

Abstract

Amyloid plaques and neurofibrillary tangles simultaneously accumulate in Alzheimer’s disease (AD). It is known that Aβ and tau exist together in the mitochondria; however, the interactions between Aβ oligomers and tau are controversial. Moreover, it is still unclear which specific domains in the tau protein can interact with Aβ oligomers and what could be the effect of these interactions. Herein, we examine three different Aβ–tau oligomeric complexes. These complexes present interactions of Aβ with three domains in the tau protein; all contain high β-structure propensity in their R2, R3, and R4 repeats. Our results show that, among these, Aβ oligomers are likely to interact with the R2 domain to form a stable complex with better alignment in the turn region and the β-structure domain. We therefore propose that the R2 domain can interact with soluble Aβ oligomers and consequently promote aggregation. EM and AFM images and dimensions revealed highly polymorphic tau aggregates. We suggest that the polymorphic tau and Aβ–tau aggregates may be largely due to repeat sequences which are prone to variable turn locations along the tau repeats.
In the Alzheimer brain, the two hallmark pathological lesions, first described by Dr. Alzheimer in 1906, are senile plaques(1) and neurofibrillary tangles (NFTs).(2) In Alzheimer’s disease (AD), senile plaques and NFTs simultaneously aggregate. These two aggregation processes appear to occur independently because the senile plaques are extracellular deposits which develop around nerve endings and consist primarily of Aβ oligomers, while the NFTs are intracellular lesions which develop in neuronal cell bodies and comprise primarily of aggregates of the microtubule (MT)-binding protein tau. Yet, recently it has been shown in vivo that Aβ binds to tau to form soluble stable complexes, and this binding can promote aggregation.(3) Importantly, the presence of both Aβ and tau cause a rapid dissociation of tau from the microtubules and a collapse of axonal structure—leading first to the malfunction of the synapses and ultimately to the neurons’ death.(4)
Further studies have shown synergy between the tau and Aβ pathology in the mitochondria,5−9 and it was suggested that Aβ may accelerate tau NFT formation.10−12 Recent experiments led to four different mechanisms through which Aβ can promote the tau pathology:13,14 (1) in vitro neuronal exposure to Aβ induces activation of GSK3β which increases tau phosphorylation,15−17 thus triggering cell death by destabilizing the microtubule network; (2) the inflammatory response may provide a link between Aβ and tau pathology in AD;(18) (3) the increased levels of Aβ may lead to proteasomal impairment within the brain and the degradation of tau;19,20 and finally (4) transport deficits may lead to tau pathology as readily as tau pathology may lead to transport deficit.21,22
These studies emphasize that to understand the mechanism through which Aβ–tau complexes lead to AD pathology the interactions between Aβ and tau should be probed. Tau is a natively disordered protein which binds and stabilizes microtubules and shares structural features with amyloid fibrils. Amyloid oligomers, amyloid fibrils, and fibrils of tau protein(23) have a predominantly cross-β pleated sheet conformation in which the polypeptide backbone is perpendicular to the fibril axis. The longest human tau isoform htau40 was observed to exist in the central nervous system. It consists of 441 residues and contains several domains, including the proline rich flanking regions P1 and P2 (residues 151–244) and four repeats R1–R4 (residues 244–369) (Figure 1). All four repeats constitute the microtubule-binding domain, and residues in repeats R2–R4 possess a high propensity to adopt a β-structure.24−27 Figure 1 illustrates the secondary structure of the three repeats indicating the propensity of β-structure. AFM, FT-IR, CD, and X-ray fiber diffraction experiments have shown that fragments of the β-structure in these repeats can stably interact with each other and with themselves and can aggregate into fibrils.28−30 Petterson and co-workers(31) suggested that the hexapeptides motifs 275VQIINK280 and 306VQIVYK311 in repeats R2 and R3, respectively, act as mediators of intermolecular interactions between tau monomers which were stabilized by the anionic factor heparin. EPR spectra and EM measurements also indicated that fragments in the R2 repeat of tau have a parallel in-register arrangement of β-strands.(32) Monte Carlo simulations(33) of the 306VQIVYK311 in repeat R3 have shown that the tau molecule not only promotes the formation of stable oligomers but also is capable of stabilizing these structures because such fragments are characteristically rich in β-sheet structures. The simulations(33) have shown that the 306VQIVYK311 fragment in repeat R3 has a mostly parallel arrangement of β-strands.
Figure 1.

Domain organizations of the tau isoform htau40 consists of 441 residues and contains several important domains: four pseudorepeats R1–R4 and two proline-rich flanking regions P1 and P2. The colors in the primary sequence of the pseudorepeats R2, R3, and R4 highlight the secondary structure of the repeats: β-structure (red), turn structure (blue), and disordered structure (green).
According to the experimental studies above, tau aggregation occurs via interactions between β-strand fragments in the tau protein. Furthermore, they indicate that tau interacts with Aβ. Since all amyloidogenic peptides, including the tau protein, polymerize into fibrils, have very similar properties and share similar sheet intermediates, it is conceivable that they may interact with these β-pleated sheet domains via a similar fibril formation mechanism; however, how and which interactions between amyloidogenic peptides and tau protein could be involved in fibril formation are not completely understood. Here we test interactions between tau and Aβ by constructing Aβ–tau oligomer complexes: we combine fragments of tau R2, R3, and R4 repeats with Aβ1–42 and Aβ17–42 oligomers to figure out the preferred tau repeat–Aβ interactions and organization. Our study revealed a stable Aβ–tau oligomer complex where the fragment in tau protein that interacts with Aβ to form a stable association derives from repeat R2, while those fragments from repeats R3 and R4 which interact with Aβ exhibit less stable oligomeric conformations. Nonetheless, amyloids are inherently polymorphic,34,35 and Aβ–tau complexes are also expected to present a rugged landscape, e.g., through variants of turn shapes and sizes and shifts in residue registration in β-strands via side-chain orientations. Understanding the range of structural features of the aggregates is important for effective drug design to reduce aggregate formation.
