

Abstract
We applied a shared genomic segment (SGS) analysis, incorporating an error model, to identify complete, or near complete, selective sweeps in the HapMap phase II data sets. This method is based on detecting heterozygous sharing across all individuals within a population, to identify regions of sharing with at least one allele in common. We identified multiple interesting regions, many of which are concordant with positive selection regions detected by previous population genetic tests. Others are suggested to be novel regions. Our finding illustrates the utility of SGS as a method for identifying regions of selection, and some of these regions have been proposed to be candidate regions for harboring disease genes.
Keywords: identity by state, identity by descent, positive selection
Introduction
Dense single nucleotide polymorphisms (SNP) data sets provide an immense opportunity for studying population genetics, allowing the development of catalogs for signatures of selection, structural variants, and haplotype assortment. In particular, the HapMap project, launched in 2002, provides a public database of common SNPs across the genome in hundreds of individuals from geographically diverse populations. There have been multiple genome-wide scans attempting to identify recent and ongoing regions of selection using HapMap data.1, 2, 3, 4, 5, 6 However, such studies tend to only report the most extreme outliers, and it is therefore conceivable that there are regions omitted from these published reports. Furthermore, it has been suggested that regions identified in multiple analyses are more likely to be true positives.6, 7
Regions of the genome that are part of ongoing selective sweeps may include loci involved in phenotypic outcomes, including both deleterious and protective variants. Thus, haplotypes across regions with evidence of recent selection are of interest to be compared with association signals in genetic mapping.4 The incorporation of population genetic concepts into genetic research may therefore be important. Understanding patterns of genetic variation and evolutionary selection in the human genome may help to shed light on genetic epidemiology studies.
In general, there are five types of established tests being used for identifying regions of selection based on varied signatures produced by natural selection,8 they include the function-altering mutations test, the heterozygosity test, as well as the derived allele test,9, 10 the population differential test,5, 11 and the long haplotype test.4, 5, 12, 13 The time scale for each type of test reduces, respectively. We are most interested in genome-wide scan methods for detecting the last three types of selection signatures, since they could be used as an aid for disease gene mapping.
The composite likelihood ratio (CLR)9 test is a type of derived allele test. The CLR test searches for a signature of recent ‘complete' selective sweep using pooled data from various populations. This test applies genomic window approach, comparing spatial patterns of allele frequencies against the whole genome and coalescent simulation model.9, 10 The long haplotype tests have several variations, including long-range haplotype (LRH) test,12 haplotype similarity, and other haplotype-sharing methods. The extended haplotype homozygosity (EHH) test is a type of LRH test that is based on extended haplotype length and high linkage disequilibrium. Two extensions have been made to EHH: the integrated Haplotype Score (iHS) test4 and the cross population extended haplotype homozygosity (XP-EHH) test, and both of which have been used extensively.5 The iHS detects alleles that have risen to median-high frequency, and XP-EHH identifies selected alleles that have risen to near fixation in one but not all populations. XP-EHH is also considered as a population differential test. Lastly, a composite of multiple signals (CMS)13 test that incorporates iHS, XP-EHH, Wright's F test (FST)11 and two additional tests, could be utilized to localize regions of selection to a more precise position.
Among these kinds of tests, the long haplotype tests are the most similar to our shared genomic segment (SGS) approach. The EHH methods detect regions of identity-by-descent through measuring LD at a distance from the core region using calculation of extended haplotype homozygosity. Our method works directly on unphased data to identify regions of reduced genetic diversity, in which the population dynamic gave rise to the reduction and produced regions of long haplotypes.
In this study, we have implemented our SGS method to identify regions of complete, or almost complete selective sweeps across the human genome. The original SGS algorithm was introduced by Thomas et al.14 In this study, we incorporate an error model to provide additional robustness to possible genotyping error in the data. We apply this new algorithm to investigate recent adaptive evolution in populations from three continents, using the second-generation HapMap data sets. Our SGS approach concentrates on identifying the longest runs of heterozygous sharing within populations, considering outlier regions as areas of possible selection. We compare our putative regions of selection with those identified by other methods and note those that are novel and replicated, as well as provide an ontological analysis of the genes in these regions, and finally list those that have been related to disease phenotypes.
