisher no. 2, 18 mm) were cleaned in a piranha solution (1:3 30% H2O2/H2SO4) at 80 °C for 30 min (Caution: Piranha solution should be handled with extreme care). Once cooled, the glass substrates were rinsed with copious amounts of mili-Q water and then sonicated for 60 min in 5:1:1 H2O/NH4OH/30% H2O2. Next, the glass was rinsed repeatedly with water and was stored in water until use. Polystyrene nanospheres (2 μL, diameter = 390 nm + 19.5 nm, Duke Scientific) were drop-coated onto the cleaned glass coverslips and allowed to dry, forming a monolayer in a close-packed hexagonal formation which served as a deposition mask. The samples were mounted into a Consolidated Vacuum Corp. vapor-deposition chamber. A Leybold Inficon XTM/2 quartz crystal microbalance was used to monitor the thickness of the metal being deposited. For all experiments, 20 nm of Ag (D. F. Goldsmith) was evaporated onto the samples. Following metal deposition, the samples were sonicated for 3–5 min in ethanol (Pharmco) to remove the polystyrene nanosphere mask, creating Ag nanoprisms on the glass substrate.
Ultraviolet-Visible Extinction Spectroscopy
Macroscale UV-vis extinction measurements were performed in standard transmission geometry with unpolarized light coupled into a photodiode array spectrometer (BWTEK, Newark, DE) using lenses. The probe diameter was approximately 1 mm. A home-built flow cell was used to control the external environment of the Ag nanoparticle substrates.
Nanoparticle Substrate Functionalization
A self-assembled monolayer (SAM) was formed on the Ag nanoprism array to stabilize the nanoparticles and allow functionalization with biotin. Nanoprism substrates were incubated in a 1 mM 3:1 ethanolic solution of octanethiol/11-mercaptoundecanoic acid (Sigma) for 12–24 h. After incubation, the substrates were rinsed with ethanol and dried with N2 prior to mounting in the flow cell. The nanoprism array was then incubated for 1 h in a 1 mM solution of amine-conjugated biotin (EZ-Link Amine-PEO3-Biotin, Pierce) in 10 mM phosphate buffered saline (PBS, Sigma) with 100 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide HCl linker (EDC, Pierce) to form an amide bond between the biotin amine group and SAM carboxylic acid group. Samples were then rinsed with 10mMPBS to remove excess EDC and biotin.
Nanoparticle-Antibody Conjugates
Antibiotin monoclonal antibodies (Sigma) were electrostatically conjugated to 20 nm Au colloids (British Biocell International). A monolayer of antibody was formed on the Au colloids by incubating a 1 mL volume of colloids, adjusted to pH 9, in 9.0 μg of antibiotin for 1 h. The colloids were then centrifuged for 1 h at 8000g and 4 °C. The supernatant was then pipetted off, and the antibody-conjugated colloids were resuspended in the volume of mili-Q H2O necessary to achieve the desired antibody concentration. Antibiotin conjugated Au colloids were then either used immediately or stored overnight at 4 °C.
Binding Affinity Assays
To determine binding affinity and LSPR shift enhancement, antibiotin or nanoparticle-antibiotin conjugates ranging from 20 pM to 1 μM in concentration were incubated for 45 min with the biotin-functionalized nanoprism arrays. Following incubation, the arrays were rinsed with mili-Q H2O and dried in N2. Extinction spectra before and after antibiotin incubation were collected in a N2 atmosphere.
RESULTS AND DISCUSSION
Biotin-specific IgG antibodies were labeled with 20 nm gold nanoparticles by taking advantage of electrostatic and covalent interactions between the antibody side chains and nanoparticle surfaces. A colloidal gold nanoparticle solution was incubated with antibiotin for an hour to allow the conjugation to occur. To verify that the antibodies had attached to the gold nanoparticles, extinction measurements of the gold nanoparticles were taken before and after the conjugation step (Figure 1). The bare gold colloids exhibited an extinction peak at 521.1 nm. Following antibody conjugation, the extinction shifted 13.1 nm to the red to give a final λmax of 534.2 nm, indicating attachment of the antibody. On the basis of the experimentally determined refractive index sensitivity of 80 nm/RIU for the 20 nm gold colloids in solution (Figure S-1), the λmax shift of 13.1 nm indicates a refractive index change of 0.16 RIU upon antibody attachment. This lies within the expected range for monolayer coverage of a protein with refractive index close to 1.5
Figure 1.