Materials and Methods
Molecular Dynamics (MD) Simulations Protocol
MD simulations of solvated tau and tau–Aβ oligomers were performed in the NPT ensemble using the NAMD program(36) with the CHARMM27 force field.37,38 The oligomers were energy minimized and explicitly solvated in a TIP3P water box39,40 with a minimum distance of 15 Å from any edge of the box to any Aβ atom. Any water molecule within 2.5 Å of the Aβ was removed. Counterions Na+ were added at random locations to neutralize the Aβ charge. The Langevin piston method36,41,42 with a decay period of 100 fs and a damping time of 50 fs was used to maintain a constant pressure of 1 atm. The temperature 330 K was controlled by Langevin thermostat with a damping coefficient of 10 ps–1.(36) The short-range van der Waals (VDW) interactions were calculated using the switching function, with a twin range cutoff of 10.0 and 12.0 Å. Long-range electrostatic interactions were calculated using the particle mesh Ewald method with a cutoff of 12.0 Å.43,44 The equations of motion were integrated using the leapfrog integrator with a step of 1 fs.
The solvated systems were energy minimized for 2000 conjugated gradient steps, where the hydrogen bonding distance between the β-sheets in Aβ17–42 is fixed in the range 2.2–2.5 Å. The counterions and water molecules were allowed to move. The hydrogen atoms were constrained to the equilibrium bond using the SHAKE algorithm.(45) The minimized solvated systems were energy minimized for additional 5000 conjugate gradient steps and 20 000 heating steps at 250 K, with all atoms allowed to move. Then, the systems were heated from 250 to 300 K for 300 ps and equilibrated at 330 K for 300 ps. All simulations ran for 30 ns. These conditions are applied to all cases.
Generalized Born Method with Molecular Volume (GBMV)
To obtain the relative structural stability of tau, Aβ, and tau–Aβ oligomers, the oligomer trajectories of the last 5 ns were first extracted from the explicit MD simulation excluding water molecules. The solvation energies of all systems were calculated using the generalized Born method with molecular volume (GBMV).46,47 In the GBMV calculations, the dielectric constant of water was set to 80. The hydrophobic solvent-accessible surface area (SASA) term factor was set to 0.005 92 kcal/(mol Å2). Each conformer is minimized using 1000 cycles, and the conformational energy is evaluated by grid-based GBMV. The minimization does not change the conformations of each conformer; it only relaxes the local geometries due to thermal fluctuation which occurred during the MD simulations.
Analysis Details
We examined the structural stability of the studied oligomers by following the changes in the number of the hydrogen bonds between β-strands with the hydrogen bond cutoff set to 2.5 Å and by monitoring the change in the intersheet distance (Cα backbone–backbone distance) in the core domain of all of the oligomers. In all models we constructed, the core domain is between residue 22 and residue 35, as previously done by Miller et al.(34)
We further investigated the averaged water molecules around each side-chain Cβ carbon within 4 Å, for the tau oligomers from repeat R2 with and without interactions with Aβ17–42 and, similarly, Aβ1–42.
Results
Construction of the Conformational Ensemble of Aβ with Tau Repeats
Interactions between the Aβ amyloid and the tau protein are more likely to appear in regions of residual β-structure in the tau protein that have the potential to serve as seeds for aggregation. Mutations in tau repeats were shown to lead to a strong tendency for aggregation which correlates with their high propensity of β-structure.(48) Our constructed Aβ oligomers, tau oligomers, and Aβ–tau oligomeric complexes are based on the NMR model of Lührs et al.(49) for the Aβ17–42 structure and on recent experimental data25,50 that provide some details of the secondary structure of the tau protein, such as β-structure and the position of the U-turn in the sequence. We also constructed Aβ–tau oligomeric complexes using the NMR model of Tycko and co-workers;(51) however, our simulations revealed unstable complexes (Figure S1). To match tau with Aβ17–42 oligomers, we extracted tau fragments that have the ability to serve as seeds for aggregation. The highest propensity of β-structure content in the tau protein is in repeats R2, R3, and R4 (Figure 1). These repeats are known to be involved in the abnormal aggregation of tau.25,50 Moreover, the high propensity of β-structures in these repeats is also known to be essential for paired helical filaments formation.28,48 In addition to the β-structure in the repeats, we also consider other similar structural properties between Aβ and each of the R2, R3, and R4 repeats. More specifically, we consider the tendency to form stable interactions between Aβ and tau repeats in both the β-structure domain and the U-turn domain, e.g., hydrophobic interactions, electrostatic interactions, polar interactions, and salt bridge that stabilize the turn. Consequently, the tau repeats consist of two β-strands connected by U-turn, similarly to Aβ peptides.
Recent EM and AFM images and dimensions of the tau fibrils(52) revealed that only the R2, R3, and R4 tau repeats can fit. It is likely that the other tau protein domains that do not have a preponderance for β-structure “hang out” and are not part of the regular fibrillar organization. In constructing the fibril, we assume that the conventional turn shape in Aβ oligomers, which is also observed in other amyloids,53,54 can act as seed for fragments of tau repeat oligomers and examine whether the simulated tau and Aβ–tau oligomers can retain the β-strand-turn-β-strand shape. Our assumption is based on the fact that amyloids from different species or with different sequences (such as in prion) have close structural similarity.(55) We further assume that the secondary structures of octamers of the tau repeats R2, R3, and R4 studied here are similar to those of Aβ oligomers, as illustrated by experimental studies25,50 and detailed below.
To construct Aβ–tau oligomeric complexes, we used tetramers of repeats R2, R3, and R4 and associated each tetramer with Aβ17–42 tetramers. We first test ensemble of alignments between Aβ17–42 and sequences in tau repeats which maximize the overlap between the hydrophobic residues. For example, hydrophobic residues in the 275VQIINK280 motif in repeat R2 can match the hydrophobic residues 17LVFFAE22 in Aβ (Figure 2), and the U-turn and C-terminal regions of Aβ can then superimpose on the continuous repeat sequence in tau. Similarly, the fitting of the hydrophobic residues in 306VQIVY310 motif in repeat R3 matches the Aβ 17LVFFA21 segment (Figure 3) and the R4 repeat 337VEVK310 motif matches the Aβ 17LVFF20 segment (Figure 4). The final alignments are indicated in Figures 2, 3, and 4 for the R2, R3, and R4 repeats, respectively. As can be seen for repeat R2, four hydrophobic interactions between tau and Aβ appear in one β-strand and two hydrophobic interactions appear in the second. For repeat R3, five hydrophobic interactions between tau and Aβ appear in the first strand and two hydrophobic interactions in the second. Finally, for repeat R4, three hydrophobic interactions between tau and Aβ appear in one strand and two hydrophobic interactions appear in the second.