Materials and methods
Data sets
We retrieved HapMap phase II October 2008 build release 24 genotype data sets,15 with alleles expressed in the forward strand of the reference human genome (NCBI build 36/UCSC hg18). We analyzed three populations: 90 unrelated individuals from two Asian panels from Tokyo, Japan and Beijing, China (ASN); 60 unrelated parents from 30 Utah trios with ancestry from Northern and Western Europe (CEU); and 60 unrelated parents from 30 Yoruba trios from Ibadan, Nigeria (YRI). The SNP genotyping data sets included over 3.7 million markers in each population: 3 879 488 SNPs (ASN); 3 849 034 SNPs (CEU); and 3 762 311 SNPs (YRI).
Shared genomic segment analysis
Consider s SNPs (with alleles coded as 1 and 2) genotyped on n individuals. Define ni11, ni22 as the number of individuals that are homozygous for alleles 1 and 2 at locus i, respectively. Define the variable Si as Si = n − min ,(n11i, n22i), which indicates the number of individuals from the total n that share at least one allele identical-by-state (IBS) at locus i. Alleles are considered IBS if they are the same type, irrespective of whether they are inherited from a recent ancestor. Missing genotypes are treated as heterozygote. Define Ri(n) as the length of the longest tract of consecutive loci containing the ith SNP at which Si=n, that is, where all of the n individuals share at least one allele IBS.
Equalizing the population sample sizes
Because of varying sample sizes in each population, we used a re-sampling method to generate consistent sample sizes of 45 individuals for each population. Specifically, we randomly sampled 45 individuals with replacement from a population and performed an SGS analysis to obtain Ri(45). In each population, we repeated the sampling 10 times and averaged the Ri(45) values for all i across the genome 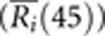 .
.
SGS error model
For genome-wide SNP genotyping assays, the estimated average per genotype accuracy is greater than 99%. However, across many samples and millions of densely genotyped SNPs, the presence of errors becomes non-negligible. For SGS analysis, a single genotyping error creating a homozygous genotype could erroneously terminate a tract of shared SNPs. Assuming a random, uniform, and independent error rate of ɛ per SNP, the number of falsely genotyped SNPs in an interval of length δ SNPs is a Poisson random variable with parameter λ=2δɛ for each individual's genome.16 In this study we use ɛ=0.01, allowing an average of one potentially erroneous non-sharing SNP per 100 SNPs in a SGS run involving n individuals. In other words, we will incorporate SNPs that are not shared by all n individuals (that is, Si<n) into the shared SGS run if this results in a non-sharing error rate of at most 1 SNP per 100. This is equivalent to a maximum genotyping error rate of 0.1% based on our sample size of 45 individuals. We also require no more than two consecutive erroneous SNPs. That is, if the non-sharing between two adjacent runs of SGS comprises no more than two SNPs and if considering them as errors leads to an error rate less than ɛ=0.01, then the two segments are merged. We post-processed the original SGS run lengths, Ri(45), to create a new run length, Oi(45), that incorporates the error modeling (averaging across 10 re-sampled data sets per population, Ōi (45)).
Comparison with other methods of positive selection analysis
We compared our putative regions of natural selection (outlier values of Ōi (45) for each population) to those published using different population genetic methods (iHS4, 6, XP-EHH5, 6, CLR9, 10, and CMS13) and HapMap data sets.
Gene ontology analysis
The BiNGO tool17 (http://www.psb.ugent.be/cbd/papers/BiNGO/) was used to perform a Gene Ontology (GO) analysis on genes residing in potential regions of selection from each population. A binomial test was used to compute P-values, and the Benjamini–Hochberg false discovery rate method was applied to account for multiple testing. We selected those GO terms that include at least four genes, with P-value cut-off of 0.05, and Benjamini–Hochberg false discovery rate q-value cut-off of 0.2. Furthermore, we retained those GO terms in the leaf node, when there are multiple GO terms account for the same cluster of genes.