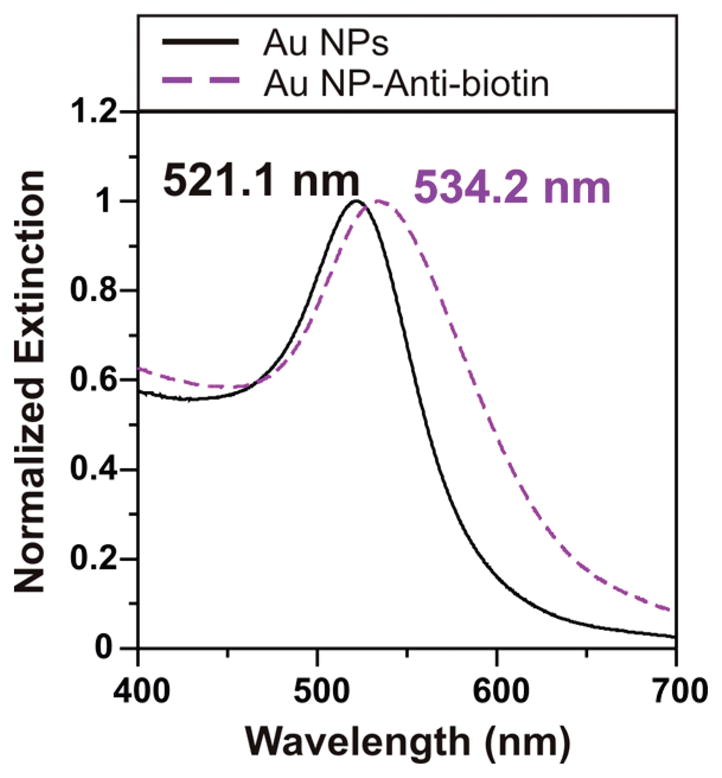
LSPR spectra confirming nanoparticle-antibody conjugation. The LSPR spectrum of bare 20 nm gold colloids shows a λmax of 521.1 nm (Au NPs, solid black). After incubation with antibiotin, the λmax shifts 13.1 nm to the red (Au NP-antibiotin, dashed purple).
To demonstrate the ability of these gold nanoparticle-conjugated antibodies (hereafter, NP-antibiotin) to bind specifically to an antigen and enhance the LSPR sensor response, we fabricated silver nanoprism arrays functionalized with biotinterminated ligands (Schematic, Figure 2A). Nanosphere lithography (NSL) was used to create ordered arrays of pyramidal silver nanoparticles on a glass surface with edge lengths of approximately 100 nm and heights of 20 nm. The nanoprisms were functionalized with a carboxylic acid-terminated self-assembled monolayer to enable amine-conjugated biotin to covalently attach to the surface. The extinction spectrum of a biotinylated LSPR sensor was recorded prior to a 45 min incubation with either NP-antibiotin or to unlabeled antibiotin. Following the antibody incubation, the extinction spectrum of the LSPR sensor was again recorded. For a solution of 100 nM unlabeled antibiotin, the λmax shifted 11 nm to the red (Figure 2B). To understand how this value compared to the enhanced LSPR shift, we performed the same experiment replacing the unlabeled antibiotin with 100 nM NP-antibiotin. The NP-antibiotin induced a Δλmax of 42.7 nm, a nearly 400% increase as compared to the unenhanced shift (Figure 2C). A nonspecific binding experiment, in which 100 nM NP-antibiotin was exposed to a SAM-functionalized LSPR sensor without biotin on the surface, showed a minimal Δλmax due to nonspecific electrostatic binding of 2.5 nm (Figure 3).
Figure 2.
