

Abstract
The purpose of our study was to better understand the relationship between mitochondrial structural proteins, particularly dynamin-related protein 1 (Drp1) and amyloid beta (Aβ) in the progression of Alzheimer's disease (AD). Using qRT-PCR and immunoblotting analyses, we measured mRNA and protein levels of mitochondrial structural genes in the frontal cortex of patients with early, definite and severe AD and in control subjects. We also characterized monomeric and oligomeric forms of Aβ in these patients. Using immunoprecipitation/immunoblotting analysis, we investigated the interaction between Aβ and Drp1. Using immunofluorescence analysis, we determined the localization of Drp1 and intraneuronal and oligomeric Aβ in the AD brains and primary hippocampal neurons from Aβ precursor protein (AβPP) transgenic mice. We found increased expression of the mitochondrial fission genes Drp1 and Fis1 (fission 1) and decreased expression of the mitochondrial fusion genes Mfn1 (mitofusin 1), Mfn2 (mitofusin 2), Opa1 (optic atrophy 1) and Tomm40. The matrix gene CypD was up-regulated in AD patients. Results from our qRT-PCR and immunoblotting analyses suggest that abnormal mitochondrial dynamics increase as AD progresses. Immunofluorescence analysis of the Drp1 antibody and the Aβ antibodies 6E10 and A11 revealed the colocalization of Drp1 and Aβ. Drp1 immunoprecipitation/immunoblotting analysis of Aβ antibodies 6E10 and A11 revealed that Drp1 interacts with Aβ monomers and oligomers in AD patients, and these abnormal interactions are increased with disease progression. Primary neurons that were found with accumulated oligomeric Aβ had lost branches and were degenerated, indicating that oligomeric Aβ may cause neuronal degeneration. These findings suggest that in patients with AD, increased production of Aβ and the interaction of Aβ with Drp1 are crucial factors in mitochondrial fragmentation, abnormal mitochondrial dynamics and synaptic damage. Inhibiting, these abnormal interactions may be a therapeutic strategy to reduce mitochondrial fragmentation, neuronal and synaptic damage and cognitive decline in patients with AD.
INTRODUCTION
Alzheimer's disease (AD) is a late-onset, devastating mental disorder characterized by the progressive decline of memory, cognitive function and changes in personality (1). In addition to the presence of neurofibrillary tangles and amyloid beta (Aβ) deposits, AD is associated with structural and functional changes in mitochondria, reduced acetylcholine, inflammatory responses and synaptic damage (2–12).
In AD, Aβ is a major component of neuritic plaques that are found in brain regions known to be responsible for learning and memory. Aβ is a cleaved product of the Aβ precursor protein (AβPP) via sequential proteolysis of β secretase and γ secretase in AD brains. Cleavage by γ secretase can generate two forms of Aβ: a shorter form with 40 amino acid residues and a longer form with 42 amino acids. The longer form is toxic and has the capability to self-aggregate, form oligomers, participate in fibrillogenesis and accumulate into deposits (13–19). In AD, levels of Aβ are controlled by the production, clearance and degradation of Aβ. A reduction in the clearance of Aβ or the overproduction of Aβ may lead to an accumulation of Aβ in subcellular compartments of neuron, including mitochondria, endoplasmic reticulum, lysosomes and multiple vesicle bodies, and may impair functions of subcellular organelles and damage neurons (8,16). Several recent studies of oligomeric Aβ in rodents reported that oligomeric Aβ impairs long-term potentiation and promotes synaptic damage (14,19–21). However, the nature of oligomeric Aβ in brains of patients with AD at different stages of progression is not completely understood.
In a recent global, time-course gene expression study of AD transgenic mice (Tg2576 line), we found an up-regulation of mitochondrial genes in 2-, 5- and 18-month-old AD transgenic mice, suggesting that mitochondrial function is impaired by mutant AβPP and Aβ, and that the up-regulation may be a compensatory response to mutant AβPP and Aβ. Recently, we (22) and others (23–25) found both monomeric and oligomeric Aβ in mitochondria, in the neurons affected by AD, and mitochondrial- Aβ—interact with mitochondrial proteins, increase free radicals production, decrease cytochrome c oxidase activity, inhibit mitochondrial ATP and damage mitochondrial structure. Further, several groups reported mitochondrial function to be defective selectively in cortical and hippocampal mitochondria from AD transgenic mice (23,25–28) and postmortem brains from AD patients (24). More recently, we (29) and others (30,31) found that Aβ reduces total mitochondrial mass; impairs mitochondrial axonal transport, particularly anterograde transport; inhibits synaptic ATP production; and causes synaptic degeneration in neurons affected by AD. However, in AD neurons, the extent and characteristics of mitochondrial structural damage in relation to Aβ are still unclear.
Increasing evidence suggests that structural and functional abnormalities in mitochondria are involved in AD (26,32,33). These structural abnormalities are caused by an imbalance in highly conserved, GTPase genes that are essential for mitochondrial fission and mitochondrial fusion. GTPase genes—dynamin-related protein 1 (Drp1), fission 1 (Fis1), mitofusins 1 and 2 (Mfn1, Mfn2) and optic atrophy 1 (Opa1)—regulate, maintain and remodel mammalian mitochondria (34–36). Mitochondria constantly divide and fuse, and travel through the neuron, from cell body to nerve terminals and synapses, where energy is in high demand (36). Mitochondrial fission is regulated and maintained by two GTPase genes: Fis1 and Drp1. Fis1 is primarily localized on the outer mitochondrial membrane and participates in mitochondrial fission. However, most of Drp1 is localized in the cytoplasm, but a small part is localized in the outer membrane, where it punctuates and promotes mitochondrial fragmentation (34). The precise role of Drp1 in fragmenting mitochondria in relation to Aβ is unclear.
Drp1 is reported to be involved in several important structural features of mitochondria, including shape, size, distribution, remodeling and the maintenance of mitochondria in mammalian cells (36). In addition, recent studies revealed that Drp1 is associated with several mitochondrial functions, including fragmentation, phosphorylation, SUMOylation, ubiquitination and cell death. Thus, two decades of intense research on mitochondrial dynamics has provided evidence, linking Drp1 to neurodegenerative diseases. Further, recent studies of Drp1 knockout in mice revealed that double knockouts are embryonic lethal (37,38). However, primary neurons from Drp1 knockout mice (−/−) forebrain showed a decrease in the number of neurites and also the formation of defective synapses. These defects highlight the importance of Drp1-dependent mitochondrial fission within neurons. Additional findings from Drp1 knockout mice (−/−) also revealed that mitochondria formed extensive networks and peroxisomes were elongated in Drp1-null embryonic fibroblasts. In addition, Drp1-homozygote embryos failed to undergo developmentally regulated apoptosis during neural tube formation in vivo (37,38). These studies suggest that Drp1 is essential for cell survival and critical for brain development, mitochondrial division and distribution of mitochondria in neurons. Recently, we found increased levels of fission proteins, Drp1 and Fis1 proteins, and decreased fusion proteins Mfn1, Mfn2 and Opa1 in affected brain tissues (frontal cortex and striatum) from grade III and IV HD patients relative to control subjects (39), indicating the presence of abnormal mitochondrial dynamics in the pathogenesis of HD. However, mRNA abundance and protein levels of mitochondrial structural proteins, including Drp1 in different stages of AD progression, are not well understood. Further, the precise connection between Drp1 and Aβ in the progression and pathogenesis of AD is not clear.
In the study reported here, we sought to determine the nature of Aβ oligomers and mitochondrial proteins in postmortem brain tissues from AD patients at different stages of disease progression. Using quantitative real-time RT–PCR, we quantified mRNA expression and protein levels of the mitochondrial fission genes Drp1 and Fis1; the fusion genes Mfn1, Mfn2, Opa1; outermembrane genes Tomm40 and VDAC; and the matrix gene CypD, in the AD brain specimens. Using immunoblotting and dotblot analyses, we also quantified Aβ oligomers in the AD brain specimens, and using immunoprecipitation and immunofluorescence analyses, we determined the interaction between monomeric and oligomeric forms of Aβ and Drp1.
RESULTS
mRNA expression of mitochondrial genes
To determine the role of mitochondrial structural genes in AD progression and pathogenesis, using quantitative real-time RT–PCR with Sybr-Green chemistry, we measured mRNA fold change for the fission genes Drp1 and Fis1; the fusion genes Mfn1, Mfn2 and Opa1; outermembrane genes Tomm40 and VADC; and matrix gene CypD in postmortem brain specimens from patients in early and late stages of AD [Braak stages I and II (early), III and IV (definite) and V and VI (severe); Table 1] and in postmortem brain specimens from patients without AD (control; Braak 0). Overall, we found increased expression of fission and matrix genes in the brain specimens from 15 AD patients and decreased expression of the fusion genes, indicating abnormal mitochondrial dynamics.
