

Abstract
Alternative splicing can produce variant proteins and expression patterns as different as the products of different genes, yet the prevalence of alternative splicing has not been quantified. Here the spliced alignment algorithm was used to make a first inventory of exon-intron structures of known human genes using EST contigs from the TIGR Human Gene Index. The results on any one gene may be incomplete and will require verification, yet the overall trends are significant. Evidence of alternative splicing was shown in 35% of genes and the majority of splicing events occurred in 5′ untranslated regions, suggesting wide occurrence of alternative regulation. Most of the alternative splices of coding regions generated additional protein domains rather than alternating domains.
The total size of human genomic DNA sequences in GenBank exceeds 100 million bases and is rising exponentially. However, the majority of human genomic sequences are uncharacterized or characterized incompletely. Thus, although a large amount of data has been published about alternative splicing of individual genes (Gelfand et al. 1999), this information remains mostly anecdotal and does not allow for any generalizations. On the other hand, it has been estimated that at least half of the human genes are represented in the existing EST collections (Schuler et al. 1996). Since these collections are created by partial sequencing of mRNAs from many different tissues and developmental stages, one would expect that the diversity of alternative splicing variants in EST data banks would be larger than in the standard samples of annotated human genes.
The problem of using ESTs for genomic DNA annotation and prediction of exon–intron structure is not trivial. It has been studied by several groups, most notably GRAIL (Xu and Uberbacher 1997). One of the main difficulties is that a considerable number of ESTs map to intergenic or intronic regions, or could be products of aberrant or incomplete splicing. It is likely that these matches constitute at least one fifth of the existing EST databases (Wolfsberg and Landsman 1997). Thus, the most informative ESTs are those that correspond to several exons. However, in this case simple matching of ESTs to genomic sequences by BLAST-like programs is not sufficient because BLAST does not map exactly the exon–intron boundaries (Altshul et al. 1990). Recently two programs were published that align EST sequences with genomic DNA (Mott 1997; Florea et al. 1998).
We have developed a program for prediction of the exon-intron structure of genomic DNA fragments using EST data. The program Procrustes-EST is based on the modified spliced alignment algorithm (Gelfand et al. 1996). When applied to known human genes and TIGR EST assemblies (Adams et al. 1995), the program found a large number of alternatively spliced genes (∼35%). Most of the alternative splicing events occurred in 5′-untranslated regions. In many cases the use of the program allowed for linking and merging multiple existing assemblies into single contigs.
RESULTS
Superstructures and EST Contigs
After aligning EST contigs to genomic DNA, the latter was used as an anchor for additional clustering and assembly of ESTs. The partial gene structures generated by spliced alignment were merged whenever they shared consecutive splicing sites spanning an intron. The superstructures so formed correspond to all possible gene structures for which each complete exon is supported by at least one of the alignments (Methods). On the EST level this leads to formation of superassemblies. Each superassembly is a merge of initial EST contigs matching a predicted superstructure. Note that linking of EST contigs to the genomic sequence and the requirement that all splicing sites in merged EST contigs coincide, precludes formation of spurious superassemblies.
Table 1 presents the distribution of the number of EST contigs that are merged to form one superassembly. In ∼50% of cases, no further merging could be done. Because the procedure for creating superstructures is local (Methods), 10% of all superstructures are chimeric in the sense that the full superstructure is not supported by any one of the original EST contigs and thus possibly includes exons from different splicing variants. The remaining 40% of superassemblies are formed by more than one contig, showing that matching ESTs to the genome allows a significant amount of additional assembly.
Table 1.
The Number of EST Contigs Corresponding to One Superstructure
| No. of contigs | chimeric | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| Percent of superstructures | 10.6 | 48.3 | 19.3 | 10.8 | 6.6 | 1.8 | 1.4 | 0.8 | 0.4 |
| No. of superstructures | 84 | 382 | 152 | 86 | 52 | 14 | 11 | 6 | 3 |
Table 2 describes the number of superassemblies of which each EST contig is a part (equivalently, the number of superstructures to which each EST contig maps). More than half of contigs (55%) are a part of only one superassembly, and slightly more than one fourth of contigs (27%) are orphans (having no common complete exons or introns with any of the genes in the starting set). Approximately 3% of contigs are part of complex alternative splicing events (being a part of four or more variant superassemblies).