Figure 2.

Initial structural models: (A) Aβ17–42 tetramer interacts with tau tetramer and (B) tau octamer. The tau protein fragment of each monomer is taken from the repeat R2. Blue arrows illustrate hydrophobic interactions between Aβ17–42 and repeat R2.
Figure 3.

Initial structural models: (A) Aβ17–42 tetramer interacts with tau tetramer and (B) tau octamer. The tau protein fragment of each monomer is taken from repeat R3. Blue arrows illustrate hydrophobic interactions between Aβ17–42 and repeat R3.
Figure 4.

Initial structural models: (A) Aβ17–42 tetramer interacts with tau tetramer and (B) tau octamer. The tau fragment of each monomer is taken from repeat R4. Blue arrows illustrate hydrophobic interactions between Aβ17–42 and repeat R4.
Considering the β-structures and the turn regions in repeats R2, R3, and R4 in tau,(50) we constructed three parallel octamers of 26 continuous residues from each repeat which form organizations similar to those obtained previously by Miller et al.(34) for Aβ17–42 octamers: the first octamer consists of a fragment in repeat R2, residues V275–V300 (Figure 2), the second of a fragment in R3, residues V306–K331 (Figure 3), and the third octamer consists of a fragment in R4, residues V337–H362 (Figure 4).
We further constructed the full-length Aβ1–42 dodecamer by applying the Lührs model(49) (PDB code ID: 2BEG) for Aβ17–42. Then, for each monomer we linked residues 1–16 in a manner similar to our recent construction of the tubular Aβ1–42 fibrils.(56) For the tau dodecamer, only the R2 repeat was used; however, we extended the sequence in the N-terminal for the structure shown in Figure 2A, i.e., each monomer in the tau oligomer consists of residues K259–V300. Finally, we constructed the Aβ1–42–tau dodecamer complex by interacting hexamers of Aβ1–42 with hexamers of the tau fragment K259–V300. Figure 5 illustrates the tau, Aβ1–42 and Aβ1–42–tau oligomers.
Figure 5.

Initial structural models: (A) Aβ1–42 tetramer interacts with tau tetramer and (B) tau octamer. The tau protein fragment of each monomer is taken from repeat R2.
Reaction Coordinates for the Formation of Aβ–Tau Oligomeric Complexes from Aβ and Tau Oligomers Demonstrate That the R2 Repeat in Tau Is More Likely to Interact with Aβ
To investigate the stability of each soluble Aβ–tau oligomeric complex for each of the fragments in repeats R2, R3, and R4 interacting with Aβ, the conformational energies were computed for Aβ oligomers, tau oligomers, and the Aβ–tau oligomeric complexes (Table 1). The conformational energies for each oligomer are based on the energy computed with the generalized Born method with molecular volume (GBMV).46,47 We estimate the relative stability of each Aβ–tau oligomeric complex by comparing its energy with the two separate components, Aβ and tau oligomers, as illustrated by the following chemical “reaction”:
where n indicates the number of monomers within an oligomer. Figure 6 demonstrates the reaction coordinates for the three repeats. As seen from Figure 6, the Aβ–tau R2 repeat oligomeric complex is more stable energetically than its two separate components, Aβ and tau oligomers. One can view the reaction coordinates for R2 as an exothermic reaction, where the product (Aβ–tau oligomeric complex) is more stable than the reactants (Aβ and tau oligomers). However, for the repeats R3 and R4, the picture is the opposite: the two separate Aβ and tau oligomer components are more stable than the Aβ–tau oligomeric complex. Hence, these relative stabilities calculations indicate that repeat R2 in the tau protein is more likely to have the ability to interact with Aβ to form a stable oligomeric complex.
Table 1. Conformational Energy of the Constructed Oligomers.
| oligomer model | conformational energya(kcal/mol) | Figure |
|---|---|---|
| Aβ17–42 | –2244.66 (103.01) | Fig. 6 and Fig. 1B in Ref. (34) |
| Aβ17–42–tau R2 | –4238.12 (99.15) | Fig. 2A |
| Aβ17–42–tau R3 | –2757.63 (104.40) | Fig. 3A |
| Aβ17–42–tau R4 | –5319.35 (116.07) | Fig. 4A |
| Tau R2 | –6206.32 (127.23) | Fig. 2B |
| Tau R3 | –3387.86 (106.66) | Fig. 3B |
| Tau R4 | –8580.03 (128.77) | Fig. 4B |
Conformational energies were computed using the GBMV calculations. The standard deviation values are presented in parentheses.
Figure 6.

Relative energies of the two separate components Aβ and tau and of Aβ–tau complex for repeats R2, R3, and R4.
Tau Repeat R2 Oligomers Structurally Stabilize the Aβ Oligomers
While parallel Aβ oligomers are stable in experiment and simulations, experimental structural data of soluble Aβ–tau oligomeric complexes are unavailable. Herein, we test models for parallel Aβ–tau oligomeric complexes and parallel tau oligomers. We investigate their conformational stability by analysis of the hydrophobic association among the β strands.