Table 1.
mRNA fold change of mitochondrial fission/fusion and matrix genes in the frontal cortex brain specimens from patients with early, definite, and severe AD
| Braak stages of AD progression | Mitochondrial fission genes |
Mitochondrial fusion genes |
Outermembrane genes |
Matrix gene, CyPD | ||||
|---|---|---|---|---|---|---|---|---|
| Drp1 | Fis1 | Mfn1 | Mfn2 | Opa1 | TOMM40 | VDAC | ||
| I and II | 1.6 | 1.9 | 2.1 | 1.1 | −1.1 | 1.3 | 2.2 | 11.6 |
| I and II | 3.4 | 1.0 | −2.8 | −3.6 | −1.3 | −5.6 | 4.1 | 4.0 |
| I and II | 3.6 | 1.4 | −1.5 | −1.1 | −1.3 | 1.1 | −2.1 | 13.3 |
| I and II | 1.3 | 1.0 | −1.4 | 1.0 | −5.5 | 1.5 | −4.0 | 7.5 |
| III and IV | −1.2 | 1.1 | −1.2 | 1.1 | −5.4 | −2.3 | 1.4 | 1.0 |
| III and IV | 1.8 | 1.0 | 1.0 | −1.4 | 1.0 | 2.5 | 1.7 | 6.5 |
| III and IV | 1.2 | 1.2 | −1.5 | −2.3 | −1.8 | 1.8 | −1.3 | 3.7 |
| III and IV | 2.3 | 1.2 | −2.4 | −1.7 | −5.9 | −2.9 | −1.6 | 4.4 |
| III and IV | 1.6 | −1.5 | −3.0 | −1.4 | −3.5 | −4.1 | −1.8 | 4.5 |
| V and VI | 1.8 | 1.3 | −4.0 | 1.4 | −1.2 | −1.7 | 1.1 | 2.3 |
| V and VI | 1.0 | 1.0 | −2.1 | 1.2 | 1.3 | 1.2 | 5.3 | 1.3 |
| V and VI | 1.3 | 1.4 | −4.8 | −2.2 | −1.2 | −3.6 | 3.4 | 1.4 |
| V and VI | −1.8 | 1.0 | −1.9 | −4.7 | −1.1 | 1.3 | 1.3 | 2.1 |
| V and VI | 1.6 | 1.1 | −1.4 | −4.3 | −1.3 | 1.4 | 1.2 | 3.0 |
Fission genes
Overall, mitochondrial fission was up-regulated in the AD brain specimens, but there was some heterogeneity. mRNA fold changes were increased for Drp1 in brain specimens at Braak stages I and II (four out of four), III and IV (four out of five) and V and VI (four out of five) compared with the specimens from the control brains (Braak stage 0). However, the range was low, from 1.0-fold to 3.6-fold. In two cases, Drp1 was down-regulated. Similar to Drp1, Fis1 was increased in 13 out of 14 specimens from the AD brains. However, the range of up-regulation for Fis1 was not as great as it was for Drp1 (Table 1).
Fusion genes
As shown in Table 1, mRNA fold changes were down-regulated for Mfn1, Mfn2 and Opa1 in the brain specimens from all AD patients, at all stages of AD progression, relative to the mRNA fold changes in the brain specimens from the control subjects. However, interestingly, these genes exhibited a heterogeneity of down-regulation: Mfn1, from −1.2 to −4.8; Mfn2, from −1.1 to −4.7; and Opa1, from −1.1 to −5.5.
Outermembrane genes
Tomm40 was down-regulated in 6 of the 14 AD brain specimens, and VDAC was down-regulated in 5 of the 14 AD brain specimens. Tomm40 was up-regulated in the remaining eight AD brain specimens, and VDAC, in the remaining nine AD brain specimens (Table 1).
Matrix gene
As shown in Table 1, CypD was up-regulated in the brain specimens from all 14 patients with AD, from 1.3 to 13.3 fold changes, indicating that increased CypD expression may be associated with mitochondrial structural damage in AD neurons.
Immunoblotting analysis of mitochondrial proteins
To determine whether mitochondrial structural proteins are altered as AD progresses, using densitometry we quantified Drp1, Fis1, Mfn1, Mfn2, Opa1, Tomm40, VDAC and CypD in frontal cortical tissues from AD patients at different stages of AD (Fig. 1). Our immunoblotting findings agreed with our real-time RT–PCR analysis. Overall, we found increased levels of fission proteins and decreased levels of fusion proteins in all of the brain tissues from AD patients, indicating the presence of abnormal mitochondrial dynamics in patients with early, definite and severe AD.
Figure 1.

Immunoblotting analysis of mitochondrial fission, fusion and matrix proteins in early, definite and severe AD patients. Drp1 levels were significantly increased in AD patients at Braak stages I and II (P < 0.005), III and IV (P < 0.02) and V and VI (P < 0.002) compared with the Drp1 levels in control subjects. Fis1 protein levels were also significantly increased [Braak stages I and II (P < 0.003), III and IV (P < 0.01) and IV and V (P < 0.01)] relative to the control subjects. Fusion proteins: in contrast to fission proteins, mitochondrial fusion proteins were significantly decreased in AD patients relative to fission proteins in control subjects. Tomm40 levels were significantly decreased in AD patients at Braak stages IV and V (P < 0.004) relative to the controls. Matrix protein: CypD was significantly increased in AD patients at Braak stages III and IV (P < 0.04) and IV and V (P < 0.02) compared with controls. *Statistical significance at P < 0.05; **statistical significance at P < 0.005.
Fission proteins
Our protein data agreed with our real-time RT–PCR findings. Drp1 levels were significantly increased in the brain specimens from the AD patients at Braak stages I and II (P< 0.005), III and IV (P< 0.02) and V and VI (P< 0.002), compared with the Drp1 levels in the control brain specimens (Braak stage 0). Fis1 protein levels were also significantly increased in the brain specimens from the AD patients [Braak stages I and II (P< 0.003), III and IV (P< 0.01) and V and VI (P< 0.01)], relative to the control brain specimens (Fig. 1).
Fusion proteins
In contrast to fission proteins, mitochondrial fusion proteins were significantly decreased in the brain specimens from the AD patients. In the brain specimens from the controls, the fission proteins were decreased. Mfn2 protein levels were also significantly decreased in AD patients at Braak stages I and II (P< 0.01), III and IV (P< 0.01) and V and VI (P< 0.01) relative to the levels in the control subjects (Braak stage 0). Mfn1 levels were significantly decreased in the AD patients at Braak stages I and II (P< 0.03) and V and VI (P< 0.001), and Opa1 levels at Braak stages I and II (P< 0.02) and III and IV (P< 0.02) (Fig. 1).
Outermembrane protein
Tomm40 levels were significantly decreased in patients with AD at Braak stages IV and V (P< 0.004) relative to control brain specimens (Fig. 1).
Matrix protein
CypD was significantly increased in patients with AD at Braak stages III and IV (P < 0.04) and V and VI (P < 0.02), compared with control subjects (Fig. 1).
Immunoblotting analysis of Aβ monomers and oligomers
To determine the amount of oligomeric Aβ in AD brains, using the oligomer-specific antibody A11, we performed western blot analysis of frontal cortex tissues from patients with AD at Braak stages I and II, III and IV and V and VI. We also conducted immunoblotting analysis, using the Aβ antibody 6E10 that recognizes both monomeric and oligomeric Aβ.
We found full-length AβPP, derivatives of AβPP including 4 kDa monomeric Aβ and several oligomeric Aβ in AD patients at early (Braak stages I and II), definite (Braak stages III and IV) and severe (Braak stages V and VI) disease progression (Fig. 2A). The intensity of monomeric and oligomeric Aβ increased in the brain specimens from the definite and severe AD patients, indicating that Aβ formation and production are dependent on disease progression.
Figure 2.

Immunoblotting analysis of Aβ monomers and oligomeric proteins in early, definite and severe AD patients. (A) Immunoblot using 6E10 antibody. Intensity of monomeric and oligomeric Aβ increased in definite and severe AD patients, indicating that Aβ formation and production are dependent on disease progression. Two distinct bands of oligomers (50 and 60 kDa) were found, in addition to two faint bands (25 and 15 kDa; B). Both the 50 kDa (P< 0.001) and the 60 kDa oligomeric Aβ (P< 0.005) were significantly increased in the brains of AD patients at Braak stages I and II relative to the control subjects. (C) Dotblot analysis. (D) Quantification of oligomeric Aβ. *Statistical significance at P< 0.05; **statistical significance at P< 0.005.
Two distinct bands of oligomers (one with 50 kDa and the other with 60 kDa), in addition to faint bands of 25 and 15 kDa (Fig. 2B), were found in the brain specimens from the AD subjects. Both the 50 kDa (P< 0.001) and the 60 kDa oligomeric Aβ (P< 0.005) were significantly increased in the brain specimens from the AD patients at Braak stages I and II, relative to the control subjects (Fig. 2B and 2D).
Dotblots
Also using the oligomer-specific Aβ antibody A11, we performed quantitative dotblot analysis of brain specimens from patients at the three different stages of AD progression. As shown in Figure 2C, significantly increased levels of oligomers were found in the specimens from patients at Braak stages I and II (P< 0.002), III and IV (P< 0.03) and V and VI (P< 0.002), relative to the levels in the specimens from control subjects (Braak stage 0) (Fig. 2C and 2D).
Overall, these immunoblotting findings indicate that oligomeric Aβ levels are higher in the brain specimens from patients with AD and may be involved in abnormal protein interactions and neuronal damage.
Interaction of Aβ with Drp1
To determine whether Aβ monomers and/or oligomers interact with Drp1, we conducted immunoprecipitation analysis, using cortical protein lysates from AβPP/PS1 transgenic mice and AD patients; primary neurons from AβPP transgenic mice (Tg2576 line); and antibodies of Aβ (6E10 monoclonal) and Drp1 (polyclonal antibody). As shown in Figure 3A, our immunoprecipitation and immunoblotting analyses using 6E10 revealed five bands of AβPP derivatives (110, 50, 25, 16 and 4 kDa) in the AβPP/PS1 transgenic mice and the primary neurons from the AβPP mice, and four bands of AβPP derivatives (110, 50, 25 and 4) in the brain specimens from the AD patients. These findings indicate that 6E10 antibody precipitates a 4 kDa monomeric, 16, 25, 50 kDa of oligomeric Aβ and a 110 kDa of full-length AβPP.