Table 2.
The Number of Superassemblies of Which One EST Contig Is a Part
| No. of superassemblies | orphans | 1 | 2 | 3 | 4 | 5 | 6 | ≧7 |
|---|---|---|---|---|---|---|---|---|
| Percent of contigs | 27.1 | 55.5 | 12.2 | 2.0 | 1.6 | 0.2 | 0.7 | 0.7 |
| No. of contigs | 338 | 694 | 152 | 3 | 2 | 3 | 8 | 8 |
Alternative Splicing
Table 3 presents the number of alternative exon–intron structures predicted per gene. More than one-third of genes have at least two variants of exon–intron structures. The alternative structures were classified initially from the point of view of mature mRNAs. Thus we distinguish alternatives at the 5′ end (5′ forks), alternatives at the 3′ end (3′ forks) and internal alternatives (loops including bulges). 5′ forks occurred in 73 genes (54% of alternatively spliced genes), loops in 41 genes (30%), and 3′ forks in 64 genes (47%) (the total exceeds 100% because these cases are not mutually exclusive). Gautheret et al. (1998) found that in 1000 EST clusters, 189 showed clear evidence of alternative polyadenlyation. These results are not directly comparable to ours, as we did not attempt to determine the location of polyadenylation sites.
Table 3.
Distribution of the Number of Alternative Superstructures per Gene
| No. of superstructures | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ≧9 |
|---|---|---|---|---|---|---|---|---|---|
| Percent of genes | 65.6 | 18.6 | 4.6 | 5.4 | 1.3 | 1.5 | 0.5 | 1.0 | 1.5 |
| No. of genes | 259 | 71 | 18 | 21 | 5 | 6 | 2 | 4 | 6 |
Column 1 corresponds to genes without alternative splicing.
We then analyzed the distribution of particular variants of alternative splicing, where 23% of loops were generated by alternative acceptors, 16% were generated by alternative donors, and 27% were exons that were present in one of the two structures and absent in the other one. There were rare instances of retained introns, alternative introns, and alternative exons. Of those examined, 25% were complex cases that could not be classified because they combined several elementary events of alternative splicing. Furthermore, 22% of 5′-forks were alternative 5′ exons, 18% had different transcription start points and an additional intron in one of the variants, and the rest were complex cases. Finally, 11% of 3′ forks were alternative terminal exons, 35% had different end points (polyadenylation sites) and an additional intron in one variant, and the rest were complex cases.
Classifying the alternatives by functional region rather than by location in the alignment, we saw that 80% of alternatively spliced genes had an alternative in the 5′-untranslated region, whereas only 20% had alternatives in the coding region as described in GenBank, and 19% had alternatives in 3′-untranslated region (the total exceeds 100% since alternatives may occur in two or all three of these regions).
True Alternatives or Splicing Errors?
Intron retention, through either genomic contamination or incomplete/incorrect splicing, is perhaps the most likely artifact that could cause misleading results. However, we placed strict conditions on the inclusion/formation of superstructures (Methods) and in the final data observed only four cases where comparison of superstructures showed one retaining an intron relative to the other (considering not only coding regions, but the entire transcript). Thus, the possibility of intron contamination can be ruled out in the vast majority of the gene structures we considered.
We also performed additional analysis, considering the influence of discovered alternatives on reading frame for those cases (161 genes) where the alternative regions were situated completely within the annotated coding region. In 95 cases (59%) the alternative influenced an integer number of codons. Of these, 23 cases involved multiple (usually two) compensated events, for example alternative exon and alternative site in the next exon. Noncompensated frameshifting (40 cases of added/lost exons, 74 cases of alternatives choice of sites) usually happens near the 3′ end of the coding region, and thus it affects only the carboxyl terminus of the protein. It is interesting to note that more than one third of frameshifts in predicted structures can be eliminated, preserving a strong EST contig to genome alignment, if we allow splicing at noncanonical sites and do not force the introns to start at GT and end at AG.