Results
We performed SGS with our error model in each of the three populations described above: ASN, CEU, and YRI. The mean physical lengths of the SGS distribution (basepairs determined from Ōi (45)) for these populations were 2.1 kb for ASN, 2.02 kb for CEU, and 1.83 kb for YRI. It is known that both CEU and ASN have experienced recent population bottlenecks, whereas the YRI population may have experienced less recent selective forces, and consequently the average SGS sharing within ASN and CEU are longer than that seen in YRI.
Outlier regions were identified as those residing in the extreme tail of the distribution for each population. Based on the shapes of SGS length distribution, we determined SGS run length thresholds of 500 SNPs for ASN, 350 SNPs for CEU, and 100 SNPs for YRI, which correspond to approximately the top 0.1–0.2% of each distribution. Figure 1 shows the Ōi (45) SGS run lengths observed in the three populations. In total, 20 regions were defined as outliers (11 in ASN, 7 in CEU, and 2 in YRI). Sixteen of these regions have additional supporting evidence from the literature,4, 5, 6, 9, 10, 13, 15 and the remaining four are novel, including both of the YRI regions. Table 1 shows the details of all the regions we identified, including the following categories: what population the region was identified in; whether the region had previously been proposed, and all reference sequence genes that reside in the region. The centimorgan distance was calculated based on HapMap provided recombination rate and distance.
Figure 1.

SGS observation in three HapMap populations. Y-axis is Ōi (45). (a) HapMap CEU data set. (b) HapMap ASN data set. (c) HapMap YRI data set. The color reproduction of this figure is available on the html full text version of the manuscript.
Table 1. Regions of extreme sharing identified by SGS.
| Chomosome numbers | Position (MB) (NCBI b36/hg18) | Size (cM) | Methods | Genes in the region | |
|---|---|---|---|---|---|
| CEU | |||||
| Chr 1 | 35.449 | 35.717 | 0.075 | iHS, CLR, XP-EHH | ZMYM4, KIAA0319 L |
| Chr 1 | 143.67 | 143.841 | 0.485 | novel | PDE4DIP, SEC22B |
| Chr 5 | 109.638 | 110.131 | 0.115 | CLR, XP-EHH, CMS | TMEM232, SLC25A46 |
| Chr 7 | 151.348 | 151.774 | 0.132 | XP-EHH | GALNT11, MLL3, FABP5P3 |
| Chr 10 | 74.416 | 75.139 | 0.138 | CLR, CMS | P4HA1, ECD, NUDT13, FAM149B1, MRPS16, TTC18, ANXA7, ZMYND17, PPP3CB, SYNP02L, AGAP5 |
| Chr 18 | 64.802 | 64.963 | 0.186 | CLR, XP-EHH | CCDC102B |
| Chr 21 | 10.024 | 13.458 | 0.6 | XP-EHH | BAGE2, BAGE |
| YRI | |||||
| Chr 1 | 143.67 | 143.755 | 0.219 | novel | PDE4DIP |
| Chr 21 | 0.019 | 0.064 | 0.637 | novel | TPTE, BAGE2, BAGE |
| ASN | |||||
| Chr 1 | 92.605 | 92.94 | 0.091 | iHS | RPAP2, GFI1, EVI5 |
| Chr 1 | 103.215 | 103.416 | 0.079 | iHS, CLR | COL11A1 |
| Chr 2 | 17.421 | 17.897 | 0.149 | XP-EHH | VSNL1, SMC6, GEN1 |
| Chr 2 | 72.211 | 73.016 | 0.278 | CLR, XP-EHH | CYP26B1, EXOC6B, SPR, EMX1 |
| Chr 2 | 177.337 | 177.599 | 0.217 | XP-EHH | |
| Chr 3 | 25.905 | 26.376 | 0.386 | iHS, XP-EHH | |
| Chr 3 | 48.665 | 49.708 | 0.04 | CLR | CELSR3, PRKAR2A, NCKIPSD, IP6K2, SLC25A20, P4HTM, WDR6, DALRD3, NDUFAF3 IMPDH2, QRICH1, QARS LAMB2, CCDC71, KLHDC8B, CCDC36, USP4, GPX1, RHOA AMT, TCTA, DAG1, APEH, BSN MST1, RNF123 |
| Chr 5 | 117.375 | 117.697 | 0.183 | CLR, XP-EHH | |
| Chr 7 | 142.266 | 142.397 | 0.192 | novel | EPHB6, TRPV6, TRPV5, KEL |
| Chr 15 | 61.568 | 62.069 | 0.339 | CLR, XP-EHH | USP3, FBXL22, HERC1, DAPK2 |
| Chr 18 | 30.411 | 30.602 | 0.172 | CLR | DTNA |
The iHS result is from the study of Voight et al4 and HapMap II.15 The CLR result is from the study of Williamson et al10 and Pickrell et al6 The XP-EHH result is from the study of Sabeti et al12 and Pickrell et al6 The CMS result is from the study of Grossman et al.13
All of regions are identified in the same population as in the cited literature. The underlined genes have previously been identified to be associated with human disease or trait. The genomic positions are according to NCBI Build 36/UCSC hg18. The centimorgan distance was calculated based on HapMap provided recombination rate and distance.