Figure 3.

Immunoprecipitation analysis of Aβ and Drp1. (A) Immunoprecipitation with 6E10 and immunoblotting with 6E10 antibody. 6E10 recognized all derivatives of the mutant AβPP. (B) Immunoprecipitation with the Drp1 monoclonal antibody (Santa Cruz) and immunoblotting with the Drp1 monoclonal antibody.
To determine the Drp1 protein in AD brains, we performed immunoprecipitation and immunoblotting analyses, using a Drp1 antibody and proteins from early and severe AD patients. As shown in Figure 3B, we found 82 kDa in immunoprecipitation elutes and also in lysates from early and severe AD patients, indicating that the Drp1 antibody used for immunoprecipitation is specific for Drp1.
To determine whether the interaction of Aβ with Drp1 increases with the progression of AD, we performed co-immunoprecipitation analysis, using the Drp1 antibody, and immunoblotting analysis, using 6E10 antibody and protein lysates of cortical tissues from control subjects and from patients at Braak stages I and II (early) AD, III and IV (definite) AD and V and VI (severe) AD. As shown in Figure 4A, we found a 4 kDa Aβ in IP elutes from all AD patients and 110 kDa full-length AβPP in severe AD patients. Further, we found increased levels of 4 kDa Aβ with disease progression in brain specimens from AD patients (Fig. 4A). However, we found no physical interaction between Aβ and Drp1 in the control subjects.
Figure 4.

Immunoprecipitation analysis of Aβ and Drp1 antibodies in brain specimens from patients at different stages of AD progression. (A) Immunoprecipitation with Drp1 (polyclonal antibody; Santa Cruz) and immunoblotting with 6E10. 6E10 immunoreacted with full-length AβPP and 4 kDa monomeric Aβ in Drp1, the immunoprecipitated elute from early, definite and severe AD patients, indicating that Drp1 interacts with both full-length AβPP and monomeric Aβ. (B) Immunoprecipitation with Drp1 and immunoblotting with A11. A11 immunoreacted with a band of 50 kDa protein, indicating that Drp1 interacts with oligomeric Aβ.
To determine whether Drp1 interacts with oligomeric Aβ, we strip-eluted the blot in the 6E10 antibody analysis, incubated the blot with A11 antibody and then performed immunoblotting analysis. As shown in Figure 4B, we found a band of 50 kDa, indicating that Drp1 interacts with oligomeric Aβ in Braak stages I and II (early AD), III and IV (definite AD) and V and VI (severe AD) and intensity of 50 kDa is increased with disease progression in AD patients. However, we also noticed a faint band of 50 kDa oligomeric Aβ in control subjects (Fig. 4B).
Localization of Drp1
To determine the localization of Drp1 in the frontal cortex of AD patients and control subjects, we performed immunostaining/immunofluorescence analysis. As shown in Figure 5A, Drp1 expression was found primarily in frontal cortex neurons, but also in the astrocytes and microglia. Our comparative immunofluorescence analysis of Drp1 revealed increased immunoreactivity of Drp1 in frontal cortex sections from the AD patients relative to the control subjects (Fig. 5B). These findings concurred with our quantitative real-time RT–PCR findings and our immunoblotting data of Drp1.
Figure 5.

Drp1 localization in the AD brain. (A) Drp1 localization in (a) neurons (red arrows), (b) astrocytes (GFAP) (white arrows) and (c) (merged). Drp1 in (d) neurons (red arrows), (e) microglia (white arrows) and (f) merged. (B) Drp1 expression in neurons from AD patients and control subjects. Levels of Drp1 were higher in neurons from AD patient (b and d) compared with the levels in control subjects (a and c).
Immunofluorescence analysis of Aβ in AD brains
To localize intraneuronal Aβ and Aβ deposits in the brains of patients with AD, we performed immunofluorescence analysis using 6E10, which recognizes both intraneuronal and extracellular Aβ deposits. To determine the levels of Aβ1–40 and 1–42 in the AD brains, we conducted immunofluorescence analysis using 1–40- and 1–42-specific antibodies. As shown in Figure 6A, we found intraneuronal Aβ in the brains of AD patients at Braak stages I and II, III and IV and V and VI. However, the Aβ deposits were abundant only in Braak stages III and IV and V and VI.
Figure 6.

Aβ in patients with AD. Both intraneuronal and extracellular Aβ were seen in AD patients (A, 5×; B, 10×; and C, 40× original magnifications). Aβ1–40 immunoreactivity and deposits were seen in AD patients (D, 5×; E, 10×; and F, 40× original magnifications). Aβ1–42 immunoreactivity deposits were seen in AD patients (G, 5×; H, 10×; and I, 40× original magnifications).
Immunostaining of the Aβ 1–40 antibody revealed mostly diffuse and extracellular Aβ deposits, but no intraneuronal immunoreactivity in the brains from the AD patients (Fig. 6B). Immunostaining of the Aβ 1–42 antibody revealed robust, dense and compact deposits of Aβ, but no intraneuronal expression of Aβ (Fig. 6C).
To determine the presence of Aβ oligomers, using A11 antibody we conducted immunofluorescence analysis of brain sections from patients with early and definite AD. As shown in Figure 7A, we found some oligomeric Aβ immunoreactivity in the cytoplasm, but it was mostly perinuclear. We also found nuclear localization of oligomeric Aβ (Fig. 7B and C).
Figure 7.

Oligomeric Aβ in neurons from patients with AD. Immunofluorescence analysis of early patients at early stages of AD progression, using A11. [A (40× the original), B and C] The perinuclear accumulation of Aβ oligomers in the AD neurons (100× the original).
Double-labeling immunofluorescence analysis of Drp1 and Aβ in AD brains
To determine whether Drp1 localizes and interacts with Aβ, we conducted double-labeling analysis of Drp1 and Aβ in frontal cortex sections from AD patients. As shown in Figure 8A, the immunoreactivity of Drp1 was colocalized with Aβ immunoreactivity (monomeric), indicating that Drp1 interacts with Aβ. These observations matched our immunoprecipitation findings of Drp1 and 6E10 (Fig. 4). Further, in agreement with our immunoprecipitation findings, double-labeling analysis of Drp1 and A11 in the frontal cortex sections from the AD patients showed colocalization of Drp1 and oligomeric Aβ (Fig. 8B).
Figure 8.

(A) Double-labeling immunofluorescence analysis of 6E10 and Drp1 in patients with AD. The localization of Drp1 (a) and Aβ (b) and the colocalization of Drp1 and Aβ (c, merged) at 40× the original magnification, and images of Drp1 (d), Aβ (e) and merged (f) at 100× the original magnification. (B) Double-labeling immunofluorescence analysis of A11 and Drp1 in patients with AD. The localization of Drp1 (a) and oligomeric Aβ (b), and the colocalization of Drp1 and Aβ (c, merged) at 40× the original magnification.
Immunofluorescence analysis of Aβ in primary neurons
To determine the localization of monomeric and oligomeric forms of Aβ expression in primary neurons from AβPP transgenic mice, we conducted immunofluorescence analysis of 6E10 and A11 antibodies in primary hippocampal neurons. As shown in Figure 9A, immunoreactivity of 6E10 was present in the cell body, neuronal processes and terminals of primary neurons (Fig. 9Aa–d). A 4 kDa monomeric Aβ was secreted and released into media, as detected by immunoblotting and ELISA analyses (data not shown). As shown in Figure 9A, immunoreactivity of oligomeric Aβ was found mainly in the cell body, but also in neuronal projections. In neurons that were found with over-accumulated oligomeric Aβ had lost branches and were degenerated, indicating that an over-accumulation of oligomeric Aβ may cause neuronal degeneration (Fig. 9Ba and b).
Figure 9.