Of course, to distinguish with certainty between true alternative splicing and artifactual sequences, one has to perform detailed case by case analysis including experimental work, for example, if some variant persists in a particular tissue, it is likely to be functional. However, all of the above evidence, even if circumstantial, does suggest that we are observing true splice variants in most cases.
Examples of Individual Cases
Sixteen genes from our sample (<5%) had alternative splicing variants found by preliminary analysis described explicitly, or at least mentioned in GenBank annotations (Gelfand et al. 1996; Sze and Pevzner 1997). In four cases no alternatives were constructed, in four cases the predicted set of alternative structures coincided with the GenBank annotation, and in eight cases additional splicing variants were found. The latter group is described in Table 4.
Table 4.
Known Alternatively Spliced Genes with Additional Variants
| Gene | AC | Known variants | Additional variantsa |
|---|---|---|---|
| Presomatotropin | V00520 | alternative acceptor sites of exon 3 | more alternative acceptor sites of exon 3; additional intron in 3′ UTR |
| Presomatotropin variant | K00470 | alternative acceptor sites of exon 3 | more alternative acceptor sites of exon 3; additional intron in 3′ UTR |
| Pulmonary surfactant protein C | J03890 | alternative donor site of last exon | exon 2 can be spliced out; additional intron in 3′ UTR (last exon) |
| High mobility group protein | L17131 | alternative first exons | more alternative splicing variants in 5′ UTR |
| Fragile X mental retardation syndrome protein | L29074 | alternative acceptor sites of exons 15 and 17 | exon 12 can be spliced out |
| Serum albumin | M12523 | alternative last exon | alternative acceptor site in 3′ UTR (one of the variant last exons) |
| Nonmuscle/smooth muscle myosin light chain | M22919 | exon 3 can be spliced out; alternative last exon | additional intron in 3′ UTR (one of the variant last exons) |
| Sex hormone-binding globulin | M31652 | alternative first exons | exon 7 can be spliced out |
(UTR) untranslated region.
In particular, we have observed three alternative acceptor sites of exon 3 of somatotropin and somatotropin variant genes. The sequences of these two genes are very close. Two variants of this site were annotated for each gene and we have observed only one of them (Fig. 1). The last exon of both these genes has an alternative intron in the 3′-untranslated region. Pulmonary surfactant protein C gene has an alternative donor site of the last exon. In addition, its exon 2 can be spliced out (its length, 159 nucleotides, is a multiple of 3), and there is an alternative intron with alternative donor sites in the 3′-untranslated region (Fig. 2). In the fragile X mental retardation syndrome gene, in addition to known variants generated by alternative acceptor sites of exons 15 and 17, exon 12 can be spliced out. In the sex hormone-binding globulin known variants are generated by alternative first exons; newly discovered alternative splicing is the result of splicing out of exon 7. Other new variants of genes with known alternative splicing result from alternative splicing of untranslated regions (Table 4).
Figure 1.

Alternative acceptor sites of exon 3 of somatotropin and somatotropin variants. (V) Presomatotropin (V00520); (K) presomatotropin variant (K00470); (A) EST contig (THC207918); (B) EST contig (THC195752); (C) EST contig (THC195753). (<, >) annotated and observed sites (resp. donor and acceptor); (]) annotated but not observed acceptor sites; [)] observed but not annotated acceptor sites; (uppercase letters) mismatching nucleotides; (===) intron shadows.
Figure 2.