In what follows we will assess the novel regions we identified as outliers in our SGS analysis. A region on chromosome 1, containing the gene PDE4DIP, is a novel region of potential selection that were identified in both CEU and YRI populations, and hence this region may correspond to selection before the ‘out of Africa' event. In addition, the PDE4DIP gene has found to be associated with esophageal squamous cell carcinoma (SCC).18
A juxtacentromeric region on chromosome 21 that have previously been identified as a potential region of selection in CEU by XP-EHH,5 was identified in both CEU and YRI by our SGS method. The common region across the two populations contains two genes: BAGE2 and BAGE. The BAGE (B melanoma antigen) gene family has been shown to be under selective pressure, with BAGE proteins considered as a potential birth site of novel genes.19 In our analysis, the region extended to TPTE gene in the YRI population. Both TPTE and BAGE2 are associated with Robertsonian Down syndrome.20
The chromosome 7 region is a novel identification in ASN, but has been identified as a region with very strong positive selection in a study using European–American sequencing samples.21 Four genes in the region, EPHG6, TRPV5, TRPV6, and KEL, were suggested to be under demographical selection in European–Americans. The genes EPHG6 and KEL were identified as candidate genes for positive selection, according to evidence of an excessive amount of rare alleles and high frequency derived alleles. A skewed polymorphism was detected in TRPV6, which also suggests of recent positive selection.22 Our results extend these findings to the ASN population. It is possible that the migration of modern humans out of Africa into Asia have been accompanied by genetic adaptations similar to that experienced by Europeans.
A total of 50 genes from regions of selection in the ASN population, 23 genes from CEU, and 4 genes from YRI were used for ontological analysis. It should be noted that there is a lack of power because of an inadequate number of genes in ontology categories for all three populations. Significant GO analysis results were only found in the ASN population, as is illustrated in Table 2.
Table 2. Gene Ontology Analysis among Genes in the Regions of Selection in ASN.
| GO ID | Term | Gene Count | Fold Increase | P-value | Benjamini q-value |
|---|---|---|---|---|---|
| Biological process | |||||
| 00090066 | Regulation of anatomical structure size | 4/50 | 4.367 | 0.0098 | 0.1447 |
| 00006519 | Cellular amino acid and derivative metabolic process | 4/50 | 3.655 | 0.0156 | 0.1662 |
| 00009628 | Response to abiotic stimulus | 4/50 | 3.609 | 0.0219 | 0.1771 |
| 00030182 | Neuron differentiation | 4/50 | 3.488 | 0.0290 | 0.1805 |
| 00000902 | Cell morphogenesis | 4/50 | 3.143 | 0.0094 | 0.1447 |
| 00035467 | Negative regulation of signaling pathway | 4/50 | 2.832 | 0.0051 | 0.1364 |
| 00043065 | Positive regulation of apoptosis | 4/50 | 2.588 | 0.0292 | 0.1805 |
| 00006461 | Protein complex assembly | 5/50 | 2.821 | 0.0106 | 0.1447 |
| 00022008 | Neurogenesis | 5/50 | 2.310 | 0.0257 | 0.1805 |
| 00033554 | Cellular response to stress | 5/50 | 2.249 | 0.0232 | 0.1805 |
| 00055114 | Oxidation reduction | 5/50 | 2.210 | 0.0274 | 0.1805 |
| 00065003 | Macromolecular complex assembly | 5/50 | 2.116 | 0.0323 | 0.1847 |
| 00007155 | Cell adhesion | 5/50 | 2.014 | 0.0387 | 0.1953 |
| 00010646 | Regulation of cell communication | 7/50 | 1.735 | 0.0305 | 0.1805 |
| 00009653 | Anatomical structure morphogenesis | 9/50 | 2.115 | 0.0039 | 0.1364 |
| 00065008 | Regulation of biological quality | 10/50 | 1.855 | 0.0058 | 0.1364 |