Previously, Miller et al.(34) demonstrated that for the Aβ17–42 parallel octamers based on the Lührs model >70% of the hydrogen bonds are retained during the 30 ns simulations, and the β-sheet distances across the U-turn are in the 12–13 Å range. Here, for the Aβ17–42–tau R2 oligomeric complex, >70% of the hydrogen bonds are also retained during the simulations, and assisted by the backbone hydrogen bonds, the association is slightly better among the β-strands compared with the Aβ17–42 parallel octamer (Figure 7A). We also examined three different β-sheet distances across the U-turn for the Aβ17–42–tau oligomeric complexes: between two β-sheets of Aβ, between two β-sheets of the tau repeat, and between a monomer of Aβ and a nearby monomer of a tau repeat (Figure 8). All three β-sheet distances across the U-turn for Aβ17–42–tau R2 oligomeric complex are in the ∼12–13 Å range, indicating that the tau R2 oligomers stabilize the Aβ17–42 oligomers. In contrast, both Aβ17–42–tau R3 and Aβ17–42–tau R4 oligomeric complexes demonstrate different values, in the range of 12–15 and 10–20 Å, respectively. Proline residues are disfavored in β-structures. In repeat R3, Pro312 may explain the relatively large β-sheet distances across the U-turn for Aβ17–42–tau R3 oligomers. The large β-sheet distances across the U-turn for Aβ17–42–tau R4 are due to the few hydrophobic interactions between the two β-strands (three hydrophobic interactions between the Aβ peptide and tau repeat in one strand and only two appear in the second). Although there are similarities in the structural properties in the turn region for Aβ17–42–tau for both repeats R3 and R4, the interactions between the β-strands are probably more important for structural stability than the turn region. Although the Aβ17–42–tau R3 and Aβ17–42–tau R4 oligomeric complexes present only slightly less than 70% of their hydrogen bonds for the last 10 ns of the simulations (Figure 7A), the differences among the β-sheet distances suggest that the repeats R3 and R4 are less preferred to interact with Aβ (Figure 9B,C).
Figure 7.
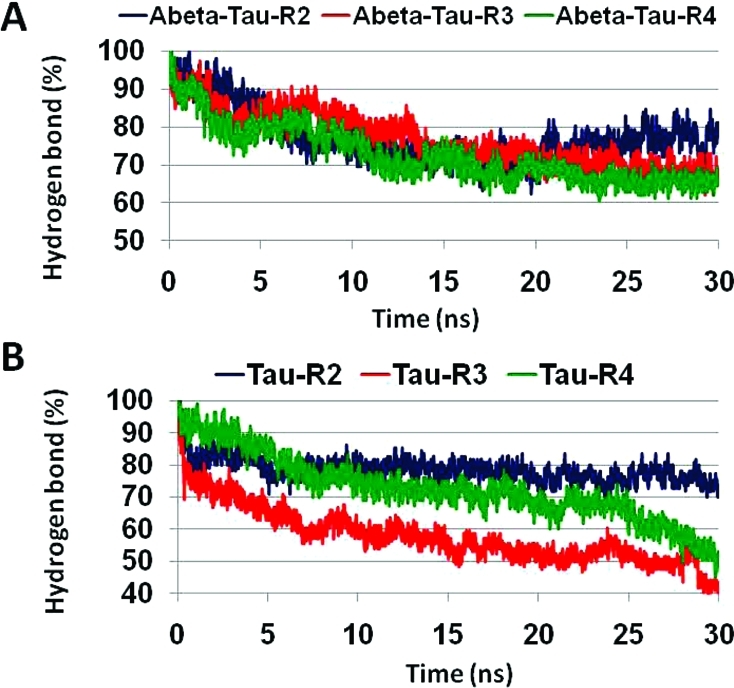
Fraction of the number of hydrogen bonds (in percentage) between all β-strands compared with the initial oligomer structures of Aβ–tau for repeats R2, R3, and R4 (A) and of tau oligomers for repeats R2, R3, and R4 (B).
Figure 8.
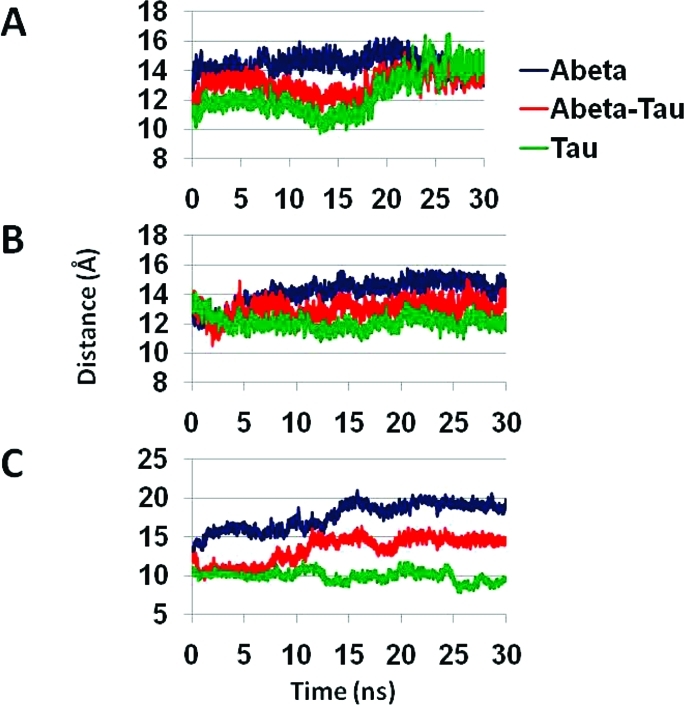
Cα backbone–backbone distance of Aβ–tau for repeats (A) R2, (B) R3, and (C) R4. Three different distances were examined: averaged distance between two Aβ peptides (blue), between Aβ peptide and tau monomer (red), and the averaged distance between two monomers of tau (green).
Figure 9.
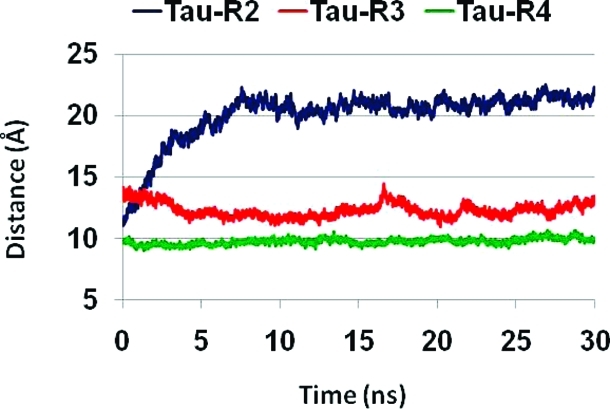
Cα backbone–backbone distance of tau oligomers for repeats R2, R3, and R4.