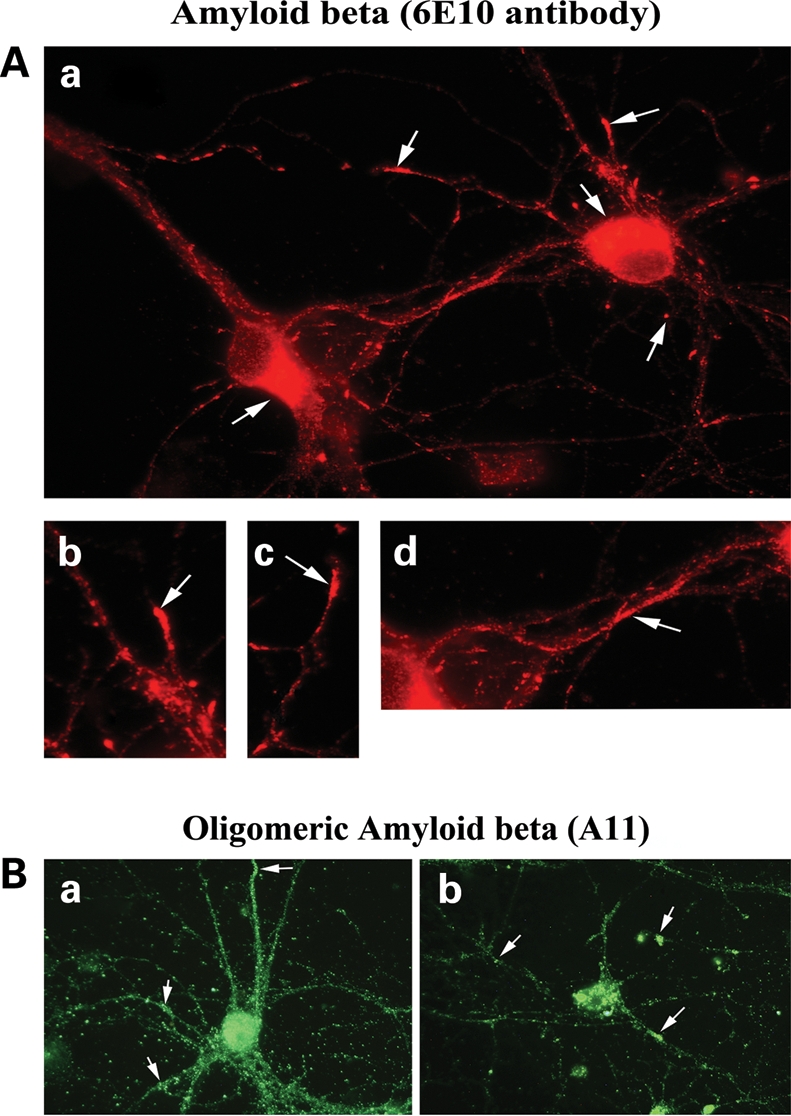
Immunofluorescence analysis of Aβ in primary hippocampal neurons from AβPP transgenic mice. (A) Aβ (6E10-immunostained) localized in the cell body, axons and nerve terminals in a hippocampal neuron (a). (b, c and d) Aβ localization in hippocampal neuron (100× original magnification). (B) Immunofluorescence analysis of oligomeric Aβ (immunostained with A11) in primary hippocampal neurons from the AβPP transgenic mice. (a) Oligomeric Aβ localization throughout the neuron. (b) Neurons with accumulated oligomeric Aβ, the loss of neuronal projections, damaged neuronal networks and other signs of neurodegeneration.
Double-labeling analysis of Drp1 and Aβ in primary neurons from AβPP transgenic mice
Using double-labeling analysis of Drp1, and 6E10 and A11, we determined the localization of Drp1 and Aβ (monomeric and oligomeric forms) in 16 DIV primary neurons from AβPP transgenic mice. As shown in Figure 10A, the immunoreactivity of Drp1 colocalized with Aβ monomers, further confirming our immunoprecipitation and immunofluorescence results that also found colocalization of Drp1 and Aβ in the AD brains. Double-labeling analysis of Drp1 and A11 in primary neurons from AβPP transgenic mice revealed colocalization of Drp1 and oligomeric Aβ, indicating that oligomeric Aβ may interact with Drp1 (Fig. 10B). These observations agree with our findings from immunoprecipitation and immunofluorescence analyses, that Drp1 and Aβ interact in AD brains.
Figure 10.

Double-labeling immunofluorescence analysis of Aβ (6E10) and Drp1 in primary neurons from AβPP transgenic mice (A). The localization of Drp1 (a) and 6E10 (b) in hippocampal neurons and the colocalization of Drp1 and Aβ (c, merged) at 100× the original magnification. (B) Double-labeling immunofluorescence analysis of A11 and Drp1 in primary neurons from AβPP transgenic mice. The localization of Drp1 (a) and A11 (b) in hippocampal neuron and the colocalization of Drp1 and Aβ (c, merged) in hippocampal neuron at 100× the original magnification.
Double-labeling analysis of Drp1 and COX1 in primary neurons from AβPP transgenic mice
We performed immunostaining for Drp1 and cytochrome c oxidase subunit 1 (COX1) in 16 DIV primary neurons from wild-type mice and AβPP mice. As shown in Figure 11, Drp1 distribution was altered in AβPP primary neurons compared with wild-type neurons. In wild-type cultures, Drp1 immunopositive puncta were distributed throughout the neuron, including significant staining in areas of active neuronal growth. However, in the AβPP primary neurons, Drp1 staining was largely localized to the cell body and neurites immediate to cell body. There were very few neuronal growth cones that labeled with Drp1 in AβPP cultures. COX1 distribution was similarly altered away from growth cones and into large axons and the cell body in neurons from AβPP mice compared with wild-type mice neurons. These findings suggest that decreased mitochondrial distribution in neuronal processes and nerve terminals in neurons are affected by AD.
Figure 11.

Double-labeling immunofluorescence analysis of Drp1 and COX1 in primary neurons from AβPP transgenic mice and wild-type mice. Drp1 and COX1 colocalize in cell body, neruites and growth cones. Hippocampal neurons from wild-type or AβPP mice were immunostained for Drp1, mitochondrial marker COX1 and DAPI. Hippocampal neurons from wild-type mice (upper panels) (Drp1, A; COX1, B; DAPI, C; and merged, D) show Drp1 immunoreactivity in growth cones and neurites, whereas neurons from AβPP mice (lower panels) (Drp1, E; COX1, F; DAPI, G; and merged, H) show Drp1 immunoreactivity mainly in the cell body and axons next to the cell body. Areas of active growth were not commonly observed in AβPP cultures. In both wild-type and AβPP cultures Drp1 colocalized with mitochondrial-encoded protein, COX1.
DISCUSSION
We studied mitochondrial structural proteins in postmortem brains in AD patients at early (Braak stages I and II), definite (Braak stages III and IV) and severe (Braak stages V and VI) disease progression and in control subjects. Using immunoprecipitation and immunoblotting and double-labeling immunofluorescence analyses, we extensively characterized monomeric and oligomeric forms of Aβ in the AD patients. We found that Aβ interacts with Drp1 in the frontal cortex of AD subjects and in the cerebral cortex of AβPP/PS1 transgenic mice. Interestingly, the interaction between Drp1 and Aβ increased as the disease progressed. These findings lead to the conclusion that the interaction of Aβ and Drp1 may initiate mitochondrial fragmentation in neurons affected by AD.
Mitochondrial genes in AD patients
Overall, we found decreased mRNA levels in the fusion genes Mfn1, Mfn2 and Opa1 in brain specimens of the frontal cortex from patients with early, definite and severe AD relative to control subjects. Among these proteins, Mfn1 was the most down-regulated, followed by Opa1 and then Mfn2. We also found the down-regulation of fusion genes to be heterogeneous, with most of the down-regulation in patients with severe AD. This is the first gene expression study to measure mRNA in fusion genes from brain specimens of AD patients at different stages of AD progression.
In contrast, we found increased mRNA levels in the fission genes Drp1 and Fis1 in the frontal cortex of patients with early, definite and severe AD. Interestingly, increased levels of Drp1 were higher in patients with early AD than in patients with definite or severe AD, indicating that Drp1 is involved in abnormal mitochondrial dynamics as an early event. The mRNA levels of the outermembrane genes Tomm40 and VDAC were higher in the frontal cortex of patients with early, definite and severe AD. However, in 6 of the 14 patients with AD, Tomm40 was down-regulated, and VDAC was down-regulated in 5 of 14 patients. Further research is needed to understand the precise connection between Tomm40 and VDAC, and disease progression of AD.
Interestingly, mRNA levels of CypD increased in all AD patients; however, the levels increased the most in patients at early AD progression, suggesting that increased mRNA levels of CypD may be an early change in AD progression. Overall, the increased mRNA levels in fission genes and CypD and the decreased mRNA levels in fusion genes suggest abnormal mitochondrial dynamics in AD patients.
Mitochondrial proteins in AD patients
Drp1 and Fis1 significantly and progressively increased in frontal cortex tissues from patients with early, definite and severe AD, indicating that Drp1 and Fis1 are affected by AD progression. These findings agree with our recent study, in which we found Drp1 and Fis1 increased in N2a cells that were treated with Aβ and in primary neurons from AβPP transgenic mice (26). Our present findings of increased Drp1 levels in patients at early AD progression may not be consistent with the results from Wang et al. (32), who found decreased levels of Drp1 in AD patients. However, it is unclear the stage of disease progression of AD postmortem brains that Wang et al. studied Drp1 in patients with AD. Further, we found decreased levels of Drp1 in brain specimens from one patient with definite AD and one with severe AD.
Our results regarding the progressive increase of Fis1 in patients with early, definite and severe AD cases are consistent with results from Wang et al. (32,33), who also reported increased Fis1 in AD patients, although the progression of AD in their patients was not specified. Overall, our findings of the progressive increase of Fis1 in AD patients at progressive stages suggest that changes in mitochondrial structure may be events in early AD progression.
Decreased levels of fusion proteins that we found concur with earlier studies of N2a cells treated with Aβ, primary neurons from AβPP transgenic mice by Manczak et al. (26) and a study of fusion proteins in AD patients in which decreased levels of fusion proteins were found (32). Both an increase in fission proteins and a decrease in fusion proteins may lead to mitochondrial structural and functional changes.
Drp1 localization in AD patients
We found Drp1 to be expressed in neurons, astrocytes and microglia in patients at all three Braak stages of AD. This result indicates that Drp1 remains involved in mitochondrial division, distribution and ATP synthesis (40). In addition, given our finding that Drp1 is increased in AD patients relative to control subjects, Drp1 may be involved in mitochondrial fragmentation, abnormal mitochondrial dynamics and decreased mitochondrial ATP in AD patients. However, further research is needed to determine whether and how increased Drp1 fragments mitochondria in relation to Aβ in neurons from AD patients.