| 00016043 | Cell component organization | 13/50 | 1.451 | 0.0121 | 0.1508 |
| 00009987 | Cellular process | 33/50 | 1.008 | 0.0029 | 0.1364 |
| Molecular function | |||||
| 00016874 | Ligase activity | 4/50 | 2.941 | 0.0230 | 0.1271 |
| 00008233 | Peptidase activity | 5/50 | 2.709 | 0.0154 | 0.1149 |
| 00016491 | Oxidoreductase activity | 5/50 | 2.251 | 0.0313 | 0.1341 |
| 00005509 | Calcium ion binding | 6/50 | 2.960 | 0.0052 | 0.0792 |
| 00030554 | Adenyl nucleotide binding | 8/50 | 1.567 | 0.0465 | 0.1507 |
| 00032555 | Purine ribonucleotide binding | 9/50 | 1.507 | 0.0432 | 0.1489 |
| 00046872 | Metal ion binding | 15/50 | 1.226 | 0.0460 | 0.1507 |
| 00043169 | Cation binding | 15/50 | 1.213 | 0.0499 | 0.1507 |
| 00003824 | Catalytic activity | 21/50 | 1.273 | 0.0084 | 0.0901 |
Discussion
We have studied three diverse populations using the HapMap phase II data set using an SGS algorithm that incorporates an error model. On the basis of outlier regions, we detected 16 genomic regions that have previously been proposed as under the influence of natural selection, demonstrating the capacity of SGS and lending more weight to those regions. In addition we have proposed four novel regions of potential selection, one of which was observed in two of the three populations studied. Because regions of selection likely contain functionally significant polymorphisms for human genes, the mapping of regions illustrating selection could aid in the effort to identify important genetic factors in recent human development or disease resistance. Also, variants that are not under direct selection, but hitch-hiking due to their proximity to the selection point will also occur on the shared segment, and may become increasingly important genetic factors in complex, polygenic diseases.
In the regions identified, we found genes from functional pathways that are known to be under selection, including pathways of immune response (KEL and P4HTM), tumor antigen (BAGE), and spermatogenesis (CYP26B1 and LAMB2), as well as sensory perception (COL11A1). In our ontological analysis, it is shown that biological processes, such as signal transduction, protein metabolism and modifications, as well as cell motility are enriched, similar as it was illustrated in other selection studies.4, 13 Furthermore, among the genomic regions identified by SGS as potential for selection, there are several genes that have been identified to be associated with complex disorders. For example, four musculoskeletal disease genes (COL11A1, DAG1, DTNA, and SLC25A20), and genes associated with disorder of oxidative phosphorylation system (MRPS16 and NDUFAF3). Several genes associated with brain development and neurological diseases were also found to be located in the SGS regions, which include 14 genes shared among ASN samples (AMT, BSN, MST1, CELSR3, CYP26B1, EMX1, EPHB6, EVI5, GFI1, GPX1, LAMB2, RHOA, VSNL1, and WDR6); and KIAA0319 L and SLC25A46 in CEU; as well as one gene (PDE4DIP) found in both CEU and YRI. In addition, there are several cancer disease genes located in our regions of selection, such as tumor suppressor gene (ANXA7), colorectal cancer gene (MLL3), and genes associated with multiple sclerosis (GFI1 and EVI5), as well as genes or gene products that are associated with multiple cancers, such as GPX1, RHOA, and APEH. In addition, the 3p21.31 region identified in ASN is harbored within a region associated with Crohn's disease. These observations are consistent with of the hypothesis that genes involved in common disease could often be targets of selection.