Interestingly, in tau oligomers consisting of repeats R3 and R4, the percentage of hydrogen bonds that are retained during the simulations are less than 60% for R3 and less than 50% for R4, while in repeat R2, >70% of the hydrogen bonds are retained. Hence, the tau oligomer with the R2 repeat has a better association among the β-strands assisted by backbone hydrogen bonds (Figure 7B), indicating that repeat R2 has a high propensity of β-structures that may interact with Aβ to form a stable oligomer. We note that the percentages of hydrogen bonds in Aβ–tau oligomeric complexes for all repeats and in particular for R3 and R4 are higher than in tau oligomers that seem to have similar structural properties. The reason for this difference could lie in the electrostatic interactions between the β-strands: for Aβ–tau R2, two (Aβ Glu22–tau Lys280; Aβ Asp23–tau K281); for Aβ–tau R3, one (Aβ Glu22–tau K311); and for Aβ–tau R4, one interaction (Aβ Asp23–tau K343). Yet, the β-sheet distances expand to 20–21 Å for tau R2 oligomers; therefore, the R2 repeat interacts more favorably with Aβ than with itself.
Solvation of Backbone Residues in Tau and Aβ–tau Oligomeric Complexes Indicate the Stability of the Complexes
We further examine the stability of the Aβ–tau R2 oligomeric complexes by investigating the backbone solvation. To this aim, we compared the backbone solvation of tau oligomers from the R2 repeat with the tau oligomers from the R2 repeat that are complexed with Aβ oligomers. Figure 10 illustrates the backbone solvation values for each residue in both tau–Aβ17–42 and tau–Aβ1–42 and the corresponding tau oligomers. For both tau oligomers, with 26 residues in the R2 repeat and the tau–Aβ17–42 complex, the backbone solvation values for all residues are similar, except for residue 37. The solvation is related also to the character of residue 37 in the sequences for the oligomers: in the Aβ sequence, it is G37 while in the tau it is D37, indicating that small residues are more solvated. Thus, overall, tau and tau–Aβ17–42 oligomers illustrate similar stabilities. This result is in agreement with the hydrogen bond analysis for these oligomers (Figure 7): both oligomers have relatively high percentage of hydrogen bonds during the simulations.
Figure 10.

Average water molecules around each side chain Cβ carbon within 4 Å.
For the full-length tau–Aβ1–42 oligomers, the backbone solvation illustrates smaller values compared with the tau oligomers (Figure 7), indicating that the tau–Aβ1–42 oligomers have better β-sheet interactions and thus higher stability than the tau oligomers.
Discussion
The aggregation of tau into NFTs, and Aβ into amlyoids, are processes long known to be related to AD progression. More recent experimental studies led to the proposition that Aβ may directly or indirectly interact with tau to accelerate NFTs formation.4−8,10−14 Yet, the mechanism and the related interactions through which tau and Aβ self-assemble together have been controversial. Herein, we investigate the interactions between tau oligomers and Aβ oligomers at the molecular level to figure out the preferred organizational state. Using all-atom MD simulations in explicit solvent, we investigated the parallel oligomeric architectures of three different repeats in the tau protein—R2, R3, and R4—and the interactions of these parallel tau architectures with Aβ17–42 oligomers. While the absence of experimental examination of the interactions of purified tau variants with Aβ oligomers limits our work to the prediction of the interactions through computational tools, our models are based on experimental data: the oligomers were based on the experimental model by Lührs et al.,(49) as previously studied by Miller et al.(34) for Aβ17–42; and the tau oligomer models are also based on experimental data which illustrate high propensity of β-structure in tau repeats R2, R3, and R4 pointing to likely intermolecular β-sheet interactions. In addition to the β-structure of the peptide 306VQIVYK311 in repeat R3,(28) other studies by NMR spectroscopy,57,58 CD measurements,(59) and FTIR spectroscopy(60) suggest α-helical structures in repeat R3. Herein, we did not test these α-helical structures, and further investigations will need to be performed.
In our proposed Aβ–tau models, we consider two main factors: hydrophobicity and flexibility of these domains. The hydrophobicity factor plays a key role in forming hydrophobic core and enhancing the interactions between Aβ oligomers and tau oligomers. Recently, hydrophobicity and flexibility were illustrated to be the primary factors for oligomer assembly.(61) A recent study(62) speculated that Aβ oligomers may be responsible for accumulation of the transactive response DNA-binding protein 43 (TDP-43). Further studies are needed to elucidate the nature of the Aβ–TDP-43 interactions.
Our study leads to a major conclusion: Aβ oligomers interact better directly with fragments of the tau repeat R2 than repeats R3 and R4. Further experiments, such as surface plasmon resonance may be useful to investigate the affinities of the various tau repeats to Aβ oligomers. The U-turn root-mean-square deviations (RMSDs) indicate that the oligomer of Aβ17–42 that interacts with fragments of repeat R2 is slightly more stable than the Aβ17–42 oligomer that interacts with fragments of repeats R3 or R4 (Figure S2). Figure S3 illustrates that the U-turn RMSDs of tau repeats R2 and R3 oligomers are similar to values observed previously(34) for Aβ17–42, and the RMSDs of oligomers of tau repeats R4 are slightly larger. Overall, the R2, R3, and R4 oligomers are structurally stable in the turn region; however, the β-sheet distances exhibit that the R2 oligomer cannot form a stable oligomer alone, while the tau repeats R3 and R4 oligomers can form stable structures (Figure 9). Nonetheless, the tau R2 oligomer is the more likely candidate to interact with Aβ17–42 as compared to R3 and R4 oligomers (Figure 8). We emphasize, however, that because amyloids including Aβ are polymorphic,(63) Aβ–tau complexes are also expected to be polymorphic, either in their gross organizations or on a minor scale. The polymorphic range can be broad, as was observed by cryo-EM, ssNMR, and modeling, with and without ion binding.34,35,56,64−66 Two likely features that can lead to polymorphism is the U-turn shape, and the organization between the sequences of tau repeats with respect to the sequence of Aβ. Additional arrangements of the Aβ and tau repeats which relate to the shift of the Aβ sequence along the tau repeats can expand the polymorphic range of the complexes. In addition, populations of the tau can be shifted: while the R2 oligomer cannot form a stable oligomer alone, it gets stabilized when interacting with Aβ oligomer, thus shifting the tau ensemble. Recent EM and AFM measurements revealed highly polymorphic fibrils for tau52,67,68 that differ significantly in fibril parameters such as cross section and twist. These measurements illustrate irregular fibril structures with no symmetry which most likely relate to the turn shapes in the tau repeats. Since the U-turn conformations in the tau repeats may differ as the sequence registration is shifted, the cross sections may be different, and consequently the number of protofilaments that wind around each other to form a mature fibril may also be different. In summary, the U-turn appears to play a key role in the amyloid polymorphic landscape, shifting the populations of the oligomers, fibrils, and gross-scale organization toward different states, as observed in the EM and AFM images and dimensions.