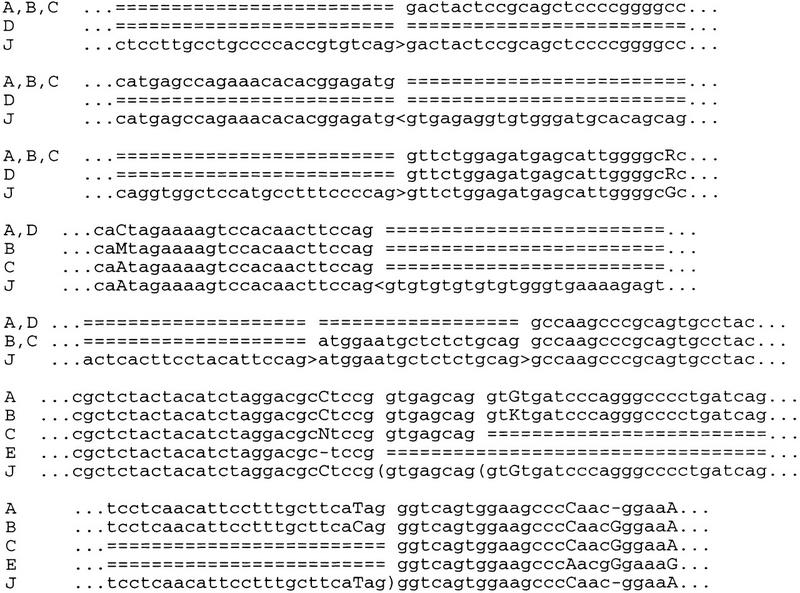
Alternative splicing of pulmonary surfactant protein C gene. (J) Genomic sequence (J03890); (A, B, C, D) EST contigs (THC211006, THC211005, THC173453, and THC173454, respectively); (E) EST(N75529). (<, >) known sites (donor and accceptor, respectively); [(] new sites; other notation as in Fig. 1.
DISCUSSION
Relatively few genes have been investigated for alternative splice forms, and it has been difficult to estimate the extent and trends of alternative splicing in human genes. We have presented a quantitative study of the prevalence of alternative splicing across many gene families. The results on any one gene may be incomplete and will require verification, yet the overall trends are significant. The results suggest that at least one-third of human genes are alternatively spliced. In particular, we have observed frequent alternative splicing in untranslated regions, specifically in the 5′ UTR. The alternative splicing at the 5′ end coupled to different starting points of transcription is probably a mechanism that allows the cell to use several differently regulated promoters for the same gene. The majority of alternative splicing events within the coding regions produces additional protein domains rather than alternating domains.
The problem of mapping ESTs to genomic sequences is addressed by several different programs, in particular EST_GENOME (Mott 1997) and SIM4 (Florea et al. 1998). The main difference between our approach and straightforward application of these and other tools is in the postprocessing step used to filter out unreliable EST hits. Moreover, the use of genomic data has allowed us to merge EST contigs in the situations where the EST overlaps alone provide insufficient evidence for contig construction. Indeed, 40% of superassemblies were produced by more than one contig.
Fraction of Genes with Alternative Splicing Is Probably Underestimated
A study such as this has many possible sources of error. However, using a very conservative approach, it is unlikely that genes for which we found alternative superstructures actually have no alternative splicing (although we may have missed some cases of genuine alternative splicing). To the best of our knowledge, we used the most conservative collection of EST contigs and found no case of an EST contig with distant genomic matches implying incorrect assembly. Alignments between EST contigs and genomic sequence were examined individually if there was any sign that the automated alignment was incorrect. When multiple EST contigs were merged, we guarded against merging of contigs from different genes by anchoring the assembly to genomic sequence. To prevent, insofar as possible, the inclusion of sequence from genomic clones or incompletely/incorrectly spliced mRNA, we only merged exons into gene structures when the overlap included splice junctions spanning an intron. The fact that only four genes showed structures with retained introns, and that alternative structures often seemed to be constrained by the reading frame, suggests that our safeguard measures were successful.
Interference among members of multigene families should not produce additional splicing variants. Indeed, since we used strict thresholds on relative alignment score in order to accept a prediction, and in addition checked local drops of similarity, interference would require extremely strong conservation of intron sequences. This can happen only for very close and recently duplicated genes (i.e., somatotropin and somatotropin variant genes, shown in Fig. 1). It is very likely that splicing alternatives in such cases are the same. The interference of nearly identical genes may have led to an overestimation, of the number of EST contigs that can be merged using genomic sequences. However, such cases are rare and the overestimation most likely small.
Our main conclusion is that alternative splicing is likely to occur in at least one-third of all genes; however, the actual fraction could be significantly higher. This is evidenced by the fact that in 4 of 16 cases with known alternative splicing, only 1 variant was found in our analysis. The underestimation is unavoidable in that many variants can have very limited tissue or stage specificity. However, in taking a number of conservative steps, we may have further reduced the estimation.