Aβ oligomers in AD
Recently, Gong et al. (14) described a 53 kDa synthetic oligomeric Aβ ligand derived from Aβ1–42 that is toxic and involved in long-term potentiation inhibition. They also found a 56 kDa oligomeric Aβ in soluble extract from postmortem brain tissues of AD patients. Recently, in brain tissues from AβPP transgenic mice, Lesné et al. (20) found 56 kDa oligomeric Aβ that appeared to have toxic effects on neurons and to impair memory. Recently, Pham et al. (17) studied membrane-bound and cytosolic Aβ oligomers, using 6E10 and 82E1 antibodies, in brains from patients at different stages of AD progression. They found several oligomeric forms of Aβ, but mostly robust 40 and 50 kDa bands with the 82E1 antibody; and 40, 50 and 60 kDa bands with the 6E10 antibody in membrane-bound lysates. However, these studies did not quantify the Aβ oligomers, using A11 antibody from AD patients, thus limiting the oligomers study in patients with AD. Ours is the first study that has quantified Aβ oligomers and has found a progressive increase in Aβ oligomers in patients at progressive stages of AD development.
Oligomeric Aβ is toxic
Our investigations of Aβ oligomers in primary neurons from AβPP transgenic mice indicated that the oligomers are expressed throughout the neuron, including the cell body, axons and dendrites (Fig. 9). Interestingly, we found that some neurons in which Aβ oligomers were over-expressed and accumulated were degenerating, in terms of losing their neuronal network, indicating that Aβ oligomers may increase in sufficient numbers to cause neuronal death (Fig. 9B). This possibility is supported by previous reports (14,19,20,41) in which researchers reported the toxicity of Aβ oligomers. We also noticed that primary neurons from AβPP transgenic mice died 7 to 9 days earlier than cultured primary neurons from non-transgenic mice (data not shown), further supporting the possibility that Aβ oligomers are toxic and may cause premature death of primary neurons.
Interaction of Aβ with Drp1
Recently, Yan and colleagues reported the interaction of Aβ with the mitochondrial matrix proteins CypD and ABAD (42,43). In the present study, using co-immunoprecipitation and immunofluorescence analyses, we found a similar interaction between Aβ and mitochondrial fission protein, Drp1. Drp1 is a cytosolic protein, enriched in the cell body, axons and dendrites. A small quantity of Drp1 are localized on the outer membrane of mitochondria and, in AD, are involved in mitochondrial fragmentation (26,36).
Abnormal mitochondrial fragmentation, damaged mitochondria and autophagy have been extensively reported in patients with AD (26,44). These mitochondrial abnormalities may be due to the interaction of Aβ with Drp1. Further, the interaction of AβPP and Aβ with mitochondrial Drp1 appears to be an early event in AD progression. This possibility is supported by a recent global gene expression study of AβPP transgenic mice by Reddy et al. (45), in which we found increased expression of mitochondria-encoded genes in 2-month-old AβPP transgenic mice. These results suggest that mutant AβPP and soluble Aβ may associate with mitochondria, induce free radical production and disrupt mitochondrial function. In addition, several recent studies of mitochondria and Aβ have described the transport of Aβ to mitochondrial membranes and the subsequent occurrence of functional abnormalities in the mitochondria (22–25,28,42,46). Also, our results here support the hypothesis that the interaction between Drp1 and Aβ results in the fragmentation of mitochondria. In the present study, we found interaction between Aβ and Drp1, and this abnormal interaction increased with disease progression. However, we found no physical interaction between 4 kDa Aβ and Drp1 in the control subjects, suggesting that Drp1 and Aβ interaction is AD-specific. In severe AD cases, full-length AβPP was found to interact with Drp1 (Fig. 4A). Further, a 50 kDa oligomeric-specific Aβ found to interact with Drp1 and this interaction is increased with disease progression in AD (Fig. 4B). For the first time, full-length AβPP and 4 kDa monomer Aβ and 50 kDa oligomeric Aβ were found to interact with Drp1, suggesting that, in AD, both monomeric and oligomeric forms of Aβ are involved in abnormal mitochondrial dynamics. Taken together, these findings suggest that the interaction of both monomeric and oligomerc Aβ with mitochondrial protein Drp1 may cause abnormal, excessive mitochondrial fragmentation and may damage mitochondrial structure and function, ultimately leading to neuronal dysfunction that characterizes AD.
Our double-labeling immunofluorescence analysis of Aβ and Drp1 in AD brains indicated that monomeric and oligomeric Aβ colocalizes and interacts with Drp1, and this interaction/colocalization increases as AD progresses. The increased interaction between Aβ and Drp1 may be due to the increased production of Aβ in AD neurons. Further, increased oligomeric Aβ may interact more robustly with Drp1 in nerve terminals, where it is enriched. Our double-labeling analysis of monomeric and oligomeric Aβ with Drp1 in the primary neurons from AβPP transgenic mice further supports our finding that Drp1 interacts with Aβ. This interaction of oligomeric Aβ with Drp1 may cause abnormal, excessive mitochondrial fragmentation and the overproduction of structurally defective mitochondria that function abnormally and may lead to neuronal damage and cognitive impairments that are hallmarks of patients with AD. Inhibiting, these abnormal interactions may be a therapeutic strategy to reduce mitochondrial fragmentation and neuronal damage in AD.
CONCLUSIONS
Although tremendous progress has been made in understanding Aβ and mitochondrial dysfunction in AD progression and pathogenesis, the mechanistic links between Aβ and mitochondria are not well understood. Further, there are few published reports that characterize mitochondrial proteins and Aβ in postmortem brain specimens from patients with AD. To develop therapies for AD, we need a clearer understanding of how Aβ and mitochondria change during AD development and progression and how they affect each other. In the present study, using molecular and cell biology techniques and gene expression analysis, we studied postmortem brain specimens from patients with AD at early (Braak stages I and II), definite (Braak stages III and IV) and severe (Braak stages IV and V) stages of progression and those from control subjects to determine the relationship between mitochondrial structural proteins, particularly Drp1 and Aβ, in the progression of AD. We also used real-time quantitative RT–PCR and immunoblotting analyses to measure the quantity of mRNA and the protein levels of mitochondrial structural genes in the frontal cortex of patients with early, definite and severe AD and in control subjects. We also characterized monomeric and oligomeric forms of Aβ in these patients. Using immunoprecipitation and immunoblotting analyses, we investigated the interaction between Aβ and Drp1. Using immunofluorescence analysis, we determined the precise localization of Drp1 and intraneuronal and oligomeric Aβ in the AD brains.
We found increased expression levels of the mitochondrial fission genes Drp1 and Fis1 and decreased expression levels of the mitochondrial fusion genes Mfn1, Mfn2, Opa1 and Tomm40. The matrix gene CypD was up-regulated in AD patients. Results from our real-time quantitative RT–PCR and immunoblotting analyses suggest that abnormal mitochondrial dynamics increase as AD progresses. Our immunofluorescence analysis of the Drp1 antibody and the Aβ antibodies 6E10 and A11 revealed the colocalization of Drp1 and Aβ. Further, Drp1 immunoblotting and immunoprecipitation of the Aβ antibody 6E10, which recognizes various derivatives of the AβPP, revealed that Drp1 interacts with Aβ monomers and oligomers in AD subjects and, interestingly, this interaction increased as AD progressed. These findings lead to the conclusion that the interaction of Aβ and Drp1 may initiate mitochondrial fragmentation in neurons affected by AD, and abnormal interaction is increased with disease progression of AD.
MATERIALS AND METHODS
Twenty postmortem brain specimens from AD patients and age-matched control subjects were obtained from the Harvard Tissue Resource Center. Table 2 presents the characteristics of the brain specimens used in this study. Fifteen specimens were from patients diagnosed with AD according to Braak criteria (47), and five were from age-matched control subjects. On the basis of quantitative pathological features, including senile plaques, neurofibrillary tangles and neuronal density, the AD brain specimens were classified as specimens from Braak stages 1 and II (early AD), III and IV (definite AD), V and VI (severe AD) or control subjects. The specimens were from the frontal cortex and were both quick-frozen and formalin-fixed (BA9).
Table 2.