Our SGS method is best suited to identifying complete, or near complete, selective sweeps, and therefore our results are most consistent with those from CLR and XP-EHH, which also have strengths for such sweeps. On the other hand, our results were less consistent with iHS, because the iHS method is designed to detect regions of selection before fixation or that have undergone balancing selection. We note that SNP ascertainment bias is an issue that hinders all methods from detecting regions of selection. Such bias in SNP selection leads to genotype data that is deviated from its normal level of variability, distribution of allele frequencies, and levels of linkage disequilibrium. This issue will be addressed when sequence data are available for each population.
In conclusion, we have demonstrated that our SGS method is capable of identifying regions of selection using dense SNP data set. Given that functional allelic variants are subject to purifying selection, this ubiquitous selection creates rare variants for functional alleles, which is considered to be a driving force of complex traits. The ability of SGS in detecting selection regions aids the search for disease susceptibility loci.
Acknowledgments
We would like to acknowledge Professors Jon Seger, Frederick Adler, Henry Harpending, Alan Rogers and Dr Chad Huff for useful discussions. Research was supported by grants NIH R01 GM81417 and DOD W81XWH-07-01-0483 to Alun Thomas, and R01CA134674 to Nicola Camp.
The authors declare no conflict of interest.
References
- The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CS, Thomas DJ, Eberle MA, et al. Genomic regions exhibiting positive selection identified from dense genotype data. Genome Res. 2005;15:1553–1565. doi: 10.1101/gr.4326505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Kodama G, Baldi P, Moyzis RK. Global landscape of recent inferred Darwinian selection for Homo sapiens. Proc Natl Acad Sci USA. 2006;103:135–140. doi: 10.1073/pnas.0509691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A Map of Recent Positive Selection in the Human Genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Coop G, Novembre J, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM. Constructing genomic maps of positive selection in humans: Where do we go from here. Genome Res. 2009;19:711. doi: 10.1101/gr.086652.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Schaffner SF, Fry B, et al. Positive Natural Selection in the Human Lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, Bustamante C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005;15:1566–1575. doi: 10.1101/gr.4252305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, Nielsen R. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007;3:e90. doi: 10.1371/journal.pgen.0030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. The genetical structure of populations. Ann Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Shylakhter I, Karlsson EK, et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010;327:883. doi: 10.1126/science.1183863. [DOI] [PubMed] [Google Scholar]
- Thomas A, Camp NJ, Farnham JM, Allen-Brady K, Cannon-Albright LA. Shared Genomic Segment Analysis. Mapping Disease Predisposition Genes in Extended Pedigrees Using SNP Genotype Assays. Ann Hum Genet. 2008;72:279–287. doi: 10.1111/j.1469-1809.2007.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Lowe JK, Stoffel M, et al. Whole population, genome-wide mapping of hidden relatedness. Genome Res. 2009;19:318–326. doi: 10.1101/gr.081398.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Shimada H, Kuboshima M, Shiratori T, et al. Serum anti-myomegalin antibodies in patients with esophageal squamous cell carcinoma. Int J Oncol. 2006;30:97–103. [PubMed] [Google Scholar]
- Ruault M, Ventura M, Galtier N, et al. BAGE genes generated by juxtacentromeric reshuffling in the Hominidae lineage are under selective pressure. Genomics. 2003;81:391–399. doi: 10.1016/s0888-7543(03)00025-9. [DOI] [PubMed] [Google Scholar]
- Shaw SW, Chen CP, Cheng PJ, et al. Gene dosage change of TPTE and BAGE2 and breakpoint analysis in Robertsonian Down syndrome. J Hum Genet. 2008;53:136–143. doi: 10.1007/s10038-007-0229-z. [DOI] [PubMed] [Google Scholar]
- Akey JM, Eberle MA, Rieder MJ, et al. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2004;2:1591–1599. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soejima M, Tachida H, Koda Y. Sequence Analysis of Human TRPV6 Suggests Positive Selection Outside Africa. Biochem Genet. 2009;47:147–153. doi: 10.1007/s10528-009-9222-x. [DOI] [PubMed] [Google Scholar]