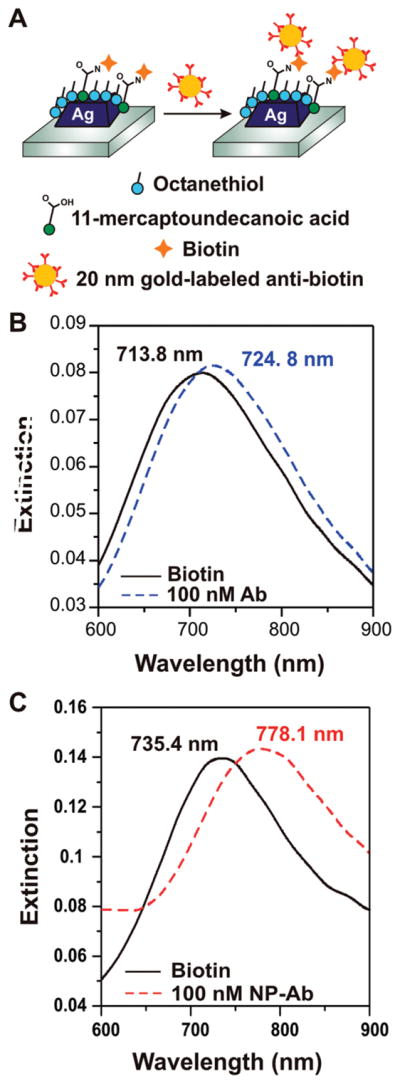
Experiment schematic and LSPR spectra. (A) Biotin is covalently linked to the nanoparticle surface using EDC coupling agent, and antibiotin labeled gold nanoparticles are subsequently exposed to the surface. LSPR spectra are collected before and after each step. (B) LSPR spectra before (solid black) and after (dashed blue) binding of native antibiotin, showing a Δλmax of 11 nm. (C) LSPR spectra before (solid black) and after (dashed red) binding of antibiotin labeled nanoparticles, showing a Δλmax of 42.7 nm.
Figure 3.
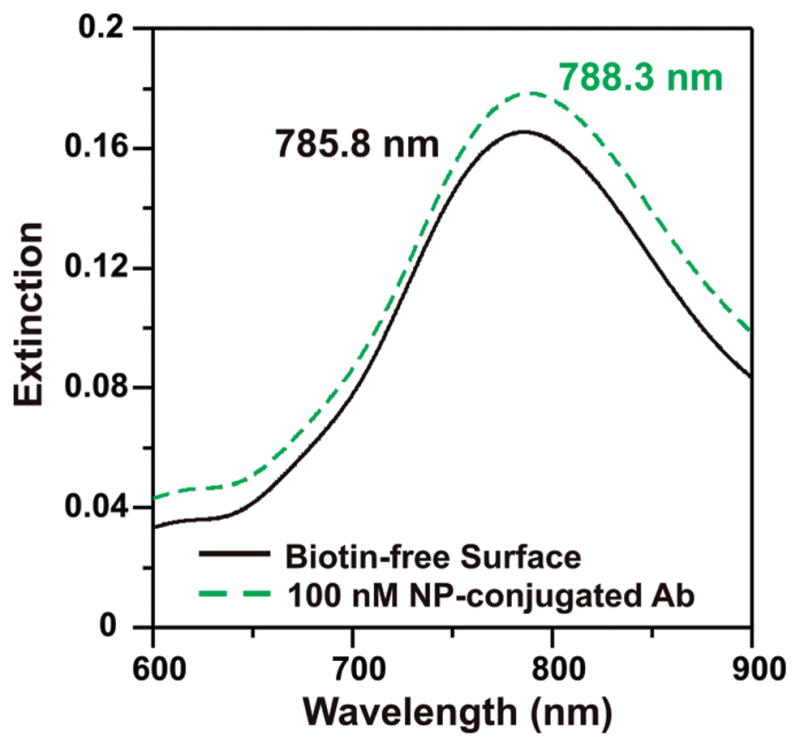
Non-specific binding assay. To test for nonspecific interactions between the gold NP-antibiotin conjugate and the LSPR sensor surface, we fabricated a nanoparticle array functionalized with a SAM of 3:1 octanethiol:11-mercaptoundecanoic acid. The SAM-functionalized array was exposed to a 100 nM concentration of NP-Ab conjugates. The resulting shift due to nonspecific interactions was 2.5 nm, indicating that the majority of the response when biotin is present on the surface is due to specific binding.
In addition to enhancing the red shift induced upon antibody binding, gold nanoparticle labels cause an increase in the LSPR bandwidth (Figure S-2). This broadening of the plasmon band does not occur in the absence of nanoparticle labels and is indicative of interparticle plasmon coupling. Thus, the nanoparticle labels act to enhance the LSPR extinction shift in two ways: (1) the nanoparticle increases the refractive index change upon antibody binding, and (2) the localized surface plasmons of the nanoparticle label couple with the plasmons of the nanoparticle substrate, causing extinction shifts that add to the refractive index-induced shifts. This coupling property depends on the distance between nanoparticle pairs and has been shown to dramatically alter the observed plasmon resonance.21–24 Gold nanoparticle labels have even made possible the detection of single DNA hybridization and cleavage events.7,25,26 Sannomiya et al. showed that DNA-mediated gold nanoparticle dimer formation induced λmax shifts of between 2 and 8 nm. Here, on the basis of surface area considerations, we expect each silver nanoprism to bind to an average of 7–8 antibody-nanoparticle conjugates at full monolayer coverage, resulting in much larger shifts as compared to simple dimer formation.