Demographic details of postmortem brain specimens from patients with AD and subjects without AD (specimens from the Harvard Tissue Resource Center)
| Case type | Age/sex | Postmortem interval (h) | Braak stage of AD brains |
|---|---|---|---|
| Control | 68/M | 21 | 0 |
| Control | 82/M | 17 | 0 |
| Control | 74/F | 23 | 0 |
| Control | 74/M | 15 | 0 |
| Control | 90/F | 12 | 0 |
| AD | 74/F | 12 | I/II |
| AD | 67/F | 10 | I/II |
| AD | 76/F | 24 | I/II |
| AD | 77/M | 18 | I/II |
| AD | 72/F | 12 | I/II |
| AD | 69/M | 18 | III/IV |
| AD | 75/M | 24 | III/IV |
| AD | 82/M | 19 | III/IV |
| AD | 80/M | 19 | III/IV |
| AD | 82/F | 18 | III/IV |
| AD | 77/M | 11 | V/VI |
| AD | 83/F | 18 | V/VI |
| AD | 83/M | 19 | V/VI |
| AD | 92/F | 5 | V/VI |
| AD | 95/F | 8 | V/VI |
Quantification of mRNA expression of mitochondrial fission, fusion and matrix genes using real-time RT–PCR
Using the reagent TRIzol (Invitrogen, Camarillo, CA, USA), we isolated total RNA in cortical tissues from AD patients and control subjects. Using Primer Express software (Applied Biosystems, Foster City, CA, USA), we designed the oligonucleotide primers for β-actin and GAPDH (housekeeping genes); for Drp1, Fis1, MFN1, MFN2, Opa1, Tomm40; and for CypD. The primer sequences and amplicon sizes are listed in Supplementary Material, Table S1. As previously described by Gutala and Reddy (48), Manczak et al. (49) and Reddy et al. (50), we measured mRNA expression of the genes mentioned above, using SYBR-Green-chemistry-based quantitative real-time RT–PCR. Briefly, 2 μg of DNAse-treated total RNA was used as a starting material, to which we added 1 μl of oligo (dT), 1 μl of 10 mm dNTPs, 4 μl of 5× first-strand buffer, 2 μl of 0.1 m DTT and 1 μl of RNAse out. The reagents RNA, dT and dNTPs were mixed first, then heated at 65°C for 5 min and finally chilled on ice until the remaining components were added. The samples were incubated at 42°C for 2 min, and then 1 μl of Superscript II (40 U/μl) was added. Then the samples were incubated again at 42°C, but for 50 min, at which time the reaction was inactivated by heating the samples at 70°C for 15 min.
Quantitative real-time PCR amplification reactions were performed with cDNA from AD patients and control subjects, using an ABI Prism 7900 sequence detection system (Applied Biosystems), in a 25 μl volume of total reaction mixture. The mixture consisted of 1× PCR buffer containing SYBR-Green; 3 mm MgCl2; 100 nm of each primer; 200 nm of dATP, dGTP and dCTP each; 400 nm dUTP; 0.01 U/μl AmpErase UNG; and 0.05 U/μl AmpliTaq Gold. A 50 ng cDNA template was added to each reaction mixture.
The CT-values of β-actin and GAPDH were tested to determine the unregulated endogenous reference gene in AD patients and controls. The CT-value, described in Manczak et al. (49), Gutala and Reddy (48) and Reddy et al. (50), is an important quantitative parameter in real-time PCR analysis. All RT–PCR reactions were carried out in triplicate, and with no template control. The mRNA transcript level was normalized against β-actin and GAPDH at each dilution. The standard curve was the normalized mRNA transcript level, plotted against the log value of the input cDNA concentration at each dilution. Briefly, the comparative CT method involved averaging triplicate samples, which were taken as the CT values for β-actin, GAPDH and mitochondrial genes. β-actin normalization was used in the present study because β-actin CT values are similar for mitochondrial structural genes, which allows us to avoid the values of GAPDH. The ΔCT-value was obtained by subtracting the average β-actin CT value from the average CT-value for the mitochondrial structural genes. The ΔCT of the controls was used as the calibrator. The fold change was calculated according to the formula 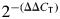 , where ΔΔCT is the difference between ΔCT and the ΔCT calibrator value.
, where ΔΔCT is the difference between ΔCT and the ΔCT calibrator value.
Aβ antibodies
To characterize Aβ in postmortem brains from AD patients and control subjects, and tissues from AD mouse models (Tg2576 and AβPP/PS1), we used Aβ monoclonal antibody (6E10) (51) and oligomer-specific antibody (popularly known as A11 (52). 6E10 is a clone of Aβ monoclonal antibody that is raised against 3–8 amino acid residues of Aβ peptide. 6E10 antibody reacts with 4 kDa Aβ, AβPP derivatives and full-length AβPP (Covance, Emeryville, CA, USA). The oligomeric Aβ antibody (A11) is extensively studied and reported that it is reactive to all forms of oligomeric Aβ.
Immunoblotting analysis of mitochondrial proteins
To determine whether mitochondrial protein levels are different in AD patients relative to control subjects, we performed immunoblotting analysis of protein lysates from cortical tissues of AD patients and control subjects. Protein lysates were prepared using a RIPA buffer [25 mm Tris–HCl (pH 7.6), 150 mm NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and protease inhibitors 0.5 g/ml pepstatin, 0.5 g/ml leupeptin, 1 mm PMSF). Fifty micrograms of protein lysates were resolved on a 10% Nu-PAGE gel (Invitrogen). The resolved proteins were transferred to PVDF membranes (Novax, Inc., San Diego, CA, USA) and then were incubated for 1 h at room temperature with a blocking buffer (5% dry milk dissolved in a TBST buffer). The nitrocellulose membranes were incubated overnight with the primary antibodies Drp1 (1:200 rabbit polyclonal; Novus Biological, Inc., Littleton, CO, USA), Fis1 (1:200, Protein Tech Group, Inc., Chicago, IL, USA), Mfn1 (1:300, rabbit polyclonal; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), Mfn2 (1:300, rabbit polyclonal; Abcam), Opa1 (1:500; mouse monoclonal; BD Biosciences, San Jose, CA, USA), Tomm40 (1:200 rabbit polyclonal; Santa Cruz Biotechnology), CypD (Santa Cruz Biotechnology) and β-actin (1:500; mouse monoclonal; Chemicon-Millipore, Temicula, CA, USA). Details of primary and secondary antibodies are given in Supplementary Material, Table S2. The membranes were washed with a TBST buffer three times at 10 min intervals and then incubated for 2 h with appropriate secondary antibodies, followed by three additional washes at 10 min intervals. Proteins were detected with chemiluminescent reagents (Pierce Biotechnology, Rockford, IL, USA), and the bands from immunoblots were quantified on a Kodak Scanner (ID Image Analysis Software, Kodak Digital Science, Kennesaw, GA, USA). Briefly, image analysis was used to analyze gel images captured with a Kodak Digital Science CD camera. The lanes were marked to define the positions and specific regions of the bands. An ID fine-band command was used to locate and to scan the bands in each lane and to record the readings. We analyzed the intensity of bands using ImageJ analysis and assessed statistical significance using Student's t-test.
Immunoblotting analysis of Aβ monomers and oligomers
To determine the quantity of Aβ monomers and oligomers in our AD patients relative to control subjects, we performed immunoblotting analysis of protein lysates from all of their cortical tissues. To detect Aβ monomers and oligomers, protein lysates were prepared using the following buffer: 250 mm sucrose, 50 mm HEPES, 25 mm MgCl2, 0.5 mm dithiothereitol and protease inhibitors, 0.5 g/ml pepstatin, 0.5 g/ml leupeptin and 1 mm PMSF. Fifty micrograms of protein lysates were resolved on a 4–12% gradient gel (Invitrogen) and then transferred to nitrocellulose membranes (Novax, Inc.). They were incubated for 1 h at room temperature with a blocking buffer (5% dry milk dissolved in a TBST buffer). The nitrocellulose membranes were incubated overnight with the 6E10 antibody (1:300; Covance, San Diego, CA, USA). Then they were washed with a TBST buffer three times at 10 min intervals and incubated for 2 h with a secondary antibody, followed by three additional washes at 10 min intervals. Aβ levels were detected with chemiluminescent reagents (Pierce Biotechnology).
To determine whether Aβ oligomers are present in the brain specimens from AD patients, we also performed immunoblotting analysis using protein lysates (described above) and the A11 antibody (anti-rabbit polyclonal, 1:200, Invitrogen) that specifically recognizes Aβ oliogomers in protein lysates from early, definite and severe AD patients. Details of primary and secondary antibodies are given in Supplementary Material, Table S2. Fifty micrograms of protein lysates were resolved on a 4–12% gradient gel (Invitrogen). Immunoblotting analysis was performed as described previously and above (22), and Aβ oligomers were detected with chemiluminescent reagents (Pierce Biotechnology). We quantified the bands from immunoblots, using densitometry and ImageJ analysis and assessed statistical significance, using Student's t-test.
Dotblot analysis
Ten micrograms of protein were spotted onto a nitrocellulose membrane and air-dried for 1 h. The membrane was soaked in 5% milk in TBST for 1 h to block non-specific sites. The membrane was then incubated with A11 (1:200; Invitrogen) in the blocking solution for 1 h at room temperature. It was washed three times with TBST and then incubated with an anti-rabbit secondary antibody that was conjugated with a horseradish peroxidase (HRP) 1:10 000 dilution for 30 min at room temperature. The membrane was again washed three times with TBS-T and incubated with an ECL reagent for 1 min before it was exposed to the X-ray film. We quantified oligomeric-specific dots from dotblots, using ImageJ analysis and assessed statistical significance using Student's t-test.
Immunoprecipitation of Aβ and Drp1
To determine the interaction of Drp1 with Aβ monomers and oligomers, we performed immunoprecipitation using cortical protein lysates from the brains of the AD patients [Braak stages I and II (early AD), III and IV (definite AD) and V and VI (severe AD)], control subjects (Braak stage 0) and AβPP/PS1 transgenic mice (53), and primary neurons from brains of the AβPP transgenic mice (54). We used a Dyna-beads kit for immunoprecipitation (Invitrogen). Fifty microliters of Dyna-beads containing protein G were incubated with 10 μg of 6E10 or 10 μg of the Drp1 antibodies (both mono- and polyclonal antibodies; Santa Cruz) for 1 h at room temperature, with rotation. The Dyna-beads–Aβ complex was washed three times with a washing buffer and was then incubated overnight with 400 μg of protein at 4°C, with rotation. The incubated ‘Dyna-beads antigen/antibody complex’ was again washed three times with a wash buffer, and the immunoprecipitant was eluted from Dyna-beads by the NuPAGE LDS sample buffer; it was loaded onto a gel where we performed immunoblotting analysis using 6E10, A11 and/or Drp1 antibodies.