Possible Overestimation of the Number of Gene Structures per Gene
Alternative splicing events in different parts of a gene may not be independent. In our study we combined all events independently, even when no single EST contig supported the full structure. Thus, our estimation of the number of alternative splice forms per gene may be high. This does not, however, affect our main conclusions regarding the extent and classification of the alternative splicing events themselves.
This problem cannot be resolved by computer analysis. Indeed, even the construction of EST contigs from relatively short ESTs can produce chimaeric contigs. Only sequencing of full-length mRNAs or directed RT-PCR-based analysis using primers to alternating regions can resolve these cases. However, this does not influence our main conclusion about the frequency of alternative splicing and its prevalence in 5′ UTRs. The latter can even be underestimated because we did not consider hanging ends of EST targets that cannot be matched to the sequenced portion of genomic DNA as alternatives.
CONCLUSIONS
Case by case analysis of many individual genes, including experimental verification, will refine our understanding of human alternative splicing significantly. We hope to begin to test our main conclusions on a set of unannotated cosmid-sized sequences. Nevertheless, we believe that the joint accumulation of EST and genomic data has provided a sufficient basis to gain some important new insights into the extent and style of human alternative splicing. On the computational side, further research will be aimed at improvement of methods for distinguishing between true alternative and aberrant splicing as well as algorithms for support of experiments on identification of alternative splicing variants.
METHODS
Human genomic DNA fragments containing complete multiexon genes were compiled by merging samples (Gelfand et al. 1996; Kulp et al. 1996). Genes were considered to be duplicates if their described exon–intron structures were identical (minor differences in intron lengths were allowed) and the longest representative from each group of duplicates was selected. The final sample consisted of 392 genes.
Repeats were filtered from the genomic sequences by RepeatMasker (Smit 1999). EST contigs corresponding to a gene were selected from the TIGR Human Gene Index (Adams et al. 1995) using BLASTN (Altschul et al. 1990). The E-value threshold was set to 10−50. For our upper limit, 10 of the highest scoring contigs (targets) per gene were retained. This limit, originally set arbitrarily to reduce the volume of output, turned out to apply rarely and did not affect the conclusions. All types of contigs were used, including singletons and contigs containing full-length cDNA.
Genes having at least one common target were grouped into clusters (i.e., two genes were linked if they had at least one common target and clusters were defined as maximal connected components in the obtained graph). The target sets for each cluster were merged and ascribed to each member of the cluster. Finally, the sequences complementary to the targets were added to the target sets.
Exon–intron structures were predicted using Procrustes-EST. This program predicts candidate splicing sites with a very weak threshold and then finds a chain of exons with the highest local similarity to the target using the spliced alignment algorithm (Gelfand et al. 1996). The following parameters were used: match weight = 1, mismatch penalty = 1, gap initiation penalty = 4, and gap extension penalty = 2. Introns are not considered as gaps by Procrustes-EST, so that gaps in the alignments produced are actually very rare and the alignments are robust with regard to a particular choice of gap penalties within a reasonable range (data not shown). Relative similarity between a predicted gene and a target was defined as the ratio of the spliced alignment score and the score of the trivial alignment of the target with itself (Mironov et al. 1998).
The threshold for accepting the prediction was set to 80% relative similarity to avoid interference of members of multigene families. Cases with local drops of similarity between predicted genes and targets (defined as ≥10 out of 25 mismatching nucleotides) were analyzed manually and 61 prediction errors caused by loss of sites or spurious short exons at prediction termini were corrected. Ends of EST contigs that did not match to the genomic sequence were ignored. Such ends could correspond to unsequenced distal ends of the gene or be the consequence of deteriorated sequence quality at 5′ ends of EST reads. As we could not distinguish between these possibilities, we did not count such cases as alternative splicing.
At the postprocessing stage predicted exon–intron structures corresponding to one gene were merged into superstructures if they intersected without local contradictions. Superstructures were constructed as follows. All triples (intron–exon–intron) from predicted structures were considered (this step did not depend on annotated coding sequence). Two triples were merged if the right intron of the first triple coincided with the left intron of the second triple. Thus, even short overlaps between exons were accepted if they were supported by reliable exon-intron junctions. On the other hand, even long simple matches within exons were not sufficient for construction of superstructures if they did not span an intron, as they are often caused by unspliced ESTs (see discussion of orphans, below).