To determine the dynamic range of this signal amplification technique, we measured the LSPR shift upon antibody binding for several different concentrations of antibiotin and NP-antibiotin. By varying the concentration of NP-antibiotin from 20 pM to 20 nM and fitting to a Langmuir isotherm 1:
| (1) |
we found the association constant (Ka) between surface-bound biotin and labeled antibiotin to be 8.8 × 108 M−1 (Figure 4). Because a majority of the antibodies in the nanoparticle-antibody conjugate are unavailable for binding due to steric constraints, the LSPR response was defined as a function of nanoparticle, rather than antibody, concentration. Also, due to the antibody-nanoparticle conjugation method used here, NP-antibiotin concentrations higher than 20 nM could not be obtained. We therefore assumed a Δλmax at saturation of 60 nm for the Langmuir isotherm model, but it is likely that this value does not truly represent the maximum shift obtainable using this enhancement scheme. For comparison, a binding isotherm for unlabeled antibiotin revealed a weaker Ka of 4.0 × 106M−1 and a maximum shift of 36 nm. With a minimum detectable Δλmax of 0.3 nm, the limit of detection was improved from 2 nM to 6 pM by the addition of a gold nanoparticle label to the detection antibody. The increase in Ka in the presence of a nanoparticle label suggests that the label not only enhances LSPR shifts through combined refractive index and plasmon coupling effects, but also strengthens the biotin-antibiotin binding interaction. This strengthened binding can be explained by polyvalency in the antibody conjugate: on the basis of surface area calculations, we predict that each nanoparticle is covered with between 8 and 30 antibodies, depending on antibody orientation. Therefore, each binding event between a silver nanoprism and an NP-antibiotin conjugate is likely mediated by more than one antibody. It is well-known that polyvalent antibodies bind to antigens with higher affinity as compared to antibody monomers, and rational design of polyvalent antibodies is being used to develop novel cancer therapeutics.27–31 The polyvalency effect from tetrameric antibody domains has previously been shown to enhance the observed binding constant from zero31 to two27 orders of magnitude, with affinity enhancement arising primarily from the slower dissociation rates of tetravalent antibodies. These observations indicate identity and orientation-dependent behavior with potentially large affinity enhancements for specific antibody antigen pairs. On the basis of the Ka enhancement of 2 orders of magnitude observed in this work for the antibiotin/biotin pair, we assume that the antibiotin antibodies are oriented in a manner that sterically favors polyvalent binding to the biotinylated surface. Further studies are needed to ascertain the exact order of polyvalency, but are beyond the scope of this work. Future work will aim to more precisely control antibody orientation in the NP-Ab conjugate to optimize antibody orientation and allow even larger enhancements in the affinity constant and lower limits of detection. In addition, rational selection of both the LSPR substrate and the plasmonic label should allow additional enhancement via the plasmon coupling mechanism and will be explored as an additional route toward optimal signal enhancement.
Figure 4.
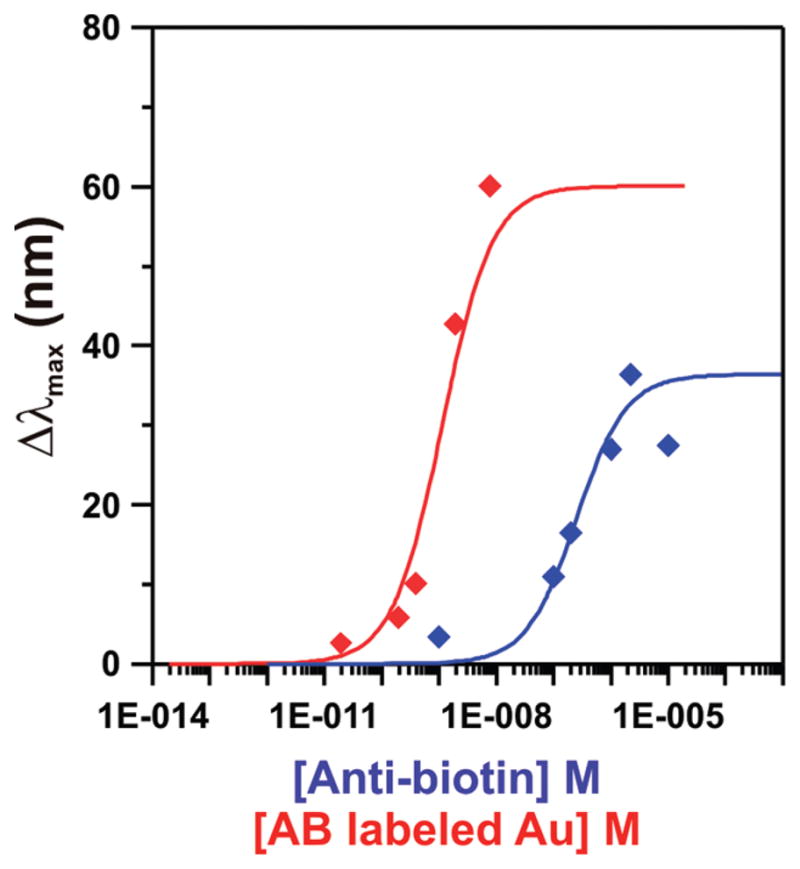
Quantitative LSPR response curves for native antibiotin (blue) and antibiotin labeled with 20 nm gold colloids (red). The extinction shift upon antibody binding was plotted as a function of either native antibody concentration or gold colloid concentration. The response curves revealed a Ka of 4 × 106 M−1 for native antibiotin and 8.8 × 108 M−1 for antibiotin labeled gold nanoparticles.