Immunohistochemistry/immunofluorescence analysis
Using immunofluorescence techniques, we localized Drp1, Aβ monomers (6E10 antibody), oligomers (A11), astrocytes (GFAP) and microglia (Iba) in frontal cortex specimens (broadman area 9) taken from postmortem brains of 15 patients in early stages of AD (Braak stages I and II) and from postmortem brains of five control subjects (Braak stage 0) (Table 2). The specimens were paraffin-embedded, and sections were cut into widths of 15 μm. We deparaffinized the sections by washing them with xylene for 10 min and then washed them for 5 min each in a serial dilution of alcohol (95, 70 and 50%). The sections were washed once for 10 min with double-distilled H2O and then for six more times at 5 min each, with phosphate-buffered saline (PBS) at pH 7.4.
To reduce the autofluorescence of brain specimens, we treated the deparaffinized sections with sodium borohydrate twice each for 30 min in a freshly prepared 0.1% sodium borohydrate solution dissolved in PBS at pH 8.0. We then washed the sections three times for 5 min each, with PBS at pH 7.4. To block the endogenous peroxidase, sections were treated for 15 min with 3% H2O2 and then with 0.5% Triton dissolved in PBS at pH 7.4. The sections were blocked for 1 h with a solution [0.5% Triton in PBS+10% goat serum+1% bovine serum albumin (BSA)]. The sections were incubated overnight at room temperature with the following antibodies: anti-Drp1 (rabbit polyclonal 1:100 dilution, Santa Cruz Biotechnology), anti-Iba (rabbit polyclonal 1:400 dilution, Wako Chemicals, Richmond, VA, USA), anti-GFAP (rabbit polyclonal, 1:400 dilution, DakoCytomation, Carpenteria, CA, USA), anti-Aβ (mouse monoclonal 1:200, Covance) and anti-oligomeric (1:50 dilution, Invitrogen). On the day after the primary antibody incubation, sections were washed once with 0.1% Triton in PBS and then incubated with appropriate biotinylated secondary antibodies (Supplementary Material, Table S3) for 1 h at room temperature. They were washed with PBS three times for 10 min each. They were incubated for 1 h with labeled streptavidin, an HRP solution (Molecular Probes). The sections were each washed three more times for 10 min each with PBS, at pH 7.4, and then treated with Tyramide Alexa 594 (red) or Alexa 488 (green) (Molecular Probes) for 10 min at room temperature. They were cover-slipped with Prolong Gold and photographed with a confocal microscope.
Double-labeling immunofluorescence analysis of Drp1 and Aβ
To determine the interaction between Drp1 and Aβ (monomers and oligomers), we conducted double-labeling immunofluorescence analysis, using an anti-Drp1antibody (rabbit polyclonal, Santa Cruz Biotechnology) and 6E10 (Covance) or A11 (rabbit polyclonal, Invitrogen). As describedearlier, the sections from AD patients and control subjects were deparaffinized and treated with sodium borohydrate to reduce autofluorescence.
For the first labeling, the sections were incubated overnight with the anti-Drp1 antibody (1:200) at room temperature. On the day after this primary antibody incubation, the sections were washed with 0.5% Triton in PBS. They were then incubated with a secondary biotinylated anti-rabbit antibody at a 1:400 dilution (Vector Laboratories, Burlingame, CA, USA) or a secondary biotinylated anti-mouse antibody (1:400) for 1 h at room temperature. They were incubated for 1 h with labeled streptavidin, an HRP solution (Molecular Probes). The sections were washed three times each with PBS for 10 min, at pH 7.4, and treated with Tyramide Alexa488 for 10 min at room temperature.
For the second labeling, the sections were blocked for 1 h with a blocking solution containing 0.5% Triton in PBS+10% donkey serum+1% BSA. Then they were incubated overnight with 6E10 (1:200 dilution, Covance) or A11 (1:50 dilution, rabbit polyclonal) at room temperature. Next, they were incubated with the donkey anti-mouse secondary antibody labeled with Alexa 594 for 1 h at room temperature. They were coverslipped with Prolong Gold and photographed with a confocal microscope.
AβPP transgenic mice, AβPP×PS1 transgenic mice and primary neuronal culture
Using AβPP transgenic mice (52), AβPP×PS1 transgenic mice (51) and age-matched wild-type littermates (controls), we studied Aβ and its relationship to mitochondria at different stages of AD progression. The mice were housed at the Oregon National Primate Research Center at Oregon Health & Science University (OHSU). The OHSU Institutional Animal Care and Use Committee approved all procedures for animal care according to guidelines set forth by the National Institutes of Health.
Primary neuronal cultures of the AβPP transgenic mice and wild-type mice were prepared using methods described by Manczak et al. (26). Briefly, mice of postnatal day 1 were decapitated, and the brains were removed into a room-temperature Hibernate®-A medium (Brain Bits, Springfield, IL, USA) supplemented with B-27 (Invitrogen) and 0.5 mm GlutaMAX™ (Invitrogen). The hippocampus was dissected and reserved for culturing, and the cerebellum was removed for genotyping. Hippocampus pairs from individual mice were minced into pieces of <1 mm3, and the pieces were transferred to a solution of papain (2 mg/ml; Worthington Biochemical Corp., Lakewood, NJ, USA) that was dissolved in Hibernate®-A without calcium, but supplemented with 0.5 mm GlutaMAX™. The tissue was digested for 30 min in a shaking water bath at 30°C. Digested tissue was then removed to 2 ml of HABG and triturated 10 times with a fire-polished, siliconized (Sigmacote; Sigma, St Louis MO, USA) 9 inch glass pipette. Samples were allowed to settle by gravity for ∼1 min. Then the supernatant containing the dissociated neurons was removed to a fresh tube. An additional 2 ml of HABG was added to the pellet, and the process was repeated until 6 ml of dissociated neurons were collected. Neurons were centrifuged at 200g for 2 min and then washed with 2 ml of HABG. The pellets were resuspended in 2 ml of Neurobasal™ (Invitrogen) supplemented with B-27 and 0.5 mm GlutaMAX™ (growth medium). The neurons were counted, plated at 500 neurons/mm2 on poly-d-lysine-coated coverslips and placed into a 37°C incubator at 5% CO2. One hour after plating, the growth medium was completely replaced. For every 3 days one-half of the growth medium was changed.
Immunofluorescence analysis of primary neurons for Drp1, Aβ, monomers and oligomers
Fourteen-day-old primary neurons from AβPP transgenic mice and wild-type mice were washed with warm PBS, fixed in a freshly prepared 4% paraformaldehyde solution for 10–15 min, washed with PBS and then permeabilized with 0.1% Triton-X100 in the PBS. All incubations were performed in a humidified environment. All neurons were blocked with 1% blocking solution (Invitrogen) for 1 h at room temperature. Primary antibodies were diluted in the blocking solution and used under the following two conditions: (i) 6E10 (1:200, Covance), room temperature, 1 h; and (ii) A11 (1:50, Invitrogen) 4°C, overnight with Drp1 (1:400). After the incubation, the neurons were washed three times for 10 min with PBS. A fluorophore-labeled, appropriate secondary antibody [1:500 in blocking solution; as appropriate: Alexa Fluor® 488 goat anti-rabbit or Alexa Fluor® 568 goat anti-mouse antibodies (Invitrogen)] was then applied for 1 h at room temperature.
For double-labeling studies, antigens were labeled sequentially, meaning that the primary antibody (A11) was applied singly and then the appropriate secondary antibody was applied. Then the second primary antibody (e.g. E610) was applied, followed by the appropriate secondary antibody. Pictures of hippocampal neurons from the AβPP transgenic and wild-type mice were taken at 40× and 100× the original magnification with a fluorescence microscope.
Immunofluorescence analysis of Drp1 and Cox1
As described above, primary neurons from wild-type or AβPP pups were stained with Drp1 and COX1. Cells were harvested at 16DIV and stained for Drp1 (1:400, rabbit polyclonal, Novus Biologicals) using an anti-rabbit Alexa 488 secondary antibody. For subsequent secondary labeling with COX1, the cells were stained with COX 1 (1:200, mouse monoclonal, Invitrogen) using an anti-mouse Alexa 568 secondary antibody. Pictures of hippocampal neurons from the AβPP transgenic and wild-type pups were taken at 40× and 100× the original magnification with a fluorescence microscope.