The hydrophobic regions in both R2 and R3 interact with the microtubules (MTs) and stabilize them. In AD, tau becomes excessively phosphorylated, loses its ability to bind to MTs, and aggregates into NFTs. One can argue that when tau loses its ability to bind to MTs, it interacts with Aβ to form a more stable oligomer, accelerating aggregation. We further suggest that when Aβ interacts directly with tau repeat R2, the tau does not interact with the MTs, and consequently MTs degradation takes place, leading to toxicity. Mutations in tau in repeats R2, R3, and R4 showed no decrease in the binding of tau to the MTs.(69) However, tau mutations lead to faster aggregation into paired helical filaments (PHFs).69−74 Some of the mutations may weaken tau’s ability to bind to MTs75−77 and thus cause degradation of the MT and toxicity. A recent study(78) indicated that Aβ oligomers induce tau aggregation and formation of β-sheet rich neurotoxic tau oligomers; yet the mechanism for the toxicity is still controversial, and further investigations will need to be performed.
Acknowledgments
All simulations had been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Glossary
Abbreviations
- NFT
neurofibrillary tangles
- MT
microtubule
- AD
Alzheimer’s disease
- MD
molecular dynamics
- R
tau repeat.
Supporting Information Available
Figures S1–S3. This material is available free of charge via the Internet at http://pubs.acs.org.
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Selkoe D. J.; Schenk D. (2003) Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu. Rev. Pharmacol. Toxicol. 43, 545–584. [DOI] [PubMed] [Google Scholar]
- Mandelkow E. M.; Mandelkow E. (1998) Tau in Alzheimer’s disease. Trends Cell Biol. 8, 425–427. [DOI] [PubMed] [Google Scholar]
- Guo J. P.; Arai T.; Miklossy J.; McGeer P. L. (2006) Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 103, 1953–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M. E.; Kan H. M.; Baas P. W.; Erisir A.; Glabe C. G.; Bloom G. S. (2006) Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J. Cell Biol. 175, 541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein V.; Song X.; Wiesner A.; Ittner L. M.; Baysang G.; Meier F.; Ozmen L.; Bluethmann H.; Drose S.; Brandt U.; Savaskan E.; Czech C.; Gotz J.; Eckert A. (2009) Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. U.S.A. 106, 20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David D. C.; Hauptmann S.; Scherping I.; Schuessel K.; Keil U.; Rizzu P.; Ravid R.; Drose S.; Brandt U.; Muller W. E.; Eckert A.; Gotz J. (2005) Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 280, 23802–23814. [DOI] [PubMed] [Google Scholar]
- Hauptmann S.; Keil U.; Scherping I.; Bonert A.; Eckert A.; Muller W. E. (2006) Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp. Gerontol. 41, 668–673. [DOI] [PubMed] [Google Scholar]
- Eckert A.; Hauptmann S.; Scherping I.; Rhein V.; Muller-Spahn F.; Gotz J.; Muller W. E. (2008) Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener. Dis. 5, 157–159. [DOI] [PubMed] [Google Scholar]
- Gotz J.; Lim Y. A.; Ke Y. D.; Eckert A.; Ittner L. M. (2010) Dissecting Toxicity of Tau and beta-Amyloid. Neurodegener. Dis. 7, 10–12. [DOI] [PubMed] [Google Scholar]
- Lewis J.; Dickson D. W.; Lin W. L.; Chisholm L.; Corral A.; Jones G.; Yen S. H.; Sahara N.; Skipper L.; Yager D.; Eckman C.; Hardy J.; Hutton M.; McGowan E. (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. [DOI] [PubMed] [Google Scholar]
- Gotz J.; Chen F.; van Dorpe J.; Nitsch R. M. (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. [DOI] [PubMed] [Google Scholar]
- Oddo S.; Caccamo A.; Shepherd J. D.; Murphy M. P.; Golde T. E.; Kayed R.; Metherate R.; Mattson M. P.; Akbari Y.; LaFerla F. M. (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M.; Laferla F. M. (2006) Pathways by which Abeta facilitates tau pathology. Curr. Alzheimer Res. 3, 437–448. [DOI] [PubMed] [Google Scholar]
- LaFerla F. M. (2010) Pathways linking Abeta and tau pathologies. Biochem. Soc. Trans. 38, 993–995. [DOI] [PubMed] [Google Scholar]
- Takashima A.; Noguchi K.; Sato K.; Hoshino T.; Imahori K. (1993) Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 90, 7789–7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J.; Lorenzo A.; Yeh J.; Yankner B. A. (1995) beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14, 879–888. [DOI] [PubMed] [Google Scholar]
- Alvarez A.; Toro R.; Caceres A.; Maccioni R. B. (1999) Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 459, 421–426. [DOI] [PubMed] [Google Scholar]
- Cruz J. C.; Tseng H. C.; Goldman J. A.; Shih H.; Tsai L. H. (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40, 471–483. [DOI] [PubMed] [Google Scholar]
- Keller J. N.; Hanni K. B.; Markesbery W. R. (2000) Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 75, 436–439. [DOI] [PubMed] [Google Scholar]
- Oh S.; Hong H. S.; Hwang E.; Sim H. J.; Lee W.; Shin S. J.; Mook-Jung I. (2005) Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech. Ageing Dev. 126, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Oddo S.; Caccamo A.; Kitazawa M.; Tseng B. P.; LaFerla F. M. (2003) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 24, 1063–1070. [DOI] [PubMed] [Google Scholar]
- Khatoon S.; Grundke-Iqbal I.; Iqbal K. (1994) Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. 351, 80–84. [DOI] [PubMed] [Google Scholar]