CONCLUSION
We demonstrated that using antibody-labeled gold nanoparticles to detect analytes bound on a nanoparticle surface can dramatically increase the observed response from LSPR sensors. Antibody-labeled gold nanoparticles can increase the LSPR shift by up to 400% as compared to comparable concentrations of native antibody. This increase in LSPR shift led to a limit of detection of 6 pM antibiotin, a 3 orders of magnitude improvement over the unlabeled format. In addition, the polyvalency gained by functionalizing a single gold nanoparticle with multiple antibodies led to an increase in observed binding affinity between antigen and antibody.
The observed enhancement in LSPR shift is produced by a combination of two mechanisms: (1) an overall increase in refractive index change upon antibody binding due to the presence of a gold nanoparticle, and (2) plasmonic coupling between the gold nanoparticle label and the nanoprism substrate. The degree of plasmonic coupling depends on several factors including nanoparticle size, interparticle distance, and nanoparticle orientation. In this study, we explored the effects of 20 nm diameter nanoparticle labels on the LSPR shift. We expect that by increasing the size of the nanoparticle label, or by controlling antibody orientation to bring the label closer to the nanoprism substrate, the observed LSPR shifts can be enhanced even further. Thus, the use of gold nanoparticle labels provides a way to meet the ever-increasing demands for sensitivity in diagnostic devices. When applied to relevant biological systems and biomarkers, this technique could lead to significant improvements in limits of detection for disease diagnosis.
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation (grants EEC-0647560, CHE-0911145, DMR-0520513, and BES-0507036), the National Cancer Institute (1 U54 CA119341-01), and a Ryan Fellowship to W.P.H.
Footnotes
Supporting Information. Nonspecific binding results and full-width half-maximum determinations for spectra in Figure 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nam JM, Thaxton CS, Mirkin CA. Science. 2003;301:1884–1886. doi: 10.1126/science.1088755. [DOI] [PubMed] [Google Scholar]
- 2.Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Nat Mater. 2008;7:442–453. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- 3.McFarland AD, Van Duyne RP. Nano Lett. 2003;3:1057–1062. [Google Scholar]
- 4.Yonzon CR, Jeoung E, Zou SL, Schatz GC, Mrksich M, Van Duyne RP. J Am Chem Soc. 2004;126:12669–12676. doi: 10.1021/ja047118q. [DOI] [PubMed] [Google Scholar]
- 5.Haes AJ, Chang L, Klein WL, Van Duyne RP. J Am Chem Soc. 2005;127:2264–2271. doi: 10.1021/ja044087q. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Svedendahl M, Kall M, Gunnarsson L, Dmitriev A. Nanotechnology. 2009;20:434015–434024. doi: 10.1088/0957-4484/20/43/434015. [DOI] [PubMed] [Google Scholar]
- 7.Sonnichsen C, Reinhard BM, Liphardt J, Alivisatos AP. Nat Biotechnol. 2005;23:741–745. doi: 10.1038/nbt1100. [DOI] [PubMed] [Google Scholar]
- 8.Hall WP, Anker JN, Lin Y, Modica J, Mrksich M, Van Duyne RP. J Am Chem Soc. 2008;130:5836. doi: 10.1021/ja7109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kreno LE, Hupp JT, Van Duyne RP. Anal Chem. 2010;82:8042–8046. doi: 10.1021/ac102127p. [DOI] [PubMed] [Google Scholar]