SUPPLEMENTARY MATERIAL
FUNDING
This research was supported by NIH grants AG028072, AG026051, RR00163 and S10RR024585; Alzheimer Association grant IIRG-09-92429; Vertex Pharmaceuticals; and Medivation, Inc.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Anda Cornea (Imaging Core of Primate Center) for her assistance with the confocal microscopy.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Keller J.N., Pang Z., Geddes J.W., Begley J.G., Germeyer A., Waeg G., Mattson M.P. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 1997;69:273–284. doi: 10.1046/j.1471-4159.1997.69010273.x. doi:10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- 3.Swerdlow R.H., Parks J.K., Cassarino D.S., Maguire D.J., Maguire R.S., Bennett J.P., Jr, Davis R.E., Parker W.D., Jr Cybrids in Alzheimer's disease: a cellular model of the disease? Neurology. 1997;49:918–925. doi: 10.1212/wnl.49.4.918. [DOI] [PubMed] [Google Scholar]
- 4.Reddy P.H., Reddy P.H. Mitochondria are a therapeutic target for neurodegenerative disease. Curr. Alzheimer Res. 2010 doi: 10.2174/156720511795745401. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattson M.P. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. doi:10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barsoum M.J., Yuan H., Gerencser A.A., Liot G., Kushnareva Y., Gräber S., Kovacs I., Lee W.D., Waggoner J., Cui J., et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. doi:10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greig N.H., Utsuki T., Ingram D.K., Wang Y., Pepeu G., Scali C., Yu Q.S., Mamczarz J., Holloway H.W., Giordano T., et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl Acad. Sci. USA. 2009;102:17213–17218. doi: 10.1073/pnas.0508575102. doi:10.1073/pnas.0508575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy P.H., Beal M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol. Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. doi:10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Querfurth H.W., LaFerla F.M. Alzheimer's disease. N. Engl. J. Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. doi:10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 10.Gouras G.K., Almeid C.G., Takahashi R.H. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol. Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. doi:10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Zawia N.H., Lahiri D.K., Cardozo-Pelaez F. Epigenetics, oxidative stress, and Alzheimer disease. Free Radic. Biol. Med. 2009;46:1241–1249. doi: 10.1016/j.freeradbiomed.2009.02.006. doi:10.1016/j.freeradbiomed.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy P.H., Manczak M., Mao P., Calkins M.J., Reddy A.P., Shirendeb U. Amyloid-beta and mitochondria in aging and Alzheimer's disease: implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 2010;20(Suppl. 2):S499–S512. doi: 10.3233/JAD-2010-100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein W.L., Krafft G.A., Finch C.E. Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. doi:10.1016/S0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 14.Gong Y., Chang L., Viola K.L., Lacor P.N., Lambert M.P., Finch C.E., Krafft G.A., Klein W.L. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl Acad. Sci. USA. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. doi:10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walsh D.M., Selkoe D.J. A beta oligomers—a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. doi:10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 16.LaFerla F.M., Green K.N., Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat. Rev. Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. doi:10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 17.Pham E., Crews L., Ubhi K., Hansen L., Adame A., Cartier A., Salmon D., Galasko D., Michael S., Savas J.N., et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. doi:10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia W., Yang T., Shankar G., Smith I.M., Shen Y., Walsh D.M., Selkoe D.J. A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 2009;66:190–199. doi: 10.1001/archneurol.2008.565. doi:10.1001/archneurol.2008.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu H.Y., Hudry E., Hashimoto T., Kuchibhotla K., Rozkalne A., Fan Z., Spires-Jones T., Xie H., Arbel-Ornath M., Grosskreutz C.L., Bacskai B.J., Hyman B.T. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. doi:10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesné S., Koh M.T., Kotilinek L., Kayed R., Glabe C.G., Yang A., Gallagher M., Ashe K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. doi:10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 21.Shankar G.M., Li S., Mehta T.H., Garcia-Munoz A., Shepardson N.E., Smith I., Brett F.M., Farrell M.A., Rowan M.J., Lemere C.A., et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. doi:10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manczak M., Anekonda T.S., Henson E., Park B.S., Quinn J., Reddy P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. doi:10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 23.Caspersen C., Wang N., Yao J., Sosunov A., Chen X., Lustbader J.W., Xu H.W., Stern D., McKhann G., Yan S.D. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 24.Devi L., Prabhu B.M., Galati D.F., Avadhani N.G., Anandatheerthavarada H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. doi:10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao J., Irwin R.W., Zhao L., Nilsen J., Hamilton R.T., Brinton R.D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc. Natl Acad. Sci. USA. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. doi:10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manczak M., Mao P., Calkins M.J., Cornea A., Reddy A.P., Murphy M.P., Szeto H.H., Park B., Reddy P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J. Alzheimers Dis. 2010;20(Suppl. 2):S609–S631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckert A., Hauptmann S., Scherping I., Rhein V., Müller-Spahn F., Götz J., Müller W.E. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener. Dis. 2008;5:157–159. doi: 10.1159/000113689. doi:10.1159/000113689. [DOI] [PubMed] [Google Scholar]
- 28.Rhein V., Song X., Wiesner A., Ittner L.M., Baysang G., Meier F., Ozmen L., Bluethmann H., Dröse S., Brandt U., et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc. Natl Acad. Sci. USA. 2009;106:20057–20062. doi: 10.1073/pnas.0905529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calkins M.J., Reddy P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim. Biophys. Acta. 2011;1812:507–513. doi: 10.1016/j.bbadis.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X., Perry G., Smith M.A., Zhu X. Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener. Dis. 2010;7:56–59. doi: 10.1159/000283484. doi:10.1159/000283484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du H., Guo L., Yan S., Sosunov A.A., McKhann G.M., Yan S.S. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc. Natl Acad. Sci. USA. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. doi:10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X., Su B., Siedlak S.L., Moreira P.I., Fujioka H., Wang Y., Casadesus G., Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl Acad. Sci .USA. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. doi:10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X., Su B., Lee H.G., Li X., Perry G., Smith M.A., Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. doi:10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H., Chan D.C. Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum. Mol. Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. doi:10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reddy P.H., Mao P., Manczak M. Mitochondrial structural and functional dynamics in Huntington's disease. Brain Res. Rev. 2009;61:33–48. doi: 10.1016/j.brainresrev.2009.04.001. doi:10.1016/j.brainresrev.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy P.H., Reddy T.P., Manczak M., Calkins M.J., Shirendeb U., Mao P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 2010 doi: 10.1016/j.brainresrev.2010.11.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wakabayashi J., Zhang Z., Wakabayashi N., Tamura Y., Fukaya M., Kensler T.W., Iijima M., Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. doi:10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S.O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y., et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. doi:10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 39.Shirendeb U., Reddy A.P., Manczak M., Calkins M.J., Mao P., Tagle D.A., Reddy P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum. Mol. Genet. 2011;20:1438–1455. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ju W.K., Liu Q., Kim K.Y., Crowston J.G., Lindsey J.D., Agarwal N., Ellisman M.H., Perkins G.A., Weinreb R.N. Elevated hydrostatic pressure triggers mitochondrial fission and decreases cellular ATP in differentiated RGC-5 cells. Invest. Ophthalmol. Vis. Sci. 2007;48:2145–2151. doi: 10.1167/iovs.06-0573. doi:10.1167/iovs.06-0573. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi R.H., Almeida C.G., Kearney P.F., Yu F., Lin M.T., Milner T.A., Gouras G.K. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. doi:10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du H., Guo L., Fang F., Chen D., Sosunov A.A., McKhann G.M., Yan Y., Wang C., Zhang H., Molkentin J.D., et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. doi:10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lustbader J.W., Cirilli M., Lin C., Xu H.W., Takuma K., Wang N., Caspersen C., Chen X., Pollak S., Chaney M., et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. doi:10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 44.Hirai K., Aliev G., Nunomura A., Fujioka H., Russell R.L., Atwood C.S., Johnson A.B., Kress Y., Vinters H.V., Tabaton M., et al. Mitochondrial abnormalities in Alzheimer's disease. J. Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reddy P.H., McWeeney S., Park B.S., Manczak M., Gutala R.V., Partovi D., Jung Y., Yau V., Searles R., Mori M., Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum. Mol. Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. doi:10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 46.Hansson Petersen C.A., Alikhani N., Behbahani H., Wiehager B., Pavlov P.F., Alafuzoff I., Leinonen V., Ito A., Winblad B., Glaser E., Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl Acad. Sci. USA. 2008;105:13145–3150. doi: 10.1073/pnas.0806192105. doi:10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braak H., Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. doi:10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 48.Gutala R.V., Reddy P.H. The use of real-time PCR analysis in a gene expression study of Alzheimer's disease post-mortem brains. J. Neurosci. Methods. 2004;132:101–107. doi: 10.1016/j.jneumeth.2003.09.005. doi:10.1016/j.jneumeth.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 49.Manczak M., Mao P., Nakamura K., Bebbington C., Park B., Reddy P.H. Neutralization of granulocyte macrophage colony-stimulating factor decreases amyloid beta 1–42 and suppresses microglial activity in a transgenic mouse model of Alzheimer's disease. Hum. Mol. Genet. 2009;18:3876–3893. doi: 10.1093/hmg/ddp331. doi:10.1093/hmg/ddp331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reddy T.P., Manczak M., Calkins M.J., Mao P., Reddy A.P., Shirendeb U., Park B., Reddy P.H. Toxicity of neurons treated with herbicides and neuroprotection by mitochondria-targeted antioxidant SS31. Int. J. Environ. Res. Public Health. 2011;8:203–221. doi: 10.3390/ijerph8010203. doi:10.3390/ijerph8010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thakker D.R., Weatherspoon M.R., Harrison J., Keene T.E., Lane D.S., Kaemmerer W.F., Stewart G.R., Shafer L.L. Intracerebroventricular amyloid-beta antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc. Natl Acad. Sci. USA. 2009;106:4501–4516. doi: 10.1073/pnas.0813404106. doi:10.1073/pnas.0813404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kayed R., Head E., Thompson J.L., McIntire T.M., Milton S.C., Cotman C.W., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. doi:10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 53.Borchelt D.R., Thinakaran G., Eckman C.B., Lee M.K., Davenport F., Ratovitsky T., Prada C.M., Kim G., Seekins S., Yager D., et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. doi:10.1016/S0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 54.Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. doi:10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.