- Berriman J.; Serpell L. C.; Oberg K. A.; Fink A. L.; Goedert M.; Crowther R. A. (2003) Tau filaments from human brain and from in vitro assembly of recombinant protein show cross-beta structure. Proc. Natl. Acad. Sci. U.S.A. 100, 9034–9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margittai M.; Langen R. (2004) Template-assisted filament growth by parallel stacking of tau. Proc. Natl. Acad. Sci. U.S.A. 101, 10278–10283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukrasch M. D.; Biernat J.; von Bergen M.; Griesinger C.; Mandelkow E.; Zweckstetter M. (2005) Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. J. Biol. Chem. 280, 24978–24986. [DOI] [PubMed] [Google Scholar]
- Mukrasch M. D.; von Bergen M.; Biernat J.; Fischer D.; Griesinger C.; Mandelkow E.; Zweckstetter M. (2007) The “jaws” of the tau-microtubule interaction. J. Biol. Chem. 282, 12230–12239. [DOI] [PubMed] [Google Scholar]
- Eliezer D.; Barre P.; Kobaslija M.; Chan D.; Li X.; Heend L. (2005) Residual structure in the repeat domain of tau: echoes of microtubule binding and paired helical filament formation. Biochemistry 44, 1026–1036. [DOI] [PubMed] [Google Scholar]
- von Bergen M.; Friedhoff P.; Biernat J.; Heberle J.; Mandelkow E. M.; Mandelkow E. (2000) Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goux W. J.; Kopplin L.; Nguyen A. D.; Leak K.; Rutkofsky M.; Shanmuganandam V. D.; Sharma D.; Inouye H.; Kirschner D. A. (2004) The formation of straight and twisted filaments from short tau peptides. J. Biol. Chem. 279, 26868–26875. [DOI] [PubMed] [Google Scholar]
- Barrantes A.; Sotres J.; Hernando-Perez M.; Benitez M. J.; de Pablo P. J.; Baro A. M.; Avila J.; Jimenez J. S. (2009) Tau aggregation followed by atomic force microscopy and surface plasmon resonance, and single molecule tau-tau interaction probed by atomic force spectroscopy. J. Alzheimers Dis. 18, 141–151. [DOI] [PubMed] [Google Scholar]
- Peterson D. W.; Zhou H.; Dahlquist F. W.; Lew J. (2008) A soluble oligomer of tau associated with fiber formation analyzed by NMR. Biochemistry 47, 7393–7404. [DOI] [PubMed] [Google Scholar]
- Margittai M.; Langen R. (2006) Side chain-dependent stacking modulates tau filament structure. J. Biol. Chem. 281, 37820–37827. [DOI] [PubMed] [Google Scholar]
- Li D. W.; Mohanty S.; Irback A.; Huo S. (2008) Formation and growth of oligomers: a Monte Carlo study of an amyloid tau fragment. PLoS Comput. Biol. 4, e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Nussinov R. (2009) Polymorphism of Alzheimer’s Abeta17–42 (p3) oligomers: the importance of the turn location and its conformation. Biophys. J. 97, 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Nussinov R. (2010) Polymorphism in Alzheimer Abeta amyloid organization reflects conformational selection in a rugged energy landscape. Chem. Rev. 110, 4820–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale L.; Skeel R.; Bhandarkar M.; Brunner R.; Gursoy A.; Krawetz N.; Phillips J.; Shinozaki A.; Varadarajan K.; Schulten K. (1999) NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312. [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. (1983) Charmm - a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem. 4, 187–217. [Google Scholar]
- Mahoney M. W.; Jorgensen W. L. (2000) A five-site J;model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 112, 8910–8922. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. (1994) Constant-Pressure Molecular-Dynamics Algorithms. J. Chem. Phys. 101, 4177–4189. [Google Scholar]
- Feller S. E.; Zhang Y. H.; Pastor R. W.; Brooks B. R. (1995) Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J. Chem. Phys. 103, 4613–4621. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle Mesh Ewald - an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 103, 8577–8593. [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Lee M. S.; Feig M.; Salsbury F. R.; Brooks C. L. (2003) New analytic approximation to the standard molecular volume definition and its application to generalized born calculations. J. Comput. Chem. 24, 1348–1356. [DOI] [PubMed] [Google Scholar]
- Lee M. S.; Salsbury F. R.; Brooks C. L. (2002) Novel generalized Born methods. J. Chem. Phys. 116, 10606–10614. [Google Scholar]
- von Bergen M.; Barghorn S.; Li L.; Marx A.; Biernat J.; Mandelkow E. M.; Mandelkow E. (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. J. Biol. Chem. 276, 48165–48174. [DOI] [PubMed] [Google Scholar]
- Luhrs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Dobeli H.; Schubert D.; Riek R. (2005) 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukrasch M. D.; Bibow S.; Korukottu J.; Jeganathan S.; Biernat J.; Griesinger C.; Mandelkow E.; Zweckstetter M. (2009) Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 7, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T.; Yau W. M.; Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S.; Jung Y. J.; Chinnathambi S.; Mandelkow E. M.; Mandelkow E.; Muller D. J. (2010) Human Tau isoforms assemble into ribbon-like fibrils that display polymorphic structure and stability. J. Biol. Chem. 285, 27302–27313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson N.; Becker J.; Tidow H.; Tremmel S.; Sharpe T. D.; Krause G.; Flinders J.; Petrovich M.; Berriman J.; Oschkinat H.; Fersht A. R. (2006) General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. U.S.A. 103, 16248–16253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladner C. L.; Chen M.; Smith D. P.; Platt G. W.; Radford S. E.; Langen R. (2010) Stacked sets of parallel, in-register beta-strands of beta2-microglobulin in amyloid fibrils revealed by site-directed spin labeling and chemical labeling. J. Biol. Chem. 285, 17137–17147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L.; Teng P. K.; Riek R.; Eisenberg D. (2010) Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. U.S.A. 107, 3487–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Tsai C. J.; Nussinov R. (2010) Hollow core of Alzheimer’s Abeta42 amyloid observed by cryoEM is relevant at physiological pH. Proc. Natl. Acad. Sci. U.S.A. 107, 14128–14133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoura K.; Tomoo K.; Ishida T.; Hasegawa H.; Sasaki M.; Taniguchi T. (2002) Amphipathic helical behavior of the third repeat fragment in the tau microtubule-binding domain, studied by (1)H NMR spectroscopy. Biochem. Biophys. Res. Commun. 294, 210–214. [DOI] [PubMed] [Google Scholar]