- 10.Dahlin AB, Tegenfeldt JO, Hook F. Anal Chem. 2006;78:4416–4423. doi: 10.1021/ac0601967. [DOI] [PubMed] [Google Scholar]
- 11.Munechika K, Smith JM, Chen Y, Ginger DS. J Phys Chem C. 2007;111:18906–18911. [Google Scholar]
- 12.Henzie J, Lee MH, Odom TW. Nat Nanotechnol. 2007;2:549–554. doi: 10.1038/nnano.2007.252. [DOI] [PubMed] [Google Scholar]
- 13.Zhao J, Das A, Schatz GC, Sligar SG, Duyne RPV. J Phys Chem C. 2008;112:13084–13088. [Google Scholar]
- 14.Lee HJ, Wark AW, Corn RM. Langmuir. 2006;22:5241–5250. doi: 10.1021/la060223o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buckle PE, Davies RJ, Kinning T, Yeung D, Edwards PR, Pollard-Knight D. Biosens Bioelectron. 1993;8:355–363. [Google Scholar]
- 16.Gu JH, LH, Chen YW, Liu LY, Wang P, Ma JM, Lu ZH. Supramol Sci. 1998;5:695–698. [Google Scholar]
- 17.Lyon LA, Musick MD, Natan MJ. Anal Chem. 1998;70:5177–5183. doi: 10.1021/ac9809940. [DOI] [PubMed] [Google Scholar]
- 18.He L, Musick MD, Nicewarner SR, Salinas FG, Benkovic SJ, Natan MJ, Keating CD. J Am Chem Soc. 2000;122:9071–9077. [Google Scholar]
- 19.Haes AJ, Van Duyne RP. J Am Chem Soc. 2002;124:10596–10604. doi: 10.1021/ja020393x. [DOI] [PubMed] [Google Scholar]
- 20.Kim HM, Jin SM, Lee SK, Kim MG, Shin YB. Sensors. 2009;9:2334–2344. doi: 10.3390/s90402334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su KH, Wei QH, Zhang X, Mock JJ, Smith DR, Schultz S. Nano Lett. 2003;3:1087–1090. [Google Scholar]
- 22.Gunnarsson L, Rindzevicius T, Prikulis J, Kasemo B, Kall M, Zou SL, Schatz GC. J Phys Chem B. 2005;109:1079–1087. doi: 10.1021/jp049084e. [DOI] [PubMed] [Google Scholar]
- 23.Jain PK, Huang WY, El-Sayed MA. Nano Lett. 2007;7:2080–2088. [Google Scholar]
- 24.Ross BM, Waldeisen JR, Wang T, Lee LP. Appl Phys Lett. 2009;95:193112–193114. doi: 10.1063/1.3254756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reinhard BM, Sheikholeslami S, Mastroianni A, Alivisatos AP, Liphardt J. Proc Natl Acad Sci USA. 2007;104:2667–2672. doi: 10.1073/pnas.0607826104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sannomiya T, Hafner C, Voros J. Nano Lett. 2008;8:3450–3455. doi: 10.1021/nl802317d. [DOI] [PubMed] [Google Scholar]
- 27.Rheinnecker M, Hardt C, Ilag LL, Kufer P, Gruber R, Hoess A, Lupas A, Rottenberger C, Pluckthun A, Pack P. J Immunol. 1996;157:2989–2997. [PubMed] [Google Scholar]
- 28.Ghetie MA, Podar EM, Ilgen A, Gordon BE, Uhr JW, Vitetta ES. Proc Natl Acad Sci USA. 1997;94:7509–7514. doi: 10.1073/pnas.94.14.7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghetie MA, Bright H, Vitetta ES. Blood. 2001;97:1392–1398. doi: 10.1182/blood.v97.5.1392. [DOI] [PubMed] [Google Scholar]
- 30.Zhao YF, Kohler H. J Immunother. 2002;25:396–404. doi: 10.1097/00002371-200209000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Miller K, Meng G, Liu J, Hurst A, Hsei V, Wong WL, Ekert R, Lawrence D, Sherwood S, DeForge L, Gaudreault J, Keller G, Sliwkowski M, Ashkenazi A, Presta L. J Immunol. 2003;170:4854–4861. doi: 10.4049/jimmunol.170.9.4854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.