This procedure was performed until no triples could be added to the constructed superstructure. All possible superstructures were constructed. Because alternative splicing in different parts of pre-mRNA may not be independent, creation of chimeric superstructures not corresponding to any mRNA are possible. However, comparison with EST contigs formed from shorter ESTs is not a good method for analysis of long-range correlations between splicing events, and in the absence of full-length mRNA sequences further conclusions cannot be reached.
Contigs or superstructures that intersected neither the annotated coding sequence nor any other superstructure in a common complete exon or intron were termed orphans and were not counted. There are two types of such superstructures. First, they could lie completely outside all other superstructures and coding sequence. These superstructures are likely to correspond to parts of unannotated genes in the analyzed fragments. Second, such superstructures, usually consisting of just one exon, could lie completely within an intron of a known gene, partially overlap with a known exon, or span without interactions several consecutive exons and introns. These cases probably correspond to mis-spliced pre-mRNAs (products of aberrant or incomplete splicing) or to antisense transcripts.
Acknowledgments
This work was supported partially by the Russian State Scientific Program Human Genome, the Russian Fund of Basic Research, and the U.S. Department of Energy. We are grateful to R. Guigo, S. Hannenhali, P. Pevzner, M. Roytberg, and S. Sze for useful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mgelfand@anchorgen.com; FAX (310) 434-0120.
REFERENCES
- Adams MD, Kerlavage AR, Fleischmann RD, Fuldner RA, Bult CJ, Lee NH, Kirkness EF, Weinstock KG, Gocayne JD, White O, et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature. 1995;377:3–17. [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheret G, Poirot O, Lopez F, Audic S, Claverie J-M. Alternative polyadenylation in human mRNAs: A large-scale analysis by EST clustering. Genome Res. 1998;8:524–530. doi: 10.1101/gr.8.5.524. [DOI] [PubMed] [Google Scholar]
- Gelfand MS, Mironov AA, Pevzner PA. Gene recognition via spliced alignment. Proc Natl Acad Sci. 1996;93:9061–9066. doi: 10.1073/pnas.93.17.9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand MS, Dubchak I, Dralyuk I, Zorn M. ASDB: Database of alternatively spliced genes. Nucleic Acids Res. 1999;27:301–302. doi: 10.1093/nar/27.1.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp D, Haussler D, Reese MG, Eeckman FH. A generalized hidden Markov model for the recognition of human genes in DNA. In: States DJ, Agarwal P, Gaasterland T, Hunter L, Smith R, editors. Proceedings of the Fourth International Conference on Intelligent Systems for Molecular Biology. Menlo Park CA: AAAI Press; 1996. pp. 134–142. [PubMed] [Google Scholar]
- Mironov AA, Roytberg MA, Pevzner PA, Gelfand MS. Performance-guarantee gene predictions via spliced alignment. Genomics. 1998;51:332–339. doi: 10.1006/geno.1998.5251. [DOI] [PubMed] [Google Scholar]
- Mott R. EST GENOME: A program to align spliced DNA sequences to unspliced genomic DNA. Comput Appl Biosci. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- Schuler GD, Boguski MS, Stewart EA, Stein LD, Gyapay G, Rice K, White RE, Rodriguez-Tomé P, Aggarwal A, Bajorek E, et al. Genome maps 7. The human transcript map (wall chart) Science. 1996;274:540–546. [PubMed] [Google Scholar]
- Smit, A. 1999. http://ftp.genome.washington.edu/RM/RM_details.html.
- Sze S-H, Pevzner PA. Las Vegas algorithms for gene recognition: Suboptimal and error-tolerant spliced alignment. J Comput Biol. 1997;4:297–309. doi: 10.1089/cmb.1997.4.297. [DOI] [PubMed] [Google Scholar]
- Wolfsberg TG, Landsman D. A comparison of expressed sequence tags (ESTs) to human genomic sequences. Nucleic Acids Res. 1997;25:1626–1632. doi: 10.1093/nar/25.8.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Uberbacher ED. Automated gene identification in large-scale genomic sequences. J Comput Biol. 1997;4:325–338. doi: 10.1089/cmb.1997.4.325. [DOI] [PubMed] [Google Scholar]