- Minoura K.; Yao T. M.; Tomoo K.; Sumida M.; Sasaki M.; Taniguchi T.; Ishida T. (2004) Different associational and conformational behaviors between the second and third repeat fragments in the tau microtubule-binding domain. Eur. J. Biochem. 271, 545–552. [DOI] [PubMed] [Google Scholar]
- Mendieta J.; Fuertes M. A.; Kunjishapatham R.; Santa-Maria I.; Moreno F. J.; Alonso C.; Gago F.; Munoz V.; Avila J.; Hernandez F. (2005) Phosphorylation modulates the alpha-helical structure and polymerization of a peptide from the third tau microtubule-binding repeat. Biochim. Biophys. Acta 1721, 16–26. [DOI] [PubMed] [Google Scholar]
- Sadqi M.; Hernandez F.; Pan U.; Perez M.; Schaeberle M. D.; Avila J.; Munoz V. (2002) Alpha-helix structure in Alzheimer’s disease aggregates of tau-protein. Biochemistry 41, 7150–7155. [DOI] [PubMed] [Google Scholar]
- Campioni S.; Mannini B.; Zampagni M.; Pensalfini A.; Parrini C.; Evangelisti E.; Relini A.; Stefani M.; Dobson C. M.; Cecchi C.; Chiti F. (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 6, 140–147. [DOI] [PubMed] [Google Scholar]
- Caccamo A.; Magri A.; Oddo S. (2010) Age-dependent changes in TDP-43 levels in a mouse model of Alzheimer disease are linked to Abeta oligomers accumulation. Mol. Neurodegener. 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teoh C. L.; Yagi H.; Griffin M. D.; Goto Y.; Howlett G. J. (2011) Visualization of polymorphism in apolipoprotein C-II amyloid fibrils. J. Biochem. 149, 67–74. [DOI] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Nussinov R. (2010) Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. U.S.A. 107, 9490–9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Y.; Ma B.; Nussinov R.. The unique Alzheimer’s β-amyloid triangular fibril has a cavity along the fibril axis under physiological conditions. J. Am. Chem. Soc. 2011, in press. [DOI] [PMC free article] [PubMed]
- Parthasarathy S.; Long F.; Miller Y.; Xiao Y.; McElheny D.; Thurber K.; Ma B.; Nussinov R.; Ishii Y.. Molecular-Level Examination of Cu2+ Binding Structure for Amyloid Fibrils of 40-Residue Alzheimer’s β by Solid-State NMR Spectroscopy. J. Am. Chem. Soc 2011, not supplied. [DOI] [PMC free article] [PubMed]
- Xu S.; Brunden K. R.; Trojanowski J. Q.; Lee V. M. (2010) Characterization of tau fibrillization in vitro. Alzheimers Dement. 6, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S.; Sahara N.; Saito Y.; Murayama M.; Yoshiike Y.; Kim H.; Miyasaka T.; Murayama S.; Ikai A.; Takashima A. (2007) Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861. [DOI] [PubMed] [Google Scholar]
- Barghorn S.; Zheng-Fischhofer Q.; Ackmann M.; Biernat J.; von Bergen M.; Mandelkow E. M.; Mandelkow E. (2000) Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 39, 11714–11721. [DOI] [PubMed] [Google Scholar]
- von Bergen M.; Barghorn S.; Biernat J.; Mandelkow E. M.; Mandelkow E. (2005) Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim. Biophys. Acta 1739, 158–166. [DOI] [PubMed] [Google Scholar]
- Goedert M.; Jakes R.; Crowther R. A. (1999) Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 450, 306–311. [DOI] [PubMed] [Google Scholar]
- Arrasate M.; Perez M.; Armas-Portela R.; Avila J. (1999) Polymerization of tau peptides into fibrillar structures. The effect of FTDP-17 mutations. FEBS Lett. 446, 199–202. [DOI] [PubMed] [Google Scholar]
- Nacharaju P.; Lewis J.; Easson C.; Yen S.; Hackett J.; Hutton M.; Yen S. H. (1999) Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 447, 195–199. [DOI] [PubMed] [Google Scholar]
- Gamblin T. C.; King M. E.; Dawson H.; Vitek M. P.; Kuret J.; Berry R. W.; Binder L. I. (2000) In vitro polymerization of tau protein monitored by laser light scattering: method and application to the study of FTDP-17 mutants. Biochemistry 39, 6136–6144. [DOI] [PubMed] [Google Scholar]
- D’Souza I.; Poorkaj P.; Hong M.; Nochlin D.; Lee V. M.; Bird T. D.; Schellenberg G. D. (1999) Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. U.S.A. 96, 5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M.; Zhukareva V.; Vogelsberg-Ragaglia V.; Wszolek Z.; Reed L.; Miller B. I.; Geschwind D. H.; Bird T. D.; McKeel D.; Goate A.; Morris J. C.; Wilhelmsen K. C.; Schellenberg G. D.; Trojanowski J. Q.; Lee V. M. (1998) Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282, 1914–1917. [DOI] [PubMed] [Google Scholar]
- DeTure M.; Ko L. W.; Yen S.; Nacharaju P.; Easson C.; Lewis J.; van Slegtenhorst M.; Hutton M.; Yen S. H. (2000) Missense tau mutations identified in FTDP-17 have a small effect on tau-microtubule interactions. Brain Res. 853, 5–14. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves C. A.; Castillo-Carranza D. L.; Guerrero-Muoz M. J.; Jackson G. R.; Kayed R